
Termodin amica fuera del equilibrio: Igualdad de Jarzynski ...

Capıtulo 3
Termodinamica y mecanicaestadıstica
Alberto Carraminana, 16 de octubre de 2018.
3.1. Las leyes de la termodinamica
La termodinamica estudia el comportamiento de sistemas macroscopicoscon su entorno. Estos sistemas se consideran acotados por una frontera quedefine su tamano y funciona como interfaz con los alrededores. El tamanodel sistema puede cuantificarse a traves de parametros dimensionales, co-mo el volumen, o el numero de partıculas que lo componen. Las partıculasque componen el sistema tienen manifiestacion en el comportamiento ma-croscopico a traves de variables como la densidad o la presion.
Las leyes de la termodinamica establecen un marco formal para el estu-dios de sistemas macroscopicos basado en el concepto de equilibrio termo-dinamico:
Un sistema esta en equilibrio termodinamico cuando las variablesque lo describen no varıan con el tiempo.
Se desprende que dos sistemas estan en equilibrio termodinamico mutuo sial establecer contacto en ellos las variables termodinamicas de cada uno novarıan. Por construccion, la termodinamica se aplica a sistemas en equilibrioy se basa en las leyes descritas someramente a continuacion.
1

Ley cero - definicion de temperatura
La ley cero define el equilibrio termodinanico entre sistemas a traves deconjuntos, o clases de equivalencia:
Dados tres sistemas termodinamicos A, B y C, si los sistemas Ay B estan cada uno en equilibrio termodinamico con C, entoncesA y B se encuentran en equilibrio entre ellos.
Partiendo de esta ley, se distinguen las distintas clases de sistemas en equili-brio definiendo el concepto de temperatura. Los estados de equilibrio propiode cada uno de dos sistemas A y B estan descritos por las variables XA, YAy XB, YB, constantes en el tiempo, siendo la temperatura una funcion deestas variables, T = f(X,Y ). La funcion f es particular a cada sistema. Elequilibrio entre sistemas se da cuando los valores numericos de T coinciden,
TA = fA(XA, YA) = TB = fB(XB, YB).
Cada funcion f define la ecuacion de estado del sistema respectivo, la cualpuede depender de muchas variables. Un punto en el plano X,Y , o es-pacio multi-dimensional correspondiente, define un estado del sistema. Unvalor de temperatura corresponde una curva isotermica en el plano X,Y .Las variables X,Y, ... se conocen como variables de estado; para un gasse consideran comunmente la presion y el volumen. Las curvas isotermicaspueden ordenarse para establecer una escala de temperaturas.
En particular, la ecuacion de estado del gas ideal clasico es,
PV = NkT, (3.1)
con P y V la presion y el volumen, las dos variables de estado; N es elnumero de partıculas que constituyen el gas, k la constante de Boltzmann1,y T la temperatura, definida por esta relacion.
Primera ley - conservacion de la energıa
El estado termodinamico de un sistema puede caracterizarse mediante laenergıa interna, E, que se mantiene constante para un sistema aislado. Cuan-do el sistema interacciona con sus alrededores, el cambio en la energıa internaesta dado por,
dE = dQ+ dW , (3.2)
1k ' 1.380650× 10−16erg K−1
2

donde Q es el calor absorbido por el sistema y W el trabajo realizado sobreel mismo como resultado de la variacion de algunos de sus parametros. Elsımbolo barrado d indica que las diferenciales respectivas no son necesaria-mente exactas. Una diferencial es exacta si se anula al integrarla sobre unatrayectoria cerrada, indicando que la variable relacionada es una variable deestado.
Los terminos en la expresion (3.2) ameritan discusion. El trabajo termo-dinamicoW describe interacciones del sistema con sus alrededores, alterandosu estado y energıa interna. El trabajo termodinamico puede referirse a unainteraccion mecanica, electrica, quımica, etc... que tienen expresiones de ti-po dW = ~F · d~λ, siendo F una fuerza generalizada y λ un desplazamientogeneralizado, que forman una pareja de variables termodinamicas conjuga-das, de las cuales una es intensiva, es decir independiente de las dimensionesdel sistema, y la otra extensiva, dependiente de las dimensiones del sistema.Ejemplos comunes son:
el trabajo mecanico ejercido al comprimir un gas, dW = −PdV ,siendo P la presion y V el volumen;
el trabajo electromagnetico ejercido al aumentar la magnetizacion,~M , de un sistema en un campo magnetico, ~H, dado por dW = ~H · d ~M .
De manera analoga, la polarizacion ~P de un material puede ser mo-dificada por un campo electrico ~E mediante un trabajo dado pordW = ~E · d~P .
el trabajo quımico relacionado a una reaccion quımica que alterael numero de partıculas del tipo mediante un trabajo dW = µdN,siendo µ la energıa necesaria para generar una partıcula, conocidacomo el potencial quımico.
Estas relaciones presuponen que estas interacciones se dan mediante sucesio-nes de estados de equilibrio, en lo que se denominan procesos cuasi-estaticos.
El calor es por defecto una diferencial inexacta y no puede representaruna variable de estado. Sin embargo, el trabajo W es una diferencial exactaen procesos adiabaticos, dQ = 0, siendo que la energıa interna es una variablede estado por definicion. La forma en que un sistema absorbe o emite calorpuede ser descrita definiendo los calores especıficos. Estos coeficientes puedendescribir procesos que mantienen alguna variable de estado fija, como elvolumen o la presion en el caso de un gas,
Cv ≡(dQ
dT
)V, Cp ≡
(dQ
dT
)p, (3.3)
3

