






Idiomas
Páginas
Jurídico


Trombocitopenia o trombopenia

Trombopenia o trombocitopenia
• Se considera trombopenia a la disminución del número de plaquetas por debajo de aproximadamente 100.000 plaquetas/mm3 .
• Disminuciones inferiores a 50.000 plaquetas/mm3 facilitan el sangrado postraumático.
• debajo de 20.000 plaquetas, se facilita la aparición del denominado sangrado espontáneo

Etiopatogenia de la trombopenia
• Hipoproducción de plaquetas (trombopenias centrales)
• Disminución de supervivencia plaquetaria (trombopenias periféricas)

Hipoproducción de plaquetas (trombopenias centrales)
Infiltración de la médula ósea, aplasia, enfermedad de Fanconi,síndrome TAR (trombopenia y ausencia de radio), trombopenia cíclica, rubéola congénita
• Disminución en el número de megacariocitos.
Enfermedad de Wiskott-Aldrich, anemias megaloblásticas, síndromes mielodisplásicos.
• Trombopoyesis ineficaz.

Disminución de supervivencia plaquetaria (trombopenias periféricas)
• Destrucción incrementada de plaquetas (la vida media plaquetaria normal es de alrededor de 10 días).
• Fármacos, púrpura trombopénica idiopática, púrpura postransfusional, púrpura inmunológica secundaria , infección por VIH .

• Se produce una destrucción periférica de plaquetas, que ocasiona un incremento en la formación de las mismas mediante un aumento del número de megacariocitos
• Es la trombopenia habitual encontrada en los adultos.Como fármacos causantes de trombopenia se encuentran: • heparina, etanol, quinidina, difenilhidantoína, sales de oro , TIACIDAS,
estrógenos, quimioterapia.• El tratamiento consiste en la suspensión del medicamento, y si latrombopenia es grave, la administración de esteroides.
Púrpura trombopénica inducida por fármacos.
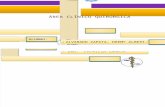
• Hiperconsumo plaquetario. Púrpura trombopénica trombótica, coagulación intravascular diseminada, hemangioma cavernoso, síndrome hemolítico urémico, infecciones agudas.
• Secuestro plaquetario.• Hiperesplenismo.

