Buenas Prácticas regulatorias de la Investigación Clínica en Colombia€¦ · en Colombia....
20
Buenas Prácticas regulatorias de la Investigación Clínica en Colombia Javier Guzmán Cruz Director General XI Encuentro EAMI Varadero - La Habana 22 de Junio 2016
Transcript of Buenas Prácticas regulatorias de la Investigación Clínica en Colombia€¦ · en Colombia....
Buenas Prácticas regulatorias de la
Investigación Clínica en Colombia
Javier Guzmán CruzDirector General
XI Encuentro EAMI Varadero - La Habana 22 de Junio 2016
Agenda
1 • Bases legales
2• ¿Qué y cómo lo
hacemos?
3 • Retos
Resolución 8430 de 1993 “Por la cual se establecen las normascientíficas, técnicas y administrativas para la investigación en salud”.
Resolución 2378 de 2008 “Por la cual se adoptan las BuenasPrácticas Clínicas para las instituciones que conducen investigacióncon medicamentos en seres humanos”.
1. Bases Legales
Moderador
Notas de la presentación
En la Resolución 8430 de 1993 se establecen las pautas generales para desarrollar investigación con seres humanos tales como: competencia de los encargados del proceso de investigación, ítems a tener en cuenta en el consentimiento informado, autorización previa de un comité de ética, manejo integral del sujeto participante en caso de eventos adversos derivados de la investigación por parte del patrocinador e investigador. Hasta el año 2010 fue aplicada esta resolución para desarrollar estudios clínicos en Colombia ya que aunque la resolución 2378 fue expedida en 2008 tuvo dos (2) años de transitoriedad Teniendo en cuenta que los estudios clínicos son indispensables para encontrar nuevas alternativas terapéuticas y que contar con los soportes de seguridad - eficacia de un medicamento son un requisito previo para aprobar su Registro Sanitario (autorización de comercialización) o para aprobar una nueva indicación surge la necesidad de contar con una norma para investigación clínica que permitiera garantizar los aspectos éticos y científicos de los estudio desarrollados en el país, así como las pautas para que los datos obtenidos de las investigaciones sean trazables y veraces. En el año 2008 se expide la Resolución 2378 por el Ministerio de la Protección Social con base al documento “Buenas Prácticas Clínicas” elaborado por los Grupos Técnicos de Trabajo delegados por los países que conforman la Red Panamericana de Armonización de la Reglamentación Farmacéutica – REDPARF.
Verificar el cumplimiento de las Buenas Prácticas Clínicas (BPC) en lasinstituciones interesadas en desarrollar estudios clínicos (EC) conmedicamentos, para lo cual expide un certificado.
Evaluar y conceptuar sobre los estudios clínicos con medicamentos enseres humanos a realizarse en Colombia, así como los documentos relacionadoscon el desarrollo de los mismos.
Realizar seguimiento a las instituciones que conducen investigación clínicacon medicamentos.
2. ¿Qué y cómo lo hacemos?
Moderador
Notas de la presentación
Conceptuar: incluye aprobación o negación de la autorización para el desarrollo de un ensayo clínico. El procedimiento de Invima habla de conceptuar para evitar que quede solo la función de aprobar como obligación. Nota importante: Esta diapositiva tiene hipervínculos. Da enlace para el resto de las diapositivas que no tienen título. Para darle continuidad hacer click en cada uno de los enlaces para el desarrollo de la presentación, durante la cual se va revisando cada una de las tres actividades competencia de INVIMA. Para regresar a esta diapositiva, usar la flecha azul ubicada en el extremo inferior izquierdo.
CERTIFICACIÓN BPC
Comité de Ética en
Investigación
Servicio Farmacéutico
Equipo Investigador
Laboratorio Clínico
Sistema de gestión de la
calidad
Patrocinador
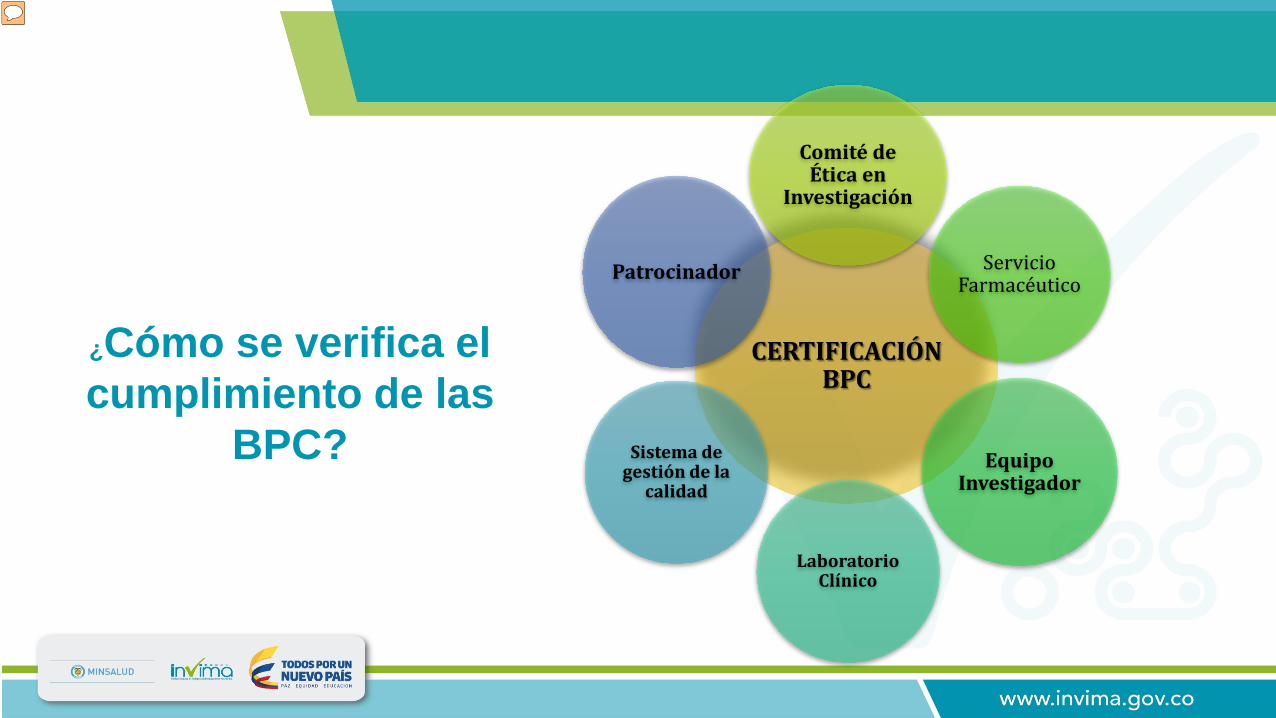
¿Cómo se verifica el cumplimiento de las
BPC?