respectivamente. Para un gas ideal (ec. 3.1) podemos relacionar estas doscantidades, considerando la primera ley de la termodinamica (ec.3.2),
Cv =
(dQ
dT
)V
=dE
dT, CP =
dE
dT+ P
(dV
dT
)P
= Cv +Nk . (3.4)
Si definimos el ındice adiabatico como el cociente de calores especıficos,γ ≡ Cp/Cv, podemos obtener la energıa interna de un gas ideal,
γ = 1 +Nk
Cv= 1 +
Nk
dE/dT⇒ E =
NkT
γ − 1.
Notese que la energıa interna de un gas ideal es independiente del volumen.
Segunda ley - entropıa
A traves del concepto de calor se puede definir la entropıa, S, que es unavariable de estado,
dQ = TdS . (3.5)
La segunda ley de la termodinamica establece que:
en un sistema cerrado la entropıa nunca decrece.
Equivalentemente,dS ≥ 0 . (3.6)
De manera muy generica, esta asercion incluye al Universo en su totalidad,un sistema cerrado cuya entropıa debe ser creciente con el tiempo. Procesosque cumplen dS = 0 en un sistema cerrado son reversibles; cualquier otroproceso en un sistema cerrado implica dS > 0 y es irreversible. Como conse-cuencia de la segunda ley, un estado de equilibrio corresponde con maximaentropıa y mınima energıa. Este principio establece que el equilibrio termo-dinamico entre sistemas requiere no solo el equilibrio termico, TA = TB, sinotambien el equilibrio mecanico, PA = PB. Si ademas consideramos interac-ciones de caracter quımico, tenemos el requerimiento adicional µA = µB.
De la definicion (3.5) podemos encontrar la expresion termodinamicapara la entropıa de un gas ideal,
S = NklnT/(γ − 1) +Nk lnV + S0 ,
con S0 una constante de integracion.
4

Tercera ley - el estado de mınima energıa
La tercera ley establece que la entropıa de un sistema se anula en el estadodefinido por, (
∂E
∂S
)V,N
= 0 .
Este estado corresponde al de mınima energıa interna, el cual se emplea paradefinir una escala absoluta de temperaturas, medida comunmente en gradosKelvin, en la que el cero absoluto corresponde con el estado en el que laentropıa se anula. Sin recurrir a una descripcion estadıstica, Nernst enuncioen 1906 el siguiente postulado:
La isoterma de un sistema a temperatura cero corresponde conuna de sus adiabaticas.
El postulado de Nernst representa un enunciado alterno de la tercera ley dela termodinamica2.
Existe un conflicto entre la expresion para la entropıa de un gas idealy la existencia de una escala absoluta de temperatura, en la que S → 0para T → 0. La ecuacion de estado del gas ideal clasico pierde validez atemperaturas bajas, que requieren una descripcion cuantica (§3.3).
3.2. Mecanica estadıstica
La mecanica estadıstica interpreta las variables macroscopicas como la mani-festacion colectiva de las propiedades individuales de las partıculas que cons-tituyen un sistema. Es necesaria hacer una descripcion estadıstica de losmicro-estados de un sistema y ligarla con el formalismo termodinamico.
3.2.1. Descripcion estadıstica de un sistema
Estado de un sistema
La descripcion de un sistema puede hacerse conforme a las leyes de la mecani-ca clasica o la mecanica cuantica, considerando que formalismo es el correcto,o el mas practico. En mecanica clasica el estado de una partıcula queda espe-cificado por su posicion y momento, ~x(t), ~p(t), en un instante t, y las fuer-zas que sobre ella actuan. Para un sistema con un numero N de partıculas,requerimos el conjunto de posiciones y momentos, ~xj(t), ~pj(t); j = 1, ..., N,
2para mas detalles consultar los libros de termodinamica de Garcıa-Colın y Callen.
5

y las interacciones entre ellas para poder especificar completamente el es-tado del sistema. Esta descripcion es en principio posible pero impractica,siendo N un numero enorme.
En mecanica cuantica el estado de un sistema se describe mediante lafuncion de onda, ψ(~x1, . . . , ~xN ), dependiente de las 3N coordenadas de laspartıculas que lo constituyen, y el hamiltoniano que describe sus interac-ciones. Frecuentemente la funcion de onda puede especificarse mediante va-riables alternas, como son los numeros cuanticos. Ası, un sistema formadopor cuatro osciladores armonicos unimensionales esta suficientemente des-crito con cuatro numeros cuanticos n1, n2, n3, n4, siendo cada ni un nume-ro natural. Notese que distintos estados pueden tener una misma energıa,E = (n1 +n2 +n3 +n4 + 2)hω. El conjunto de funciones de onda ψn1,...,n4es enumerable, facilitando la descripcion estadıstica del sistema.
Ensemble estadıstico
No todos los micro-estados son consistentes con un estado termodinamicodefinido. En particular, consideramos la energıa interna del sistema comouna limitacion a los micro-estados que son accesibles dada la restriccionmacroscopica. Los estados que cumplen con las restricciones macroscopicasse denominan estados accesibles y definen los que se conoce como ensembleestadıstico. En el caso simplista de los osciladores 1-D, al asignar a la energıainterna a un valor dado, por ejemplo E = 5hω, se definen como ensembleestadıstico los micro-estados que satisfacen la restriccion n1+n2+n3+n4 = 3.
Espacio fase
En general resulta ventajoso localizar los estados del sistema dentro delespacio-fase formado por las posiciones y momentos que pueden tener laspartıculas. Esta descripcion permite mudar el enfoque estadıstico de laspartıculas individuales al espacio-fase, el cual se discretiza en celdas dedimensiones finitas, ∆x∆p = h0 (figura 3.1), de forma congruente con elprincipio de incertidumbre que rige en mecanica cuantica, el cual impideel conocimiento perfecto de la posicion y el momento de una partıcula3,∆x∆p ≥ h. Mas restrictivo aun, el principio de exclusion de Pauli prohi-be que dos partıculas tengan el mismo estado dentro de la indeterminacioncuantica. En el caso cuantico se emplean celdas de espacio fase de dimen-siones h0 → h. El lımite clasico h0 → 0, corresponde con un espacio-fasecontinuo.
3h = h/2π con h ' 6.626× 10−27erg s la constante de Planck.
6