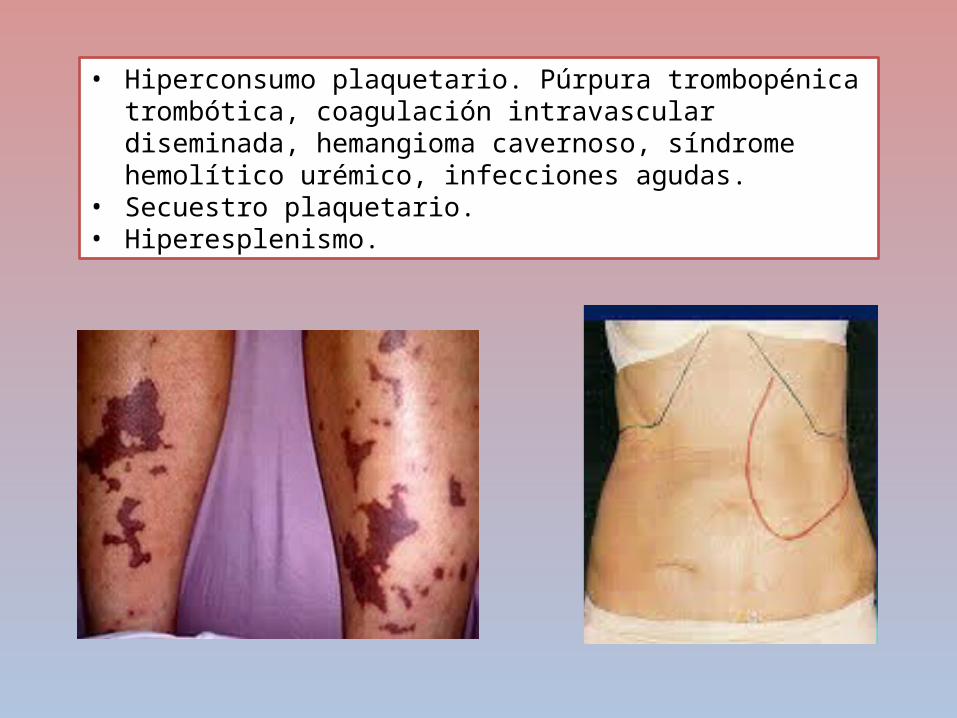
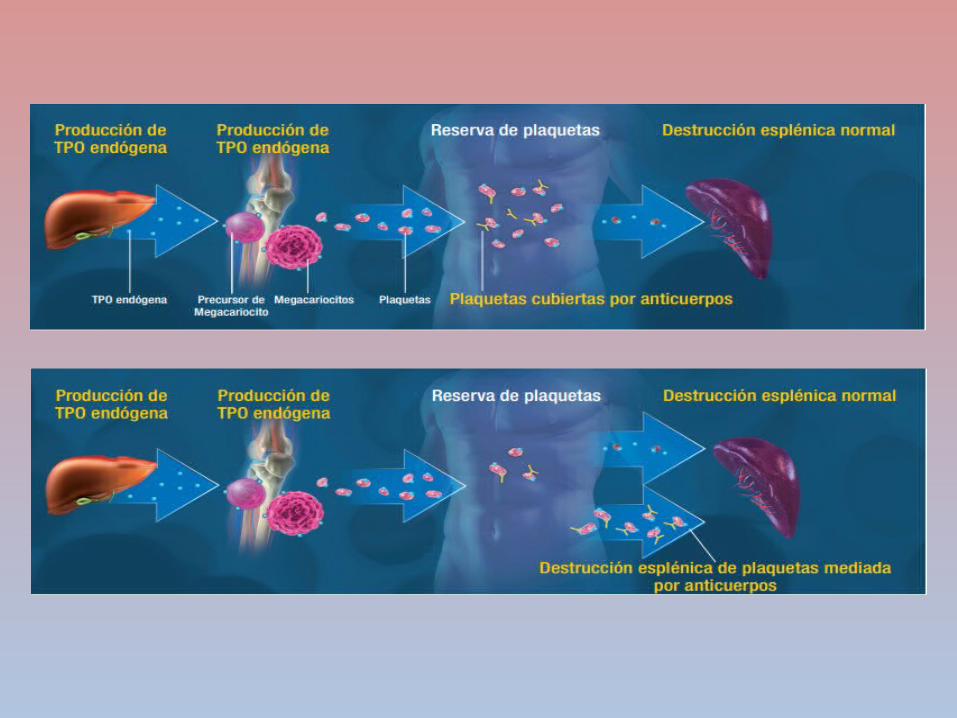
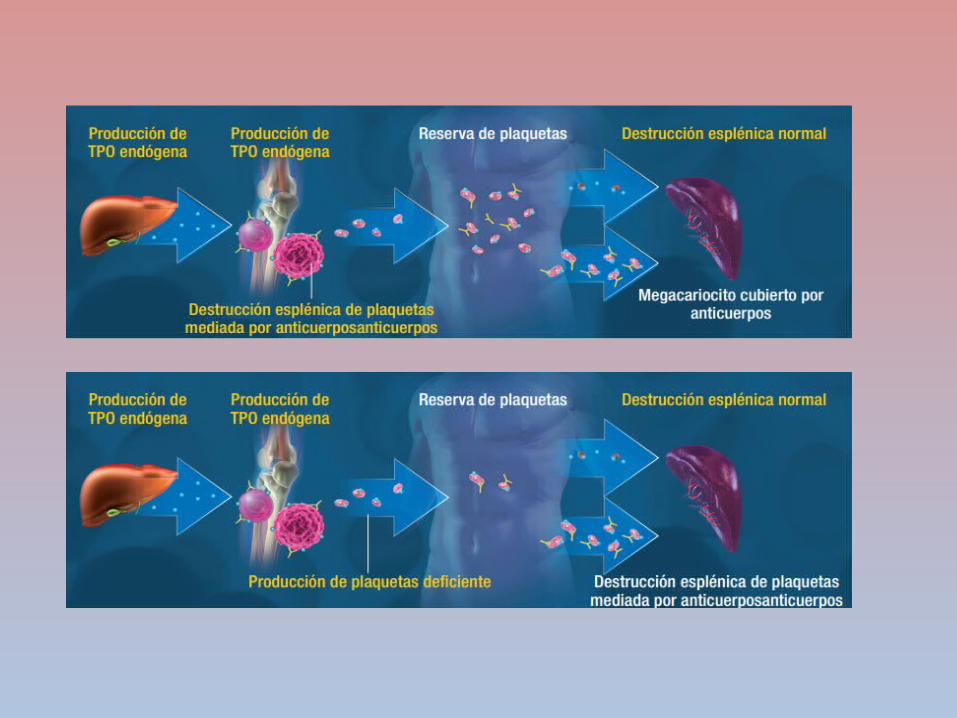
PURPURA TROMBOCITOPÉNICA INMUNE
ENFERMEDAD
HEMORRAGICA
INMUNE
DESTRUCCIÓN
PREMATURA DE
PLAQUETAS
AUTOANTICUERPOS
CLASE IGG SE UNEN A
LA GLUCOPROT
EINA PLAQUETARI
A
SISTEMA FAGOCITICO MONONUCL
EAR
CONTEO PLAQUETAR
IO ES MENOR A
50000 PTI

