







Idiomas
Páginas
Jurídico


FARMACOCINÉTICA
Modelo abierto de un compartimento, cinéticas
Datos plasmáticos
Constantes: kel, vida media, depuración, volumen de distribución, área bajo la curva

SISTEMA LADME FARMACOS EN FORMA LIBERACIÓN DEL FÁRMACO FÁRMACOS CRISTALINOS DISOLUCIÓN DE DOSIFICACIÓN EXPUESTOS A FLUIDOS EN DE CRISTALES SÓLIDA FORMA DE DOSIFICACIÓN EL TRACTO G.I. (P.EJ. DESINTEGRACIÓN DE TABLETAS) ABSORCIÓN
FÁRMACO DISUELTO FÁRMACO DISUELTO EN EL FLUJO SANGUÍNEO EN FLUÍDOS G.I. ELIMINACIÓN DEL FÁRMACO Y/O SUS METABOLITOS

Farmacocinética
Tres posibles cinéticas para explicar el proceso LADME:– Orden cero
– Primer orden
– Orden mixto (Cinética de Michelis-Menten)

Procesos
Tipo de cinética Ecuación de velocidad
Ecuación diferencial
Unidades de la constante, k
Orden uno (Primer orden)
dQ/dt = -k*Q Q = Qo*e –kt
lnQ = lnQo - kt
Tiempo-1
Cero orden dQ/dt = -ko Q = Qo – ko*t Masa/tiempo
Michaelis-Menten dQ/dt =
-Vm*Q/km + Q
T = 1/Vm (Qo-Q+Km*lnQo/Q
Michaelis-Menten, cantidad de fármaco muy inferior a Km
dQ/dt = - Vm*Q/Q = -ko (ecuación de orden cero)
Ko = Vm