Figura 3.1: Particion del espacio enceldas de tamano finito, ∆x∆p = h0;la descripcion cuantica se hace conh0 = h, mientras que h0 → 0 es ellımite clasico.
Dos ensembles comunes son el canonico, donde la restriccion esta en laasignacion de la energıa interna a un valor E; y el ensemble gran canonico,que restringe energıa y numero de partıculas, E,N, simultaneamente.
3.2.2. Definicion estadıstica de la entropıa
Sea Ω(E) el numero de estados accesibles correspondientes a una energıainterna E. Podemos establecer una conexion con la termodinamica, intro-duciendo la definicion estadıstica de la entropıa,
S(E) ≡ k ln Ω(E) , (3.7)
con k la constante de Boltzmann. Esta definicion es congruente con la se-gunda y tercera ley: Ω es, de manera natural, una funcion creciente de E;y existe por lo general un solo estado de mınima energıa, Ω(Emin) = 1 ⇒S(Emin) = 0. Podemos ahora considerar la dependencia del numero de es-tados accesibles en funcion de E y de las variables extensivas x. Esta de-pendencia, Ω = Ω(E;x = V,N, . . .), conecta la descripcion estadıstica conla termodinamica. De la primera ley obtenemos,
T =∂E
∂S⇒ β(E) ≡ 1
kT=∂ ln Ω
∂E; (3.8)
y de manera similar,
P = −∂E∂V
=1
β
∂ ln Ω
∂V, µ =
∂E
∂N= − 1
β
∂ ln Ω
∂N, . . . .
En un sistema con energıa interna E y Ω(E) estados accesibles, pode-mos definir como Ω(E, y) el numero de aquellos que cumplen una restric-cion adicional, y = y. La probabilidad de medir y = y esta dada por
7

P (y) = Ω(E; y)/Ω(E), y el valor promedio de la variable y es:
y =1
Ω(E)
∑
y Ω(E, y) . (3.9)
En la practica resulta mas conveniente calcular variables termodinamicasempleando la funcion de particion, definida mas adelante.
3.2.3. La funcion de particion
Podemos considerar el equilibrio entre dos sistemas desde el punto de vistaestadıstico. Sean A y A′ dos sistemas cerrados de energıas respectivas Ey E′ en contacto termico. El sistema conjunto A(0) = A + A′ es tambiencerrado, y tiene energıa total E(0) = E + E′. Los numeros respectivos deestados accesibles son Ω(E) y Ω′(E′), por lo que el sistema conjunto tendraΩ0(E+E′) = Ω(E)Ω′(E′) estados. Por tanto la probabilidad de que A tengaenergıa E sera de la forma,
P (E) = C Ω(E)Ω′(E0 − E),
con C una constante de normalizacion sobre todos los estados (energıas)posibles. Esta probabilidad es maxima si su logaritmo tambien lo es,
d lnP (E)
dE= 0 ⇒ d ln Ω(E)
dE+d ln Ω′(E′)
dE= 0, (3.10)
que, considerando E′ = E0 − E, implica
d ln Ω(E)
dE− d ln Ω′(E′)
dE′= β(E)− β′(E′) = 0,
es decir T = T ′. La relacion (3.10) equivale al principio de maxima entropıa.Si ahora suponemos que A′ es un reservorio de energıa, en el sentido
de que sus dimensiones son mucho mayores que las de A (figura 3.15), enparticular E′ E, la probabilidad de que el sistema A este en un estado res de la forma
Pr = C Ω′(E(0) − Er) , (3.11)
con C constante de normalizacion tal que∑rPr = 1. Podemos hacer la
expansion de ln Ω′(E′) alrededor de E(0),
ln Ω′(E(0) − Er) ' ln Ω′(E(0))−[∂ ln Ω′
∂E′
]0Er . (3.12)
8

Figura 3.2: Intercambiode energıa entre un sis-tema termodinamico, co-mo una taza de cafe, y unreservorio de energıa querepresenta el entorno, co-mo la cafeterıa, hasta al-canzar el equilibrio ter-modinamico.
El equilibrio termodinamico entre A y A′ se establece cuando
∂ ln Ω
∂E=∂ ln Ω′
∂E′⇒ β = β′. (3.13)
es decir que T se iguala con la temperatura del reservorio. Se tiene entoncesPr ∝ e−βEr , que nos permite escribir la probabilidad del estado r como
Pr =e−βEr∑
r′e−βEr′
. (3.14)
Definimos aquı la funcion de particion del ensemble canonico como:
Z ≡∑r
e−βEr . (3.15)
La funcion de particion permite calcular variables termodinamicas intensivasdirectamente de Z = Z(β, x),
∂ lnZ
∂β=
1
Z
∑r
(−Er) e−βEr ,∂ lnZ
∂x=
1
Z
∑r
(−β∂Er
∂x
)e−βEr = −βX ,
de donde
E = −∂ lnZ
∂β, X = − 1
β
∂ lnZ
∂x⇒ P = +
1
β
∂ lnZ
∂V, µ = − 1
β
∂ lnZ
∂N. (3.16)
La expresion 3.16 viene dando la ecuacion de estado del sistema. Una relacionutil entre la funcion de particion y la entropıa se obtiene identificando4
d lnZ =∂ lnZ
∂βdβ +
∂ lnZ
∂xdx = −Edβ − βXdx
4entendiendo que Xdx se refiere a∑
Xdx = −PdV + µdV + . . .
9

resultando en
S = k(lnZ + βE)⇒ F = E − TS = − lnZ ,
donde F es la funcion libre de Helmholtz.La funcion de particion del sistema compuesto A(0) = A′ + A esta da-
da por Z(0) = Z ′Z, el producto de las funciones de particion de sus sub-sistemas. Este resultado es congruente con el hecho de que tanto la energıainterna como la entropıa son propiedades aditivas.
3.2.4. Gas ideal clasico
Funcion de particion y relaciones termodinamicas
Un gas ideal consiste en un conjunto de N partıculas libres; la energıa delsistema esta dada por la suma de las energıas cineticas individuales:
E =N∑=1
|~p|2
2m. (3.17)
Al hacer h0 → 0, la sumatoria sobre estados tiende a una integral sobre losvalores de ~r y ~p de cada una de las partıculas,
Z =
∫exp
− β
2m
N∑=0
p2
d3Nrd3Np
h3N0, (3.18)
que se convierte en un producto de N integrales identicas, resultando en lafuncion de particion:
Z = V N(
2πmkT
h20
)3N/2
= ζN , (3.19)
donde ζ = V(2πmkT/h20
)3/2es la funcion de particion correspondiente a
una partıcula. La relacion (3.16) da la ecuacion de estado del gas ideal,
P = NkT/V .
Al derivar con respecto a β obtenemos
E = 3N/2β = (3/2)NkT ,
10