CLASIFICACIÓN

CAU
SAS INFECCION VIRAL
EJ: HEPATITIS, LUPUS, VIH
Generalmente <15 años
EMBARAZO
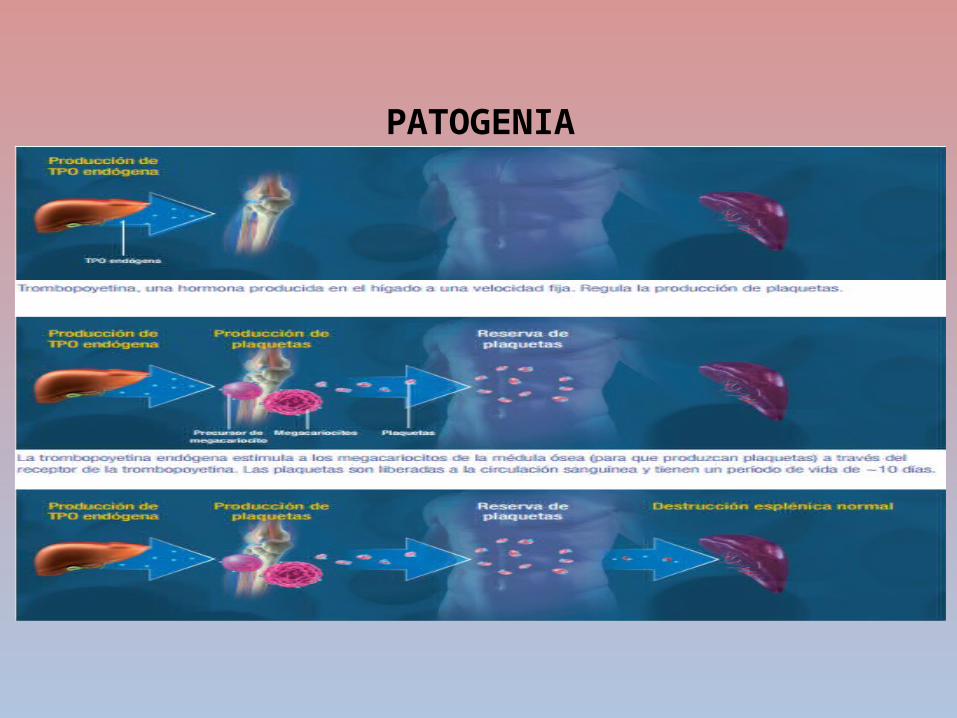
PATOGENIA
SIGNOS Y SINTOMAS
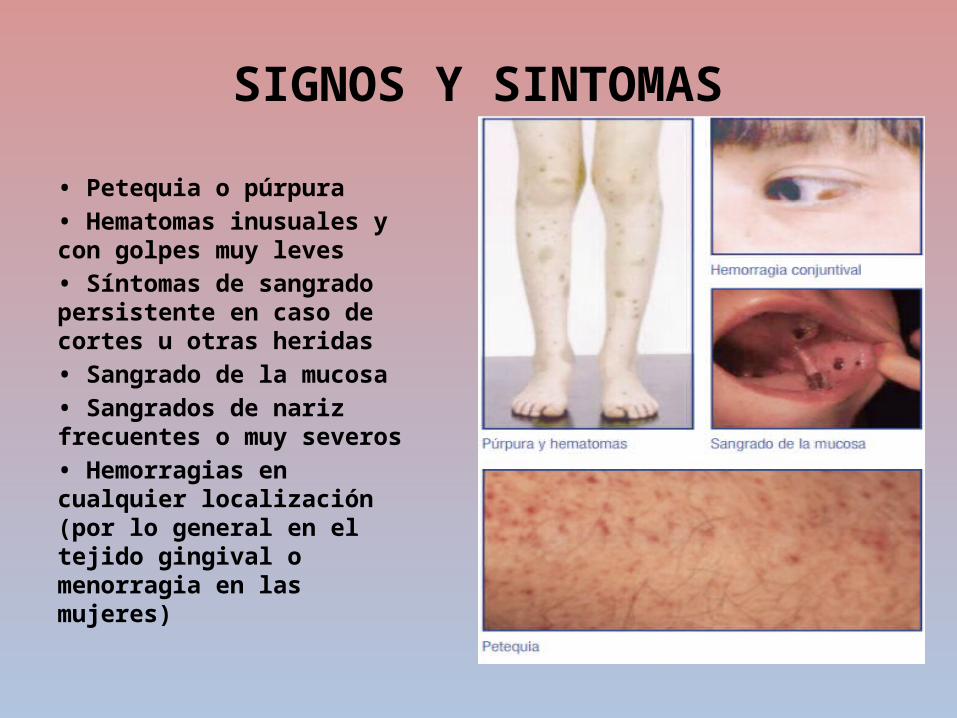
• Petequia o púrpura• Hematomas inusuales y con golpes muy leves • Síntomas de sangrado persistente en caso de cortes u otras heridas • Sangrado de la mucosa• Sangrados de nariz frecuentes o muy severos • Hemorragias en cualquier localización (por lo general en el tejido gingival o menorragia en las mujeres)

DIAGNÓSTICO

CONSECUENCIAS

TRATAMIENTO
ESPLENECTOMIA
PURPURA TROMBOCITOPENICA TROMBOTICA
DEFINICIONLa purpura trombocitopenia
trombotica es un trastorno de la sangre que actúa formándolos de
sangre coágulos en pequeños vasos sanguíneos en el cuerpo y origina una
disminución de las plaquetas.
Su nombre proviene de: purpura porque se presenta como moretones
en la piel, lo cual es característico de la enfermedad; tromocitopenica porque
causa la disminución de las plaquetas y trombotica porque forma trombos.