Moderador
Notas de la presentación
Durante la certificación de BPC, son verificados los siguientes componentes: 1. Comité de ética en investigación: Órgano multidisciplinario encargado de velar por la protección de los derechos y el bienestar de los sujetos participantes en investigación, mediante la evaluación de los ensayos clínicos y su posterior seguimiento. Se verifican las responsabilidades, conformación y procedimientos ejecutados por el comité. 2. Servicio farmacéutico: Liderado por un Químico farmacéutico, responsable del manejo del producto en investigación desde la recepción hasta la disposición final del mismo. 3. Equipo Investigador: Verificación de CV, experticia, capacitación. El equipo esta conformado por el investigador principal, sub investigadores, coordinador de estudios, bacteriólogo y químico farmacéutico. Se tienen en cuenta que cada uno de estos profesional tenga un back-up con el fin de garantizar la continuidad de los procesos en alguna eventualidad. 4. Laboratorio Clínico: Encargado de la toma, procesamiento, embalaje de las muestras biológicas, obtenidas del proceso de investigación clínica. 5. Sistema Gestión de Calidad: Son evaluados los procedimientos de investigación acoplados un sistema de gestión de calidad, programa de capacitación continuo, programa de mantenimiento y calibración de equipos. 6. Patrocinador: Se verifican las responsabilidades del mismo en cuanto a capacitaciones al equipo investigador, monitorias, cubrimiento de eventos adversos. La vigencia de la certificación es de cinco años
Departamento Número
Antioquia 16
Atlántico 4
Bogotá D.C. 28
Bolívar 2
Caldas 2
Córdoba 1
Cundinamarca 1
Nariño 2
Risaralda 1
Santander 8
Tolima 1
Valle del Cauca 8
TOTAL 74
Departamento Número
Antioquia 23
Atlántico 15
Bolívar 2
Caldas 2
Casanare 2
Córdoba 1
Cundinamarca 46
Meta 2
Nariño 2
Quindío 5
Risaralda 3
Santander 8
Tolima 1
Valle del Cauca 11
TOTAL 123Comités de Ética
Centros de Investigación
Certificado de cumplimiento de las BPC
Instituciones Certificadas BPC
Moderador
Notas de la presentación
Algunos Comités de ética prestan sus servicios de evaluación y seguimiento de ensayos clínicos a las instituciones certificadas en BPC, razón por la cual hay un menor número de Comités en comparación a instituciones certificadas. Al finalizar dar click en la flecha (anterior)
¿Cómo se solicita la aprobación de un EC?
Moderador
Notas de la presentación
El Invima ha establecido mediante Guías disponibles en la página web institucional los lineamientos para realizar la solicitud de aprobación de un ensayo clínico, para lo cual se debe: Diligenciar el respectivo formato de presentación de protocolos. Allegar los documentos que soportan los aspectos técnicos, éticos y científicos para el desarrollo del ensayo clínico (protocolo, manual del investigador, consentimiento informado, información farmacéutica (certificado BPM, certificados de análisis, estudios de estabilidad, etiquetas)).