la energıa interna de un gas ideal clasico de N partıculas, que es indepen-diente del volumen. Para la entropıa obtenemos
S = Nk
[lnV +
3
2lnT + σ
]con σ = 3/2 ln
(2πmk
h20e
). (3.20)
Esta expresion presenta dos inconvenientes: uno es el no comportarse comouna variable extensiva - la paradoja de Gibbs. Supongamos dos sistemas,A y B, donde B duplica al sistema A, es decir VB = 2VA y NB = 2NA.Siendo la entropıa una variable extensiva deberıa cumplirse SB = 2SA, locual no es consistente con (3.20). Gibbs obtuvo la solucion a esta paradojaal considerar que el intercambio de partıculas microscopicas identicas nomodifica el estado del sistema y dividir la funcion de particion por el numerode permutaciones entre partıculas, igual a N !; es decir,
Z =V N
N !
(2πmkT
h20
)3N/2
. (3.21)
Usando la aproximacion de Stirling, lnN ! ' N lnN −N , Gibbs obtuvo
S = Nk
[ln
(V
N
)+
3
2lnT + σs
], (3.22)
donde σs = σ+1. Esta expresion satisface SB = 2SA, resolviendo la paradojade Gibbs. El segundo problema, irresoluble en un contexto clasico, es laincongruencia con la tercera ley de la termodinamica, ya que S → −∞ paraT → 0. El lımite de bajas temperaturas requiere un tratamiento cuantico.
La distribucion de Maxwell-Boltzmann
La funcion de distribucion de Maxwell-Boltzmann describe la probabilidadque tiene una partıcula de tener momento entre ~p y ~p+d~p, y posicion entre ~r y~r+d~r. Esta probabilidad se relaciona directamente con la probabilidad de unestado r, dada por Pr ∝ e−βEr . Podemos definir esta funcion de distribucion,f(~r, ~p), que describe cuantas partıculas tienen momento alrededor de ~p yposicion alrededor de ~r, que para el gas ideal adquiere la expresion,
f(~r, ~p) d3r d3p =n
(2πmkT )3/2e−p
2/2mkT d3r d3p , (3.23)
que solo depende de p = |~p|. La funcion de distribucion de Maxwell-Boltzmannse define normalizada al numero de partıculas,
∫f(p)d3r d3p = N . Es comun
definirla alternativamente en terminos de la velocidad ~v de las partıculas,
f(~v) d3v = n
(m
2πkT
)3/2
e−mv2/2kT 4πv2dv ,
11

donde se emplea el hecho de que f solo depende de la magnitud de la ve-locidad y d3v = 4πv2dv. La distribucion de Maxwell-Boltzmann se empleapara calculos como el de la velocidad promedio de las partıculas, o la pre-sion del gas, donde se considera la fuerza que las partıculas ejercen sobre unelemento de superficie cuya normal al eje z:
P =
∫mv2z f(v) d3v =
1
3nm < v2 > . (3.24)
El lımite cuantico
Un gas ideal puede describirse de manera clasica siempre y cuando la ocu-pacion de las celdas de espacio-fase sea baja. Esta condicion puede escribirsecomo
[∆x]3 [∆p]3 Nh3,
donde [∆x]3 y [∆p]3 indican el rango de posiciones y momentos ocupados porlas N partıculas. Claramente [∆x]3 = V , mientras que [∆p]3 ' (mkT )3/2.La condicion para la aplicabilidad de la estadıstica clasica viene siendo,
T n−2/3 h2/mk , (3.25)
con n = N/V , la densidad numerica. El aire en condiciones ambientalestıpicas cumple n ' 2 × 1019 cm−3, con partıculas de masa m ≈ 29 uma '5× 10−23 g. La ocupacion del espacio fase es baja si T 0.047 K; o, a tem-peratura ambiente, T = 300 K, si n 1025 cm−3, como sucede en nuestraatmosfera. El regimen cuantico se aplica para temperaturas bajas y densi-dades altas.
3.3. Gases cuanticos
3.3.1. Funcion de particion de un ensemble gran canonico
El ensemble canonico no considera el intercambio de partıculas entre el siste-ma A y el reservorio A′. En el ensemble gran canonico el numero de partıcu-las en un estado r no es fijo, pudiendo variar como resultado de las inter-acciones entre ellas. Considerando nuevamente la interaccion de A con unreservorio A′, con la consideracion adicional N ′ N , el sistema compuestoA(0) = A+A′ cumple las dos restricciones:
E(0) = E + E′ , N (0) = N +N ′. (3.26)
12

De manera analoga al procedimiento seguido para el ensemble canonico, seobtiene la funcion de probabilidad del ensemble gran-canonico,
Pr ∝ exp−β(Er − µNr) , (3.27)
donde µ es el potencial quımico del sistema, que corresponde con la energıarequerida para producir una nueva partıcula. La funcion de particion grancanonico esta dada por
Z =∑r
e−β(Er−µNr) .
Podemos relacionar la energıa del sistema y el numero de partıculas con laocupacion n de las celdas de espacio fase:
Er =∑
nε , Nr =∑
n , (3.28)
para obtener:
Pr ∝ exp
∑
−β (nε − µn)
=∏
[exp (−β(ε − µ))]n , (3.29)
La sumatoria de la exponencial sobre estados r nos da la funcion de particion:
Z =∑r
(∏
wn
)=∏
∑n
wn
, (3.30)
donde abreviamos w = exp −β(ε − µ). La primera igualdad indica que lafuncion de particion es la sumatoria sobre todos los estados r del productosobre los numeros de ocupacion correspondientes a cada estado, definido
por el conjunto de numeros de ocupacion n(r)1 , n(r)2 , n
(r)3 , . . .. El termino
de la derecha indica el producto sobre todas las celdas de espacio fase dela sumatoria sobre todos los posibles numeros de ocupacion. Las energıasε dependen de la ubicacion de la -esima celda de espacio-fase, y no de suocupacion, lo cual permite permutar el producto y la suma sobre estados.Consideremos un ejemplo simple, el de un sistema con tres micro-estadosrepresentados por las celdas de espacio fase = a, b, c; estas pueden serocupadas por cero o una partıcula, n = 0, 1, resultando en ocho estadosr con distintos valores de Er y Nr. En este sistema los dos terminos de la
13