EPIDEMIOLOGIA • La incidencia en la ultima década se a estimado a 3.7 casos por año por
millo de personas, siendo mas frecuente en mujeres que en hombres 3:2
• La forma heredada de PTT es llamada Síndrome de Upshaw-Schülman, generalmente asociado a la deficiencia de enzima ADAMTS13
• Los pacientes con esta deficiencia desarrollan un PTT en ciertas situaciones clínicas (por ejemplo, infecciones) con incremento en los niveles del factor von Willebrand.
• Se ha reportado una cantidad de casos que representan alrededor de un 5-10% del total de casos de PTT

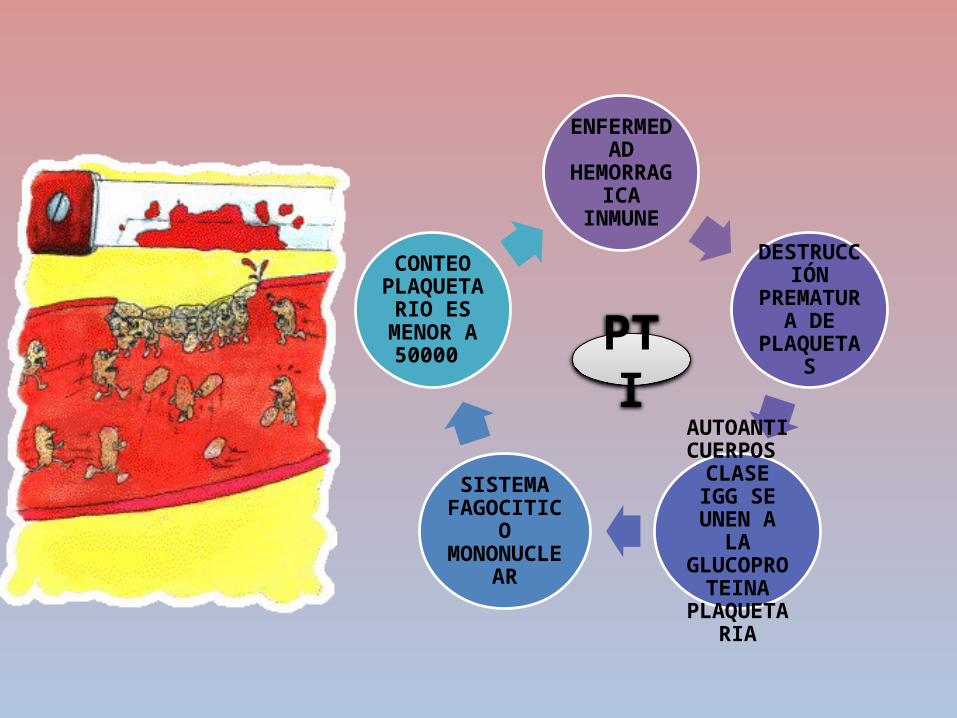
fisiopatologíaDepositos
antigenos en los vasos sanguineos
Inmunidad celular
Produccion de anticuerpos
contra antigenos
Activacion del sistema de
complementoDaño endotelial
Agregacion plaquetaria
Aumento del factor de Von
willebrand

CLINICA Anemia Trombocitopenia Alteración neurológica Fiebre y disfunción renal• Sangrado en la piel o membranas mucosas.• Anemia• · Cambios en el estado de conciencia.• · Confusión.• · Fatiga fácil. • · Mialgias y artralgias• · Fiebre.• · Dolor de cabeza.

• · Frecuencia cardíaca sobre 100 latidos por minuto.
• · Palidez• · Manchas purpurinas en la piel producidas
por pequeños vasos sangrantes cerca de la superficie cutánea (púrpura).
• · Dificultad respiratoria• · Cambios en el habla.• · Debilidad• · Color amarillento de la piel (ictericia).