CÓMITE DE ÉTICA EN INVESTIGACIÓN (CEI) • Evaluación ética y
científica
GRUPO DE INVESTIGACIÓN
CLÍNICA (GIC)
• Evaluación Técnica• Evaluación farmacéutica
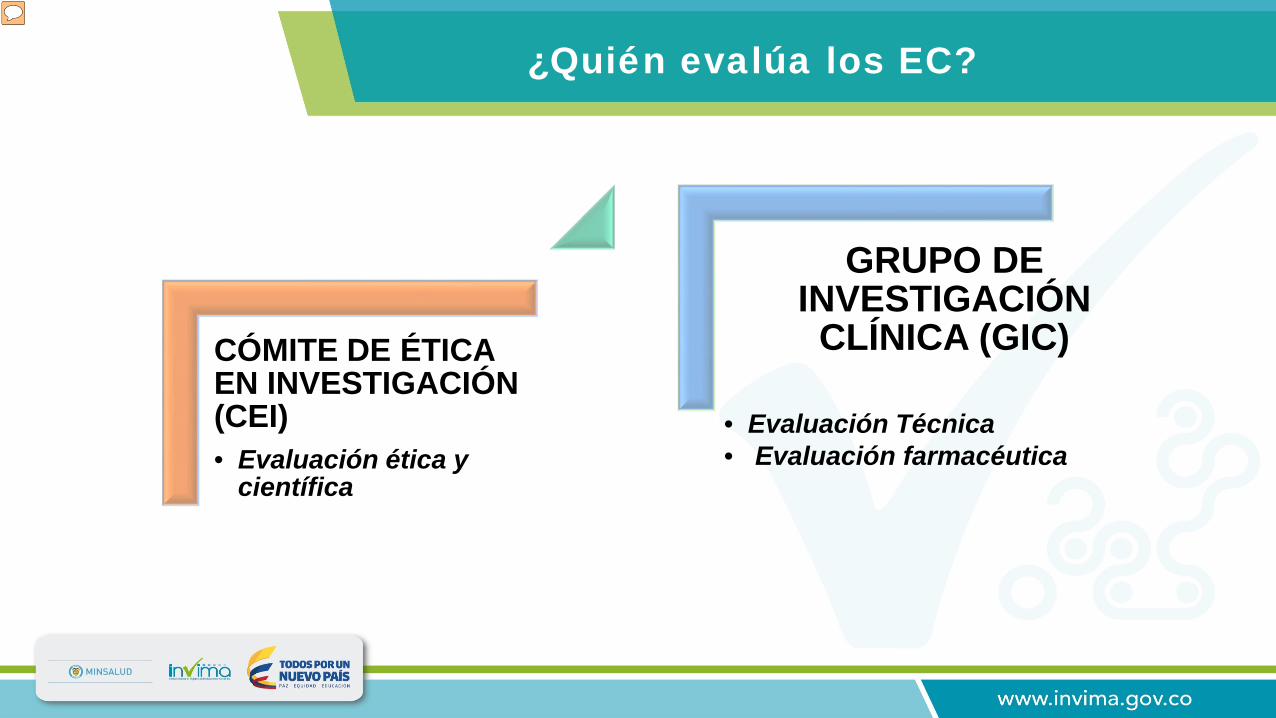
¿Quién evalúa los EC?
Moderador
Notas de la presentación
Los ensayos clínicos que son desarrollados en el país son previamente evaluados de manera ética y científica por un comité de ética en investigación, quien posteriormente emite un concepto de aprobación o negación del mismo. Una vez se cuenta con la aprobación del CEI, el grupo de investigación clínica del INVIMA realiza la evaluación técnica y farmacéutica del protocolo de investigación siguiendo procedimientos, instructivos y formatos estandarizados.
GRUPO DE INVESTIGACIÓN
CLÍNICA (GIC)
Solicitud Resolución Publicación
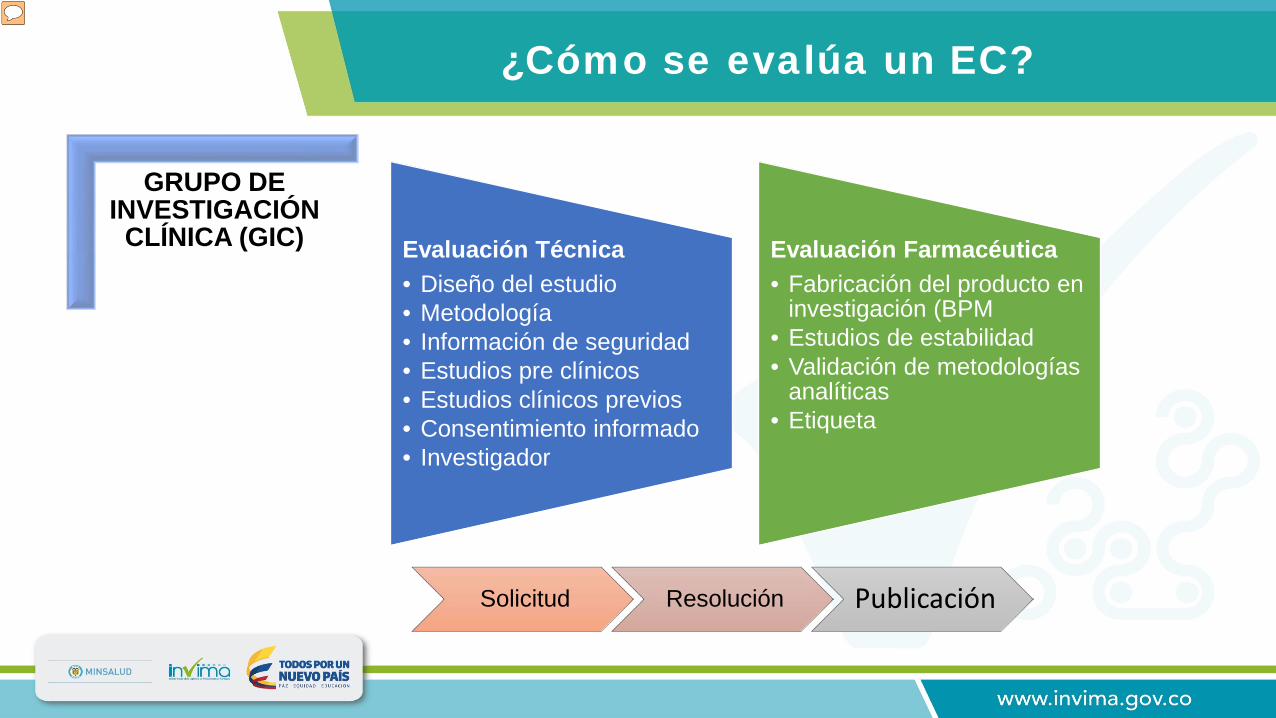
¿Cómo se evalúa un EC?