igualdad (3.30) se escriben como:
111∑r=000
1∏=0
wn
= 1 + wa + wb + wc + wawb + wawc + wbwc + wawbwc
∏=a,b,c
1∑n=0
wn
= (1 + wa)(1 + wb)(1 + wc) . (3.31)
Se puede verificar que la igualdad se cumple en este caso partıcular. Puedeinferirse que la igualdad tambien se cumplira si aumentamos el numero deceldas o las posibilidades del numero de ocupacion.
Hay tres casos de particular interes que dan lugar a las tres estadısticasmas comunes:
1. Bose-Einstein: las partıculas son indistinguibles y no existe restriccionen la ocupacion de las celdas de espacio-fase: n = 0, 1, 2, . . .. Al con-siderar al espın entre las variables microscopicas se duplica la posibleocupacion del espacio-fase. De aquı se deriva la estadıstica de un gasde fotones que se desarrolla en la siguiente seccion.
2. Fermi-Dirac: las partıculas siguen el principio de exclusion de Pauli,por lo que cada celda de espacio fase puede ser ocupada por un maximode una partıcula: n = 0, 1. Las partıculas son indistinguibles y hayque incluir el espın duplicando la ocupacion del espacio-fase. De estecaso se deriva la estadıstica de un gas degenerado que se desarrollaposteriormente.
3. Maxwell-Boltzmann: corresponde al caso clasico de numeros de ocupa-cion muy pequenos, o altas temperaturas ⇒ β(ε−µ) 1⇒ w 1,
resultando en lnZ ≈ N ln(∑
r e−βεr
).
3.3.2. Aplicacion a un gas de fotones
En un gas de bosones no hay restriccion en cuanto al numero de partıcu-las que pueden ocupar un estado dado. La funcion de particion se obtienedirectamente de la sumatoria del lado derecho de la expresion 3.30,
∞∑n=0
wn =
1
1− w⇒ Z =
∏
1
1− e−(ε−µ)/kT
. (3.32)
14

La ocupacion del espacio fase sale de derivar el numero total de bosones dela funcion de particion:
N = − 1
β
∂ lnZ
∂µ=∑
1
e(ε−µ)/kT − 1=∑
n . (3.33)
En el caso de un gas de fotones el potencial quımico5 es nulo, µ = 0, y tene-mos una ocupacion promedio dada por nν = (ehν/kT−1)−1, con la frecuenciacaracterizando el micro-estado de cada foton, de forma que ε = hν.
La ocupacion del espacio fase y el numero de fotones se relacionan atraves del numero N de estados accesibles. Este se calcula dividiendo elvolumen del espacio fase entre el de cada celda (h), incluyendo un factor dedos por los dos valores posibles de la helicidad del foton
dN = 2d3xd3p
h3⇒ dN
dV= 2
(ν2
c3
)dν dΩ , (3.34)
empleando d3p = p2dΩdp, con p = hν/c. La densidad de energıa de radiacionisotropica en equilibrio termodinamico, uν(T ), viene dada por la energıa decada estado (hν), la densidad de estados (ec. 3.34), la isotropıa (dΩ→ 4π),y la ocupacion de cada estado, dando lugar a la distribucion de Planck:
uν(T ) =8πν2
c3hν
ehν/kT − 1. (3.35)
Al integrar sobre frecuencias obtenemos u(T ) = aT 4.
3.3.3. Gas ideal de fermiones en degeneracion
En este caso un estado dado puede ser ocupado a lo mas por una partıcula,y la sumatoria sobre ocupaciones es trivial,
Z =∏
1 + e−β(ε−µ)
. (3.36)
Podemos encontrar el numero de fermiones en un estado de energıa ε, bajoun potencial quımico µ:
N = − 1
β
∂ lnZ
∂µ=∑
1
e(ε−µ)/kT + 1=∑
n , (3.37)
5la energıa necesaria para la creacion del foton.
15
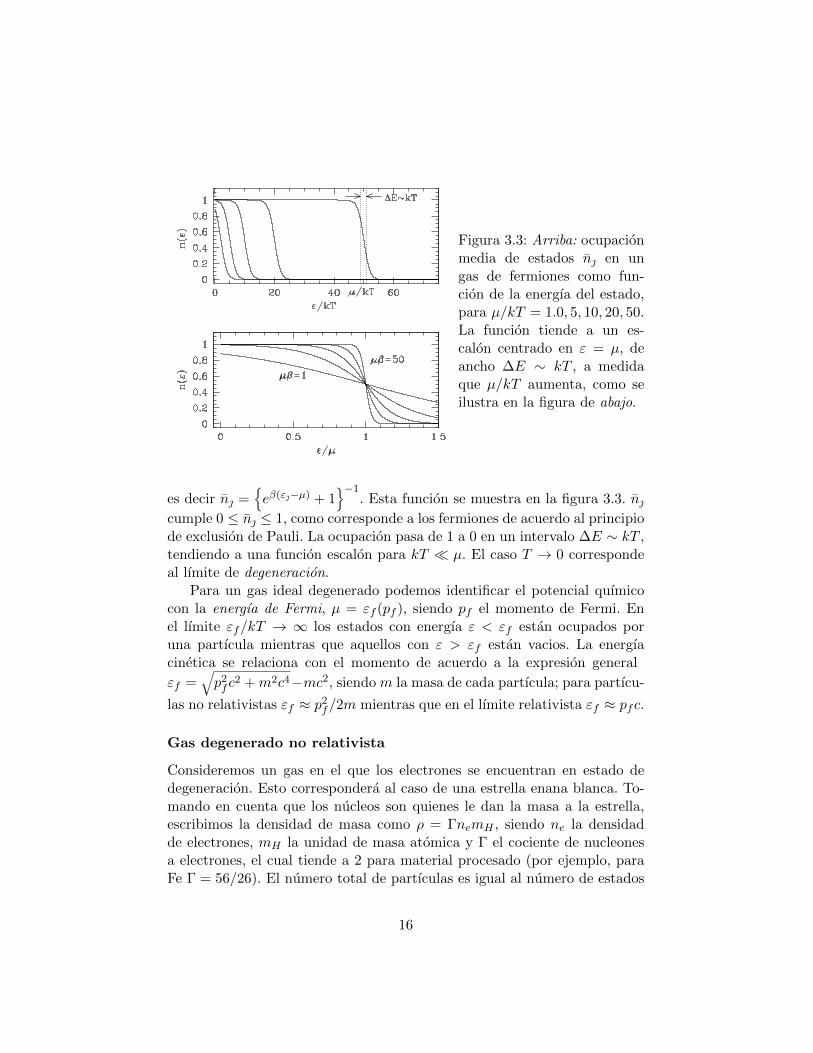
Figura 3.3: Arriba: ocupacionmedia de estados n en ungas de fermiones como fun-cion de la energıa del estado,para µ/kT = 1.0, 5, 10, 20, 50.La funcion tiende a un es-calon centrado en ε = µ, deancho ∆E ∼ kT , a medidaque µ/kT aumenta, como seilustra en la figura de abajo.
es decir n =eβ(ε−µ) + 1
−1. Esta funcion se muestra en la figura 3.3. n
cumple 0 ≤ n ≤ 1, como corresponde a los fermiones de acuerdo al principiode exclusion de Pauli. La ocupacion pasa de 1 a 0 en un intervalo ∆E ∼ kT ,tendiendo a una funcion escalon para kT µ. El caso T → 0 correspondeal lımite de degeneracion.
Para un gas ideal degenerado podemos identificar el potencial quımicocon la energıa de Fermi, µ = εf (pf ), siendo pf el momento de Fermi. Enel lımite εf/kT → ∞ los estados con energıa ε < εf estan ocupados poruna partıcula mientras que aquellos con ε > εf estan vacios. La energıacinetica se relaciona con el momento de acuerdo a la expresion general
εf =√p2fc
2 +m2c4−mc2, siendo m la masa de cada partıcula; para partıcu-
las no relativistas εf ≈ p2f/2m mientras que en el lımite relativista εf ≈ pfc.
Gas degenerado no relativista
Consideremos un gas en el que los electrones se encuentran en estado dedegeneracion. Esto correspondera al caso de una estrella enana blanca. To-mando en cuenta que los nucleos son quienes le dan la masa a la estrella,escribimos la densidad de masa como ρ = ΓnemH , siendo ne la densidadde electrones, mH la unidad de masa atomica y Γ el cociente de nucleonesa electrones, el cual tiende a 2 para material procesado (por ejemplo, paraFe Γ = 56/26). El numero total de partıculas es igual al numero de estados
16