DIAGNOSTICO· Nivel de actividad de ADAMTS 13.
· Bilirrubina.
· Frotis de sangre.
· Nivel de creatinina.
· Nivel de deshidrogenasa láctica (DHL).
· Conteo de plaquetas.
· Análisis de orina.
· Electroforesis para el factor de Von Willebrand

Diagnostico diferencial
Lupus eritematoso
Artritis
Sindrome de anticuerpo antifosfolipidicos
Vasculitis

TRATAMIENTO• Se utiliza el intercambio de plasma para remover los
anticuerpos que están afectando la coagulación de la sangre y también reponer la enzima faltante. Este tratamiento se repite diariamente hasta que los exámenes de sangre muestran mejoría.
• Esplenectomía• Corticoides:0,5 mg/Kg/día vía oral Inmunosupresores ciclofosfamida 1-5 mg/Kg/día. :
ciclosporina, tacrolimus, sirolimus
Vincristina, Antiagregantes, Inmunoglobinas a altas dosis

Otras enfermedades de la hemostasia primaria

Enfermedad de Von Willebrand
• Diátesis hemorrágica hereditaria más frecuente
• Anomalías cuantitativas y/o cualitativas del factor vW
• En el laboratorio existe alteración de la adhesión plaquetaria con ristocetina
Formas clínicas
Casos leves: aparece tras
cirugía o traumatismos
Característico un tiempo de
sangría prolongado con
plaquetas normales
Disminución de la concentración de
factor Von Willebrand
Actividad reducida del
factor VIII
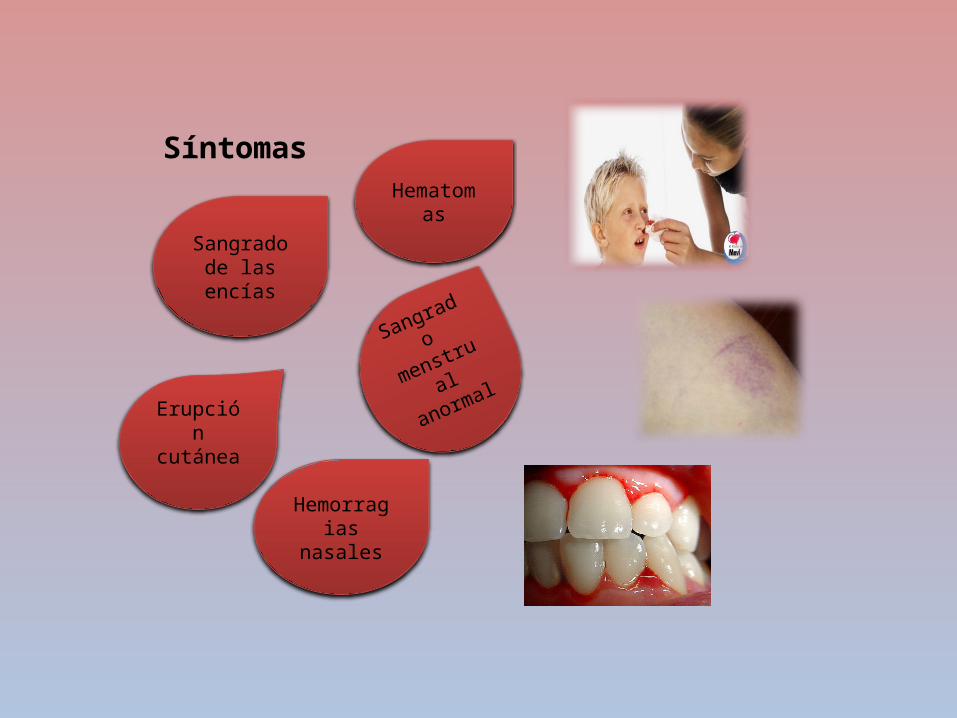
Síntomas
Sangrado
menstrual
anormal
Sangrado de las encías
Hematomas
Erupción cutánea
Hemorragias
nasales

Clasificacion:La EvW tipo 1: forma más común. FvW menores a los normales. Los síntomas generalmente son muy leves. es posible presente hemorragias graves.
La EvW tipo 2: defecto en la estructura del FvW. La proteína del FvW no funciona adecuadamente, provocando una actividad menor a la normal. Los síntomas generalmente son moderados.
La EvW tipo 3: la más grave. Tienen muy poco o no tienen FvW. Los síntomas son más graves. Pueden presentar hemorragias en músculos y articulaciones, algunas veces sin ser provocadas por una lesión.
Adquiridas
• Anticuerpos contra el factor vW• Lupus eritematoso sistémico• Gammapatías monoclonales • Procesos linfoproliferativos o
hipernefroma• La expresión clínica más
frecuente de esta enfermedad es el sangrado ORL y las equimosis.