Evaluación Técnica• Diseño del estudio• Metodología • Información de seguridad• Estudios pre clínicos• Estudios clínicos previos• Consentimiento informado• Investigador
Evaluación Farmacéutica• Fabricación del producto en
investigación (BPM• Estudios de estabilidad• Validación de metodologías
analíticas• Etiqueta
Moderador
Notas de la presentación
La evaluación de protocolos de investigación clínica esta basada en lineamientos internacionales. En Colombia se siguen los lineamientos de la Red PARF establecidos en el documento "Buenas Prácticas Clínicas: Documentos de las Américas". Los lineamientos de la Red PARF se basan en las Guías ICH Conferencia Internacional de Armonización - Guías de Buenas Prácticas Clínicas (E6) documento elaborado con la participación de las agencias regulatorias de la Unión Europea, Estados Unidos. y Japón. Una vez evaluada una solicitud de aprobación de EC, se genera por parte de Invima un acto administrativo mediante al cual se aprueba o niega el desarrollo de un EC en el país. La información generada de esas evaluaciones alimenta la publicación que se encuentra disponible en la página web.
Información publicada por el Invima
Información general del Protocolo
• Código del Protocolo• Título• Patrocinador
Conceptos emitidos
Estado de los ensayos
Registro primario en basesinternacionales
Moderador
Notas de la presentación
El Invima, cuenta con una base de protocolos pública que se encuentra disponible en la página web institucional, la cual permite verificar información general del protocolo (código de protocolo, titulo y patrocinador), así como los conceptos emitidos sobre la evaluación de los ensayos clínicos y el estado de los mismos (activo / no activo). Adicionalmente se encuentra relacionado el registro primario realizado en bases de datos internacionales como Clinicaltrials, EU Clinical Trials “Se considera que el registro de todos los ensayos clínicos de intervención es una responsabilidad científica, ética y moral debido a que: Existe la necesidad de garantizar que las decisiones sobre la atención sanitaria se notifiquen con el aval de todos los datos científicos disponibles Es difícil tomar decisiones informadas si existe sesgo de publicación y notificación selectiva La Declaración de Helsinki (parámetro internacional al cual nos adherimos) establece que “Se debe registrar cada ensayo clínico en una base de datos de acceso público antes de reclutar el primer sujeto” Propiciará la identificación de ensayos similares o idénticos de manera que los investigadores y patrocinadores eviten la duplicación innecesaria Describir los ensayos clínicos en curso puede facilitar la identificación de vacíos en la investigación clínica. Informar a los investigadores y posibles participantes sobre ensayos de reclutamiento puede facilitar la selección” Tomado de OMS.
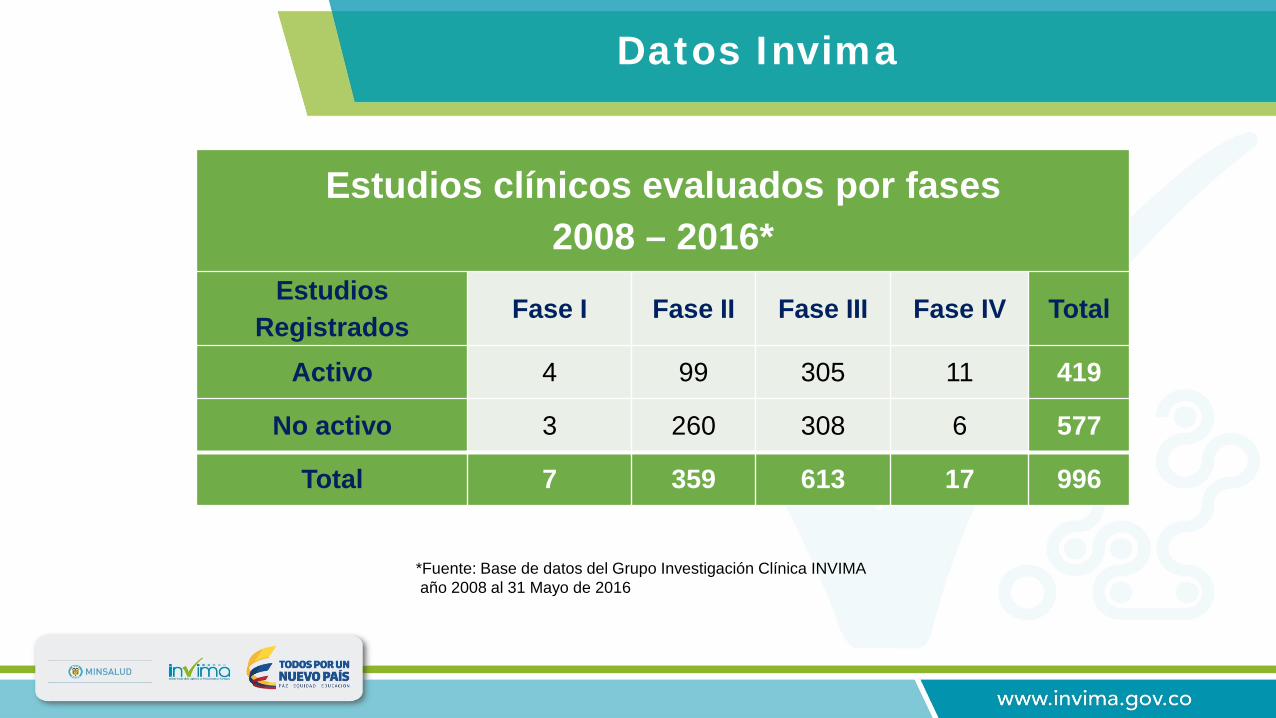
Estudios clínicos evaluados por fases 2008 – 2016*
Estudios Registrados Fase I Fase II Fase III Fase IV Total
Activo 4 99 305 11 419
No activo 3 260 308 6 577
Total 7 359 613 17 996
*Fuente: Base de datos del Grupo Investigación Clínica INVIMA año 2008 al 31 Mayo de 2016
Datos Invima
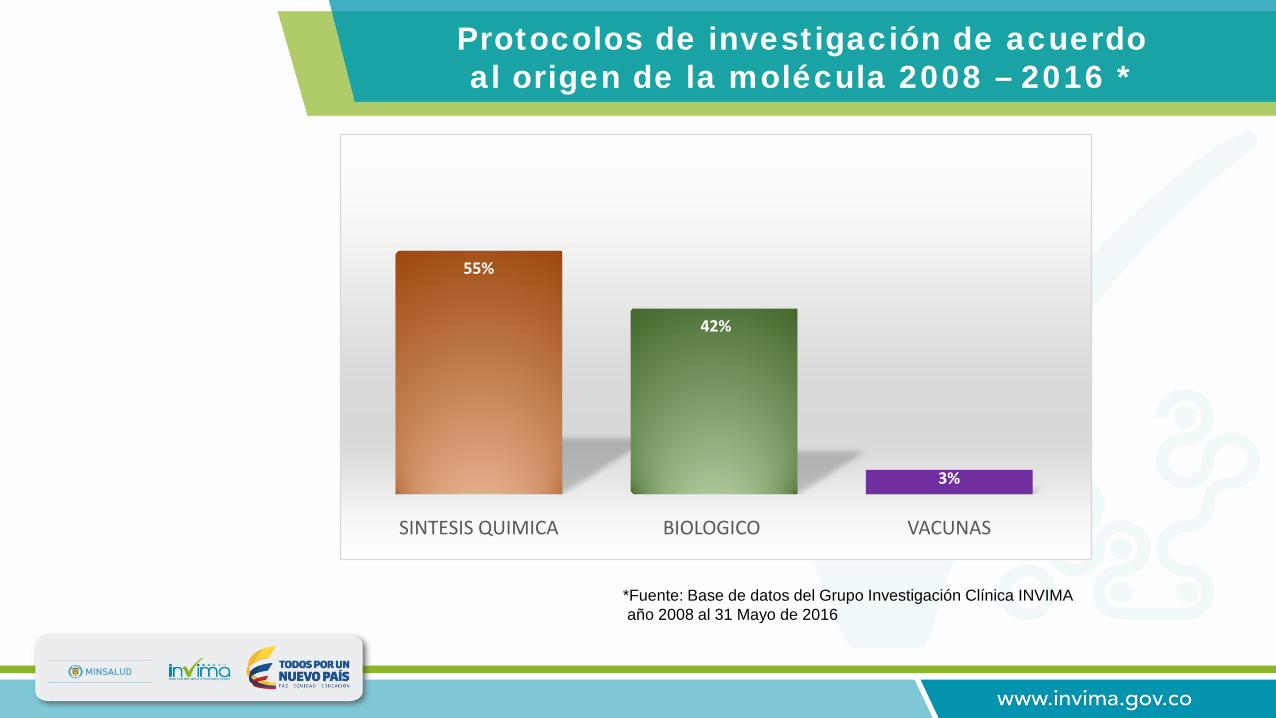
55%
42%
3%
SINTESIS QUIMICA BIOLOGICO VACUNAS
Protocolos de investigación de acuerdo al origen de la molécula 2008 – 2016 *
*Fuente: Base de datos del Grupo Investigación Clínica INVIMA año 2008 al 31 Mayo de 2016
Reumatología 23%
Oncología 20%
Cardiología 18%
Endocrinología 14%
Neumología 14%
Otras11%
Reumatología Oncología Cardiología
Endocrinología Neumología Otras
Fuente: Base de datos del Grupo Investigación Clínica INVIMA año 2008 al 31 Mayo de 2016
Especialidades investigadas
Moderador
Notas de la presentación
Al finalizar dar click en la flecha (anterior)
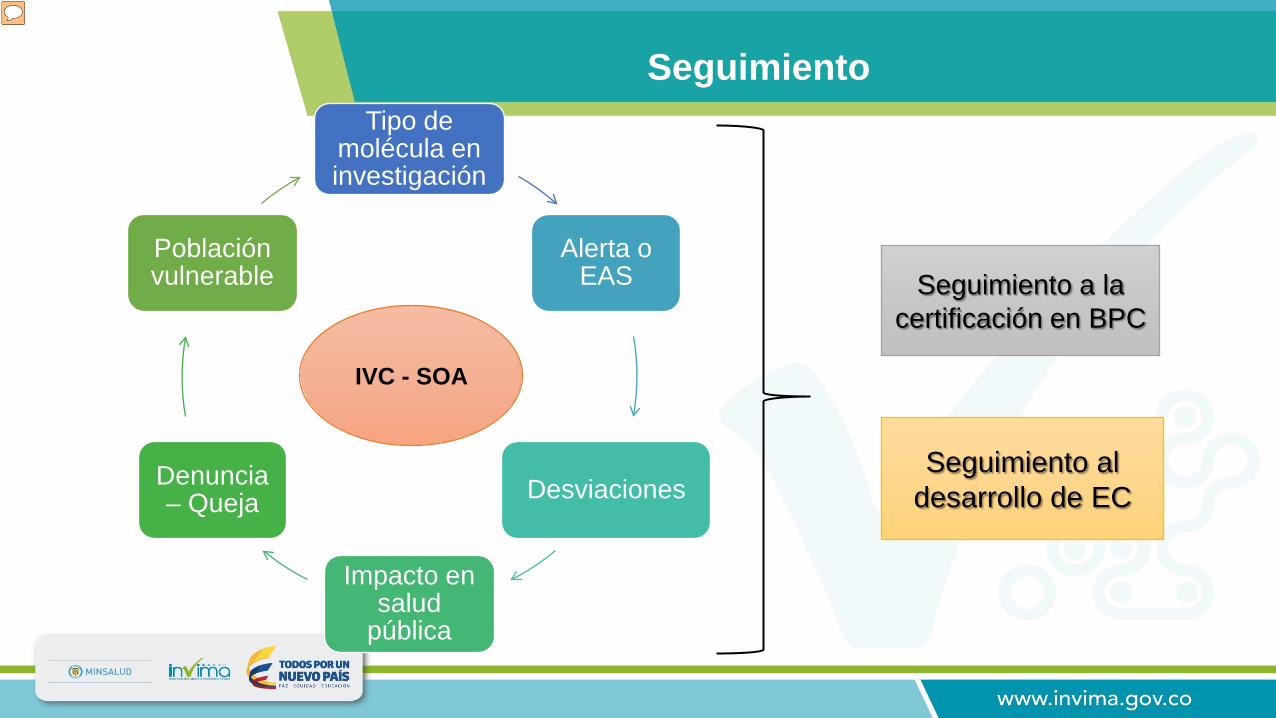
Seguimiento
Seguimiento a la certificación en BPC