contenidos en una esfera de Fermi, con el factor de dos debido al espın,
N = 2N = 2
∫d3x
∫ pf
0
d3p
h3=
2V
h3
(4π
3p3f
), (3.38)
es decir n = k3f/3π2, empleando pf = hkf como se hace en buen numero de
textos. Combinando ambas expresiones obtenemos el momento de Fermi enfuncion de la densidad:
pf =
(3h3ρ
8πΓmH
)1/3
⇐⇒ kf =
(3π2ρ
ΓmH
)1/3
. (3.39)
Suponiendo un gas ideal no relativista es posible calcular una temperaturade Fermi, por debajo de la cual el gas esta en estado de degeneracion
kTf =pf
2
2m⇒ Tf ' 2.4× 109 K
(ρ
106 g cm−3
)2/3 ( Γ
1.4
)−2/3. (3.40)
La temperatura de Fermi es muy superior a la del interior de una estrellaenana blanca.
La densidad de energıa, u, y la presion, P , para un gas degenerado estandadas por integrales sobre el espacio de momentos derivadas de las integralessobre espacio fase:
u =
∫ pf
0ε(2h−3d3p
), P =
∫ pf
0
1
3~p · ~v
(2h−3d3p
). (3.41)
Usando la forma de energıa cinetica de una partıcula relativista y la igualdad~v = ~pc2/ε, obtenemos las integrales planteadas en el libro de Weinberg, enel caso no relativista, p mc:
u =2
h3
∫ pf
0
(p2
2me
)4πp2dp, P =
2
h3
∫ pf
0
1
3
(p2
me
)4πp2dp . (3.42)
Se tiene claramente u = 3P/2, igual que para un gas ideal clasico, y,
P =
(3
π
)2/3 h2
20me
(ρ
ΓmH
)5/3
. (3.43)
P = Kρ5/3 es la ecuacion de estado de un gas de fermiones no relativista.La transicion al caso relativista se da cuando pf = mec, que implica
ρ = ρc ≡8π (ΓmH)m3
ec3
3h3' 0.97× 106 Γ g cm−3 . (3.44)
Esta es la densidad tıpica de una estrella enana blanca.
17

Gas degenerado relativista
La expresion general para densidad de energıa, u, y la presion, P , para un gasdegenerado esta dadas por integrales sobre el espacio de momentos derivadasde las integrales sobre espacio fase planteadas en el libro de Weinberg:
u =2
h3
∫ pf
0
[√p2c2 +m2
ec4 −mec
2]
4πp2dp,
P =2
h3
∫ pf
0
1
3
[p2c2√
p2c2 +m2ec
4
]4πp2dp . (3.45)
En el caso relativista, ρ ρc, tenemos que el termino de momento, pc,domina en las integrales de las ecuaciones 3.45, resultando en u = 3P , comopara un gas de fotones, y la ecuacion de estado,
P =
(3
π
)1/3 hc
8
(ρ
ΓmH
)4/3
, (3.46)
es decir P ∝ ρ4/3 para un gas degenerado de partıculas relativistas.
Estrellas degeneradas
La estructura de una estrella degenerada puede investigarse de manera cua-litativa y semi-cuantitativa con un modelo simplificado. Se puede calcularla energıa total de la estrella como la suma de la energıa interna (termica)y gravitacional,
Etot =
∫ R
0u(r) 4πr2dr −
∫ R
0ρ(r)
GM(r)
r4πr2dr , (3.47)
siendo u = Kργ/(γ − 1) la densidad de energıa, R el radio de la estrellay M(r) la masa contenida dentro de un radio r. Un ejemplo relativamentesimple es el de una estrella con densidad constante, para la cual encontramosla expresion
E(R) =KMγ
(γ − 1)
(3
4π
)γ−1R−3(γ−1) − 3
5
GM2
R, (3.48)
siendo la configuracion de equilibrio determinada por la condicion de mınimaenergıa, dE/dR = 0. Distinguimos los dos casos:
Caso no relativista: ⇒ ρ ρc ⇒ γ = 5/3:La configuracion de mınima energıa se da para
R =5K
G
(3
4π
)2/3
M−1/3 (3.49)
18