Diagnóstico• 1: Historia personal de hemorragias
mucocutaneas excesivas• 2: Historia familiar de hemorragias excesivas• 3: Pruebas de laboratorio de hemostasia
consistentes con la EVW
• Tiempo de Sangria• Nivel de factor VIII• Conteo de Plaquetas

Tratamiento
• Crioprecipitados • Acetato de desmopresina (DDAVP) aumenta la
liberación de factor vW

Telangiectasia hemorrágica hereditaria (enfermedad de Rendu-Osler-Weber)
• trastorno autosómico dominante• consistente en una malformación vascular
congénita• vasos reducidos a un simple endotelio• sin soporte anatómico ni capacidad contráctil• Producen dilataciones vasculares• telangiectasias • Fístulas arteriovenosas, que sangran
espontáneamente o tras traumatismo mínimo
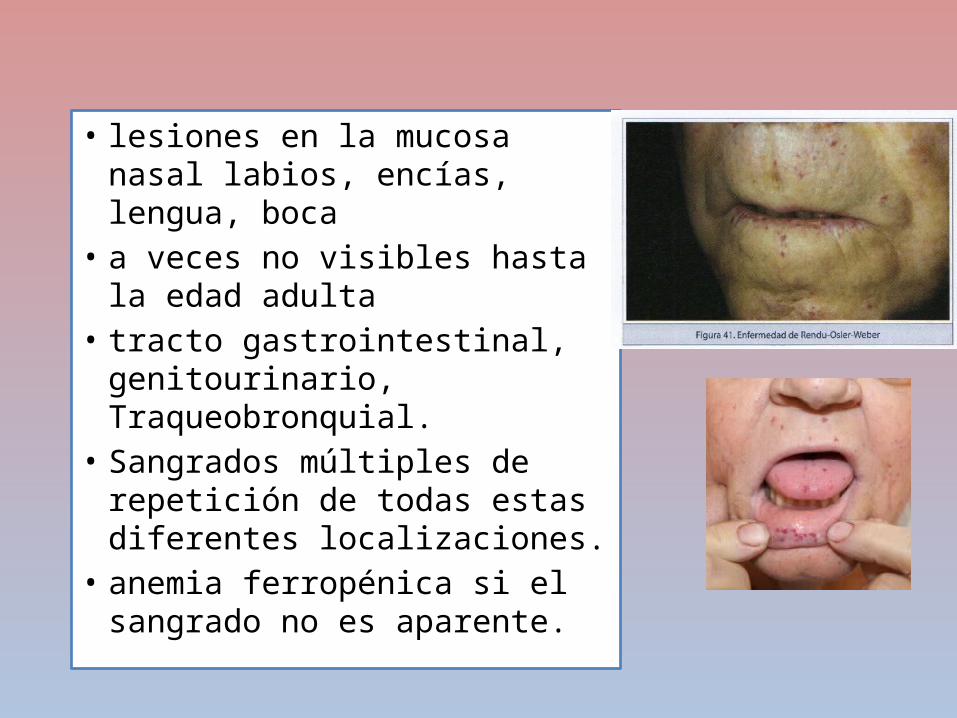
• lesiones en la mucosa nasal labios, encías, lengua, boca
• a veces no visibles hasta la edad adulta
• tracto gastrointestinal, genitourinario, Traqueobronquial.
• Sangrados múltiples de repetición de todas estas diferentes localizaciones.
• anemia ferropénica si el sangrado no es aparente.

•Gracias
Top Related