Seguimiento al desarrollo de EC
IVC - SOA
Tipo de molécula en investigación
Alerta o EAS
Desviaciones
Impacto en salud
pública
Denuncia – Queja
Población vulnerable
Moderador
Notas de la presentación
El Invima realiza seguimientos a las instituciones certificadas en BPC y seguimientos desarrollo de los ensayos clínicos, las cuales son programadas de acuerdo a parámetros (matriz) de riesgo en la cual se tienen en cuenta los siguientes aspectos: Tipo de molécula: Se debe tener en cuenta si se trata de un producto de síntesis química o de origen biológico (vacuna), estos últimos tienen priorización. Alerta de seguridad o Evento Adverso Serio (EAS) reportado a nivel nacional o internacional. Desviaciones: Procedimientos o prácticas que afectan negativamente los derechos, la seguridad o el bienestar de los sujetos o la calidad y la integridad de los datos obtenidos de la investigación. (El protocolo establece unos parámetros que se deben seguir al pie de la letra para el desarrollo del Ensayo clínico, cuando por alguna razón no se pueden cumplir esos lineamientos se presenta una desviación al protocolo, algunas con mayor relevancia como por ejemplo no firmar el consentimiento informado antes del ingreso al ensayo clínico u otras de menor relevancia como una excursión de temperatura en el almacenamiento de un producto) Impacto en salud pública: protocolos en los cuales se investigan productos para enfermedades de interés Público (dengue, leishmaniasis, etc) Denuncia o queja: realizada por un particular, sujeto de investigación u otro actor en relación al proceso de investigación clínica. (malos tratos del investigador, mal manejo del producto en investigación). Población vulnerable: niños, personas en condición de discapacidad, adultos mayores
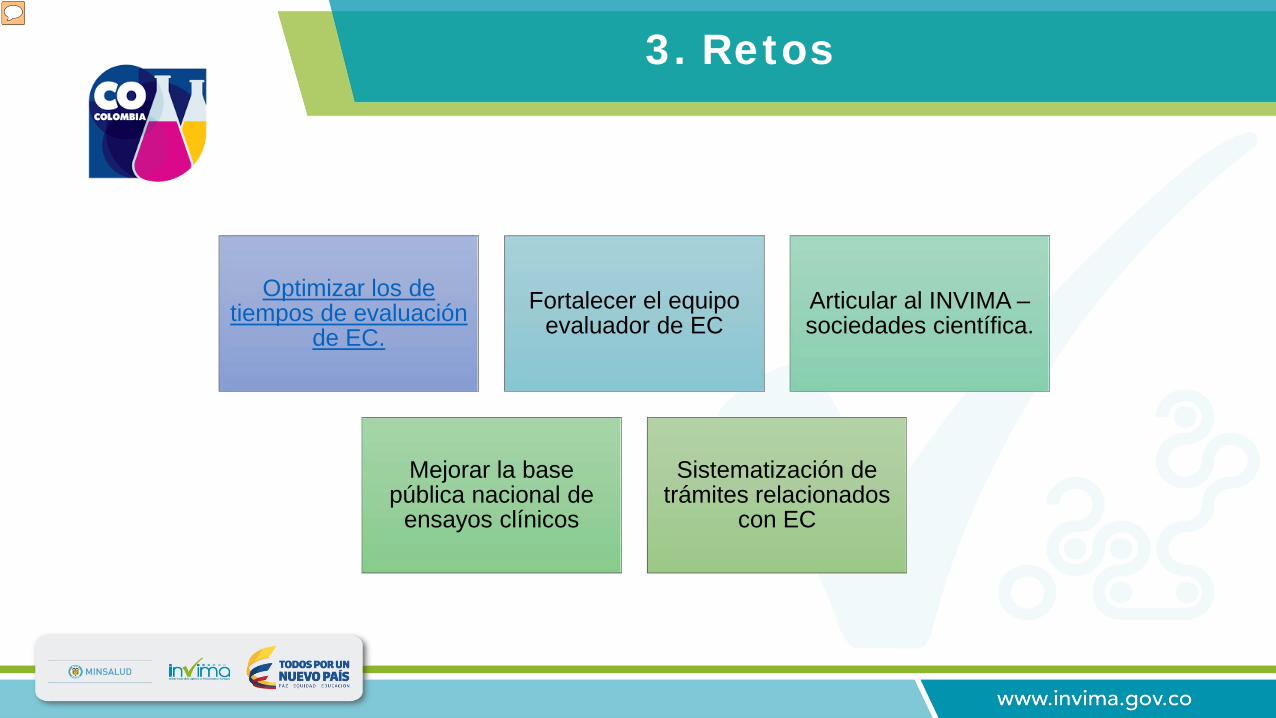
3. Retos
Optimizar los de tiempos de evaluación
de EC.Fortalecer el equipo
evaluador de ECArticular al INVIMA –sociedades científica.