resultando en una relacion R ∝ M−1/3 para estrellas degeneradas. Siempleamos la expresion para K obtenemos
R =
(3
2π
)4/3 h2M−1/3
4Gm(Γmh)5/3' 7153 km
(M
M
)−1/3.
Caso relativista: ⇒ ρ ρc ⇒ γ = 4/3:El mınimo de energıa se da para dE/dR = 0 es decir
M =
(3
4π
)1/2 (5K
G
)3/2
, (3.50)
independientemente del valor de R. Substituyendo el valor de K paraγ = 4/3 se obtiene el lımite de masa para una estrella sostenida pordegeneracion de electrones
M = Mch =3
2π
(5hc/8G)3/2
(Γmh)2' 3.42× 1033 g ' 1.72M . (3.51)
Esta viene siendo la masa de Chandrasekhar para una estrella condensidad constante. Si M = Mch tenemos E = 0, mientras que siM < Mch tenemos E > 0, lo cual corresponde a una estrella fuerade equilibrio donde la presion termodinamica domina a la gravedad ypor tanto la estrella esta en expansion; si M > Mch entonces E < 0,y la estrella esta fuera de equilibrio con la gravedad dominando y portanto en colapso.
El mismo analisis se puede hacer para una estrella de neutrones, reempla-zando me por mN ' mH y Γ = 1. La densidad crıtica queda como
ρc =m4Hc
3
3π2h3' 6.1× 1015 g cm−3 , (3.52)
obteniendose para el caso no relativista
R =
(3
2π
)4/3 h2M−1/3
4Gm8/3N
' 4.9 km (M/M)−1/3 , (3.53)
y una masa lımite
Mns =3 (5hc/8G)3/2
2πm2h
' 6.9M . (3.54)
El radio de la estrella queda subestimado con respecto a un analisis mas rigu-roso, en particular porque hemos despreciado la interaccion entre partıculas.Los valores actualmente aceptados para estrellas las dimensiones de las es-trellas de neutrones son M∗ ' 1.44 M y R∗ ' 10 km. Para una discusiondetallada ver el libro de Weinberg (1973).
19

3.4. Reacciones en equilibrio
La mecanica estadıstica permite tratar problemas que involucran la co-existencia de varias componentes distintas, o especies quımicas, en equili-brio. Ejemplos son el equilibrio entre especies neutras, iones y electrones; ola creacion y destruccion conjunta de partıculas en reacciones quımicas, oen reacciones nucleares. El proceso de destruccion y creacion puede darse enun estado de equilibrio con la reaccion inversa, por ejemplo:
la ionizacion del hidrogeno y el proceso de recombinacion: H←→ H+ + e−;
la produccion de pares y su aniquilacion: γ + γ ←→ e+ + e−;
la fusion de dos nucleos de helio en un nucleo de berilio y el decaimientodel berilio, 4He + 4He←→ 8Be.
Cuando se tiene un equilibrio entre una reaccion y su inversa se establece unarelacion entre las funciones de particion de las partıculas involucradas quepermite establecer relaciones entre las densidades de las distintas especies.
3.4.1. La ley de accion de masas
Para varias especies en co-existencia tenemos,
dE = TdS − PdV +m∑`=1
µ`dN` ,
donde el ındice ` enumera m especies. En equilibrio se minimiza la energıay se maximiza la entropıa, lo cual requiere∑
`
µ`dN` = 0 . (3.55)
La ley de accion de masas surje al considerar la funcion de particion queviene dada por el producto de las funciones de particion de las distintasespecies, Z =
∏` Z`. Mas aun, podemos escribir la funcion de particion
como el producto de funciones de particion de partıculas individuales, ζ,que pueden agruparse por especie,
Z =m∏`=1
Z` =m∏`=1
ζN``
N`!, (3.56)
20

con el factor N`! considerando la indistinguibilidad de las N` partıculas dela especie `. El potencial quımico de cada especie se calcula de la funcion departicion,
µ` = − 1
β
∂ lnZ
∂N`≈ −kT ln
(ζ`N`
), (3.57)
que, junto con la condicion de equilibrio 3.55 y la aproximacion de Stirlingpara N` 1, da,∑
`
ln
(ζ`N`
)dN` = 0 ⇒
∏`
(ζ`n`V
)ν`=∏`′
(ζ`′
n`′V
)ν′`, (3.58)
con ` denotando las especies del lado izquierdo de la reaccion, `′ las del ladoderecho, y ν` la proporcion relativa de la especie ` en la reaccion, que seintroduce como factor de proporcionalidad, dN` ∝ ν`. Tambien se emplea ladensidad de partıculas, en lugar del numero, N` = n`V .
En el caso de partıculas libres podemos emplear la expresion de ζ deriva-da para un gas ideal, ζ = V (2πmkT/h2)3/2. En el caso de una partıcula conmas grados de libertad que el translacional, por ejemplo un sistema ligadocon potencial de ionizacion χ, tenemos que la funcion de particion puedesepararse en una parte translacional y otra interna, ζ = ζt ζint, donde ζt esuna funcion de partıcula libre y ζint = g e−E/kT .
Dos aplicaciones importantes en astrofısica son la ley de Boltzmann y laecuacion de Saha, mismo que vemos a continuacion.
3.4.2. Ley de Boltzmann
Pensando en el equilibrio entre el numero de atomos en un estado n1 y otroestado n2, podemos escribir la reaccion como (1) (2), de donde la ley deaccion de masas (ec. 3.58) da:
n2/n1 = ζ2/ζ1 . (3.59)
El potencial quımico para un foton es cero y por tanto podemos despreciarlosen estas expresiones. Para un sistema cuantico ζ = ge
−E/kT , con g elnumero de estados de energıa E (estados degenerados de misma energıa),por lo que obtenemos la ley de Boltzmann:
n2n1
=g2g1e(E1−E2)/kT . (3.60)
Para un sistema con un numero arbitrario de niveles tenemos:
nn
=ge−E/kT∑
g`e−E`/kT. (3.61)
21

En la practica el rapido aumento de la degeneracion g con el numero deestados eventualmente se ve compensado por el termino exponencial y unopuede realizar el calculo considerando un numero finito de estados.
3.4.3. Ecuacion de Saha
Para procesos como la ionizacion y recombinacion del hidrogeno,
H(0) H(1) + e , (3.62)
donde el numero entre parentesis denota el grado de ionizacion, la ley deaccion de masas queda como:
ζ0n0V
=
(ζ1n1V
)(ζeneV
). (3.63)
Tanto el electron como el proton, H(1), son partıculas libres, por lo que laley de accion de masas, al considerar las distintas funciones de particion, da:
ζ0/V = (1/λ30)g0 eχ/kT , ζ1/V = 2/λ31 , ζe/V = 2/λ3e , (3.64)
donde introducimos λ ≡ h/√
2πmkT = h/p, la longitud de onda de Brogliepara partıculas termicas, con g0 la degeneracion y χ > 0 el potencial deionizacion del atomo considerado. Los factores de dos representan los estadosde espın posibles para el electron y el proton. En estos terminos obtenemos:
n1n0
=2
neλ3e
(2
g0
)e−χ/kT . (3.65)
Consideremos las condiciones de una atmosfera estelar, de acuerdo a laecuacion 3.65. Las longitudes de onda de De Broglie se pueden estimar en
λe = 0.75× 10−7cm(T/104K
)−1/2, λp = 1.74× 10−9cm
(T/104K
)−1/2,
para electrones y protones respectivamente en un gas a 104K. En el ca-so de los electrones, para una densidad tıpica ne ∼ 1010cm−3 tenemosneλe ∼ 10−10, lo cual refleja el bajo nivel de ocupacion del espacio-fase encondiciones normales comparado con el lımite de degeneracion, neλ
3e → 1.
Dicho de otro modo, los electrones tienen una ocupacion ∆x∆p h. Escomun expresar esta relacion de forma exponencial, neλ
3e/2 = e−ξ, con ξ
tıpicamente entre 10 y 30. La ec. 3.65 puede generalizarse a otros estadosde ionizacion ı 6= 0 y reescribirse como:
nı+1
nı=gı+1
gıexp ξ − χı/kT , (3.66)
22
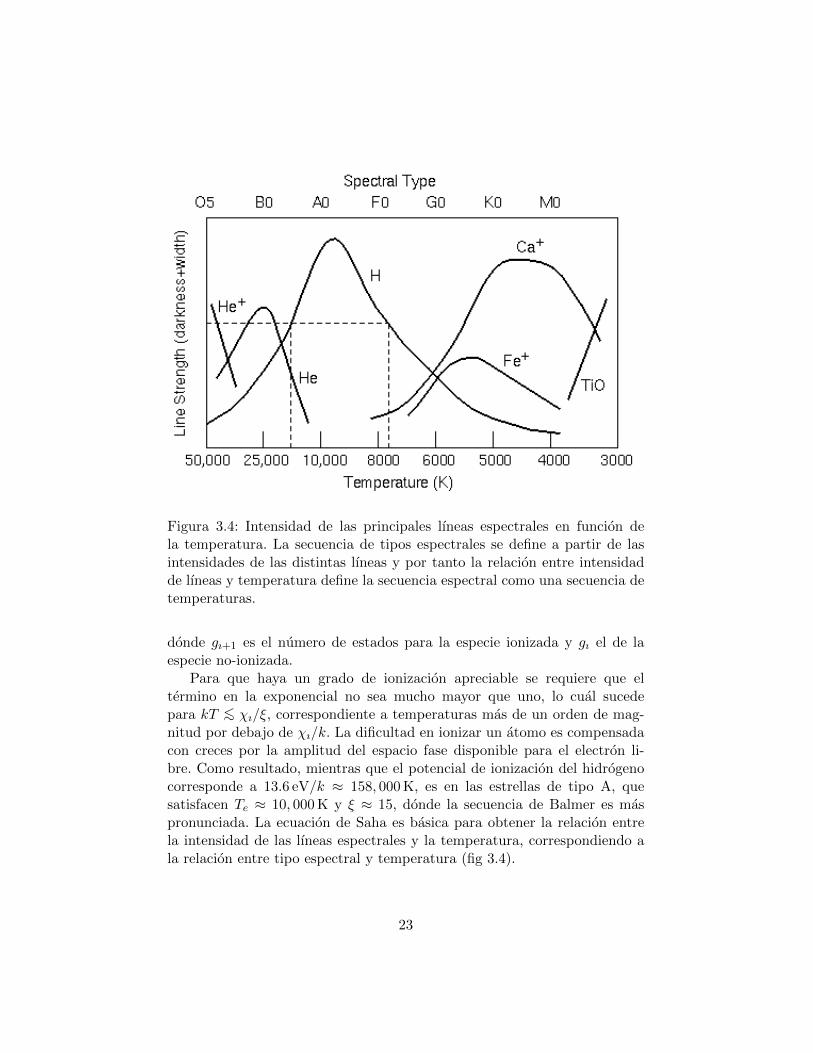
Figura 3.4: Intensidad de las principales lıneas espectrales en funcion dela temperatura. La secuencia de tipos espectrales se define a partir de lasintensidades de las distintas lıneas y por tanto la relacion entre intensidadde lıneas y temperatura define la secuencia espectral como una secuencia detemperaturas.
donde gı+1 es el numero de estados para la especie ionizada y gı el de laespecie no-ionizada.
Para que haya un grado de ionizacion apreciable se requiere que eltermino en la exponencial no sea mucho mayor que uno, lo cual sucedepara kT <∼ χı/ξ, correspondiente a temperaturas mas de un orden de mag-nitud por debajo de χı/k. La dificultad en ionizar un atomo es compensadacon creces por la amplitud del espacio fase disponible para el electron li-bre. Como resultado, mientras que el potencial de ionizacion del hidrogenocorresponde a 13.6 eV/k ≈ 158, 000 K, es en las estrellas de tipo A, quesatisfacen Te ≈ 10, 000 K y ξ ≈ 15, donde la secuencia de Balmer es maspronunciada. La ecuacion de Saha es basica para obtener la relacion entrela intensidad de las lıneas espectrales y la temperatura, correspondiendo ala relacion entre tipo espectral y temperatura (fig 3.4).
23


