Mejorar la base pública nacional de
ensayos clínicos
Sistematización de trámites relacionados
con EC
Moderador
Notas de la presentación
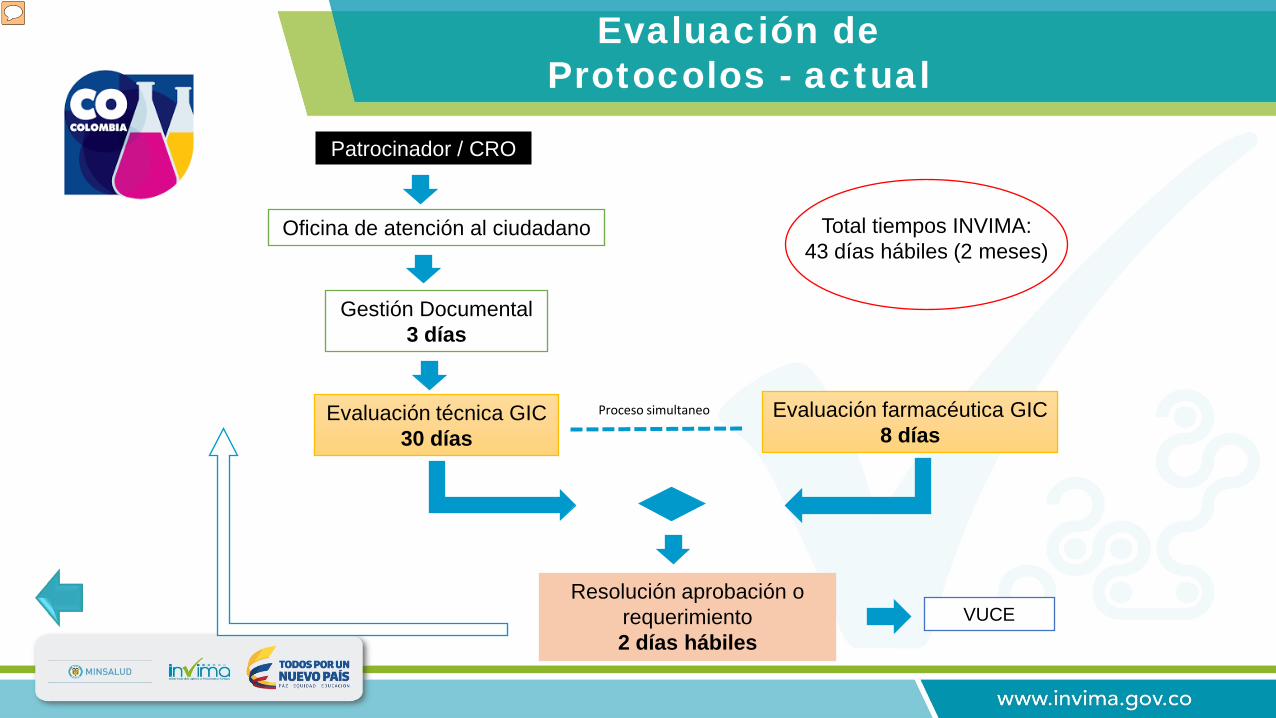
Nota importante: Dar click en el recuadro “optimizar los tiempos de evaluación de EC” Este hipervínculo te lleva a las 3 ultimas diapositivas Cuando llegues a la diapositiva No. 20 ( la de los nuevos tiempos) debes dar click a la flecha (anterior) UBICADA en el extremo inferior izquierdo.
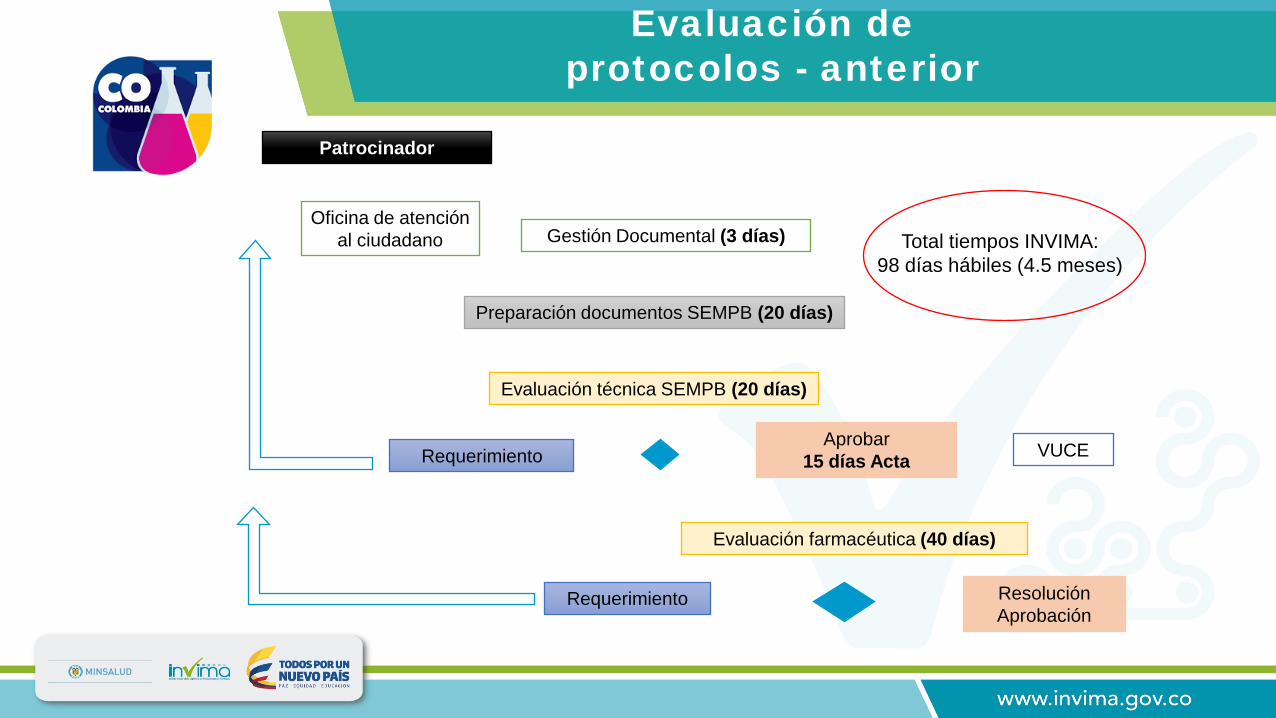
Oficina de atención al ciudadano Gestión Documental (3 días)
Aprobar 15 días Acta
Evaluación farmacéutica (40 días)
Requerimiento
Patrocinador
Evaluación técnica SEMPB (20 días)
Preparación documentos SEMPB (20 días)
Resolución Aprobación
Requerimiento
VUCE
Total tiempos INVIMA:98 días hábiles (4.5 meses)
Evaluación de protocolos - anterior
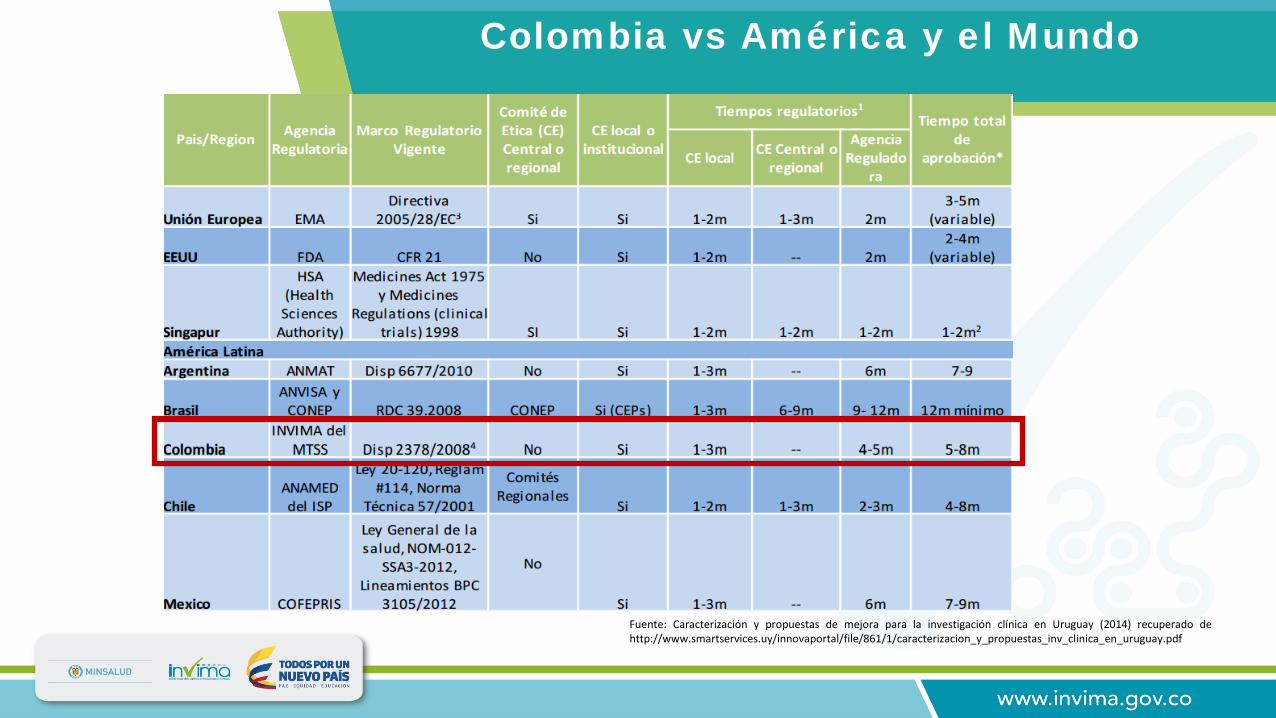
Colombia vs América y el Mundo
Fuente: Caracterización y propuestas de mejora para la investigación clínica en Uruguay (2014) recuperado dehttp://www.smartservices.uy/innovaportal/file/861/1/caracterizacion_y_propuestas_inv_clinica_en_uruguay.pdf
Resolución aprobación o requerimiento2 días hábiles
Evaluación farmacéutica GIC 8 días
Evaluación técnica GIC30 días
Gestión Documental3 días
Proceso simultaneo
Patrocinador / CRO
Oficina de atención al ciudadano Total tiempos INVIMA:43 días hábiles (2 meses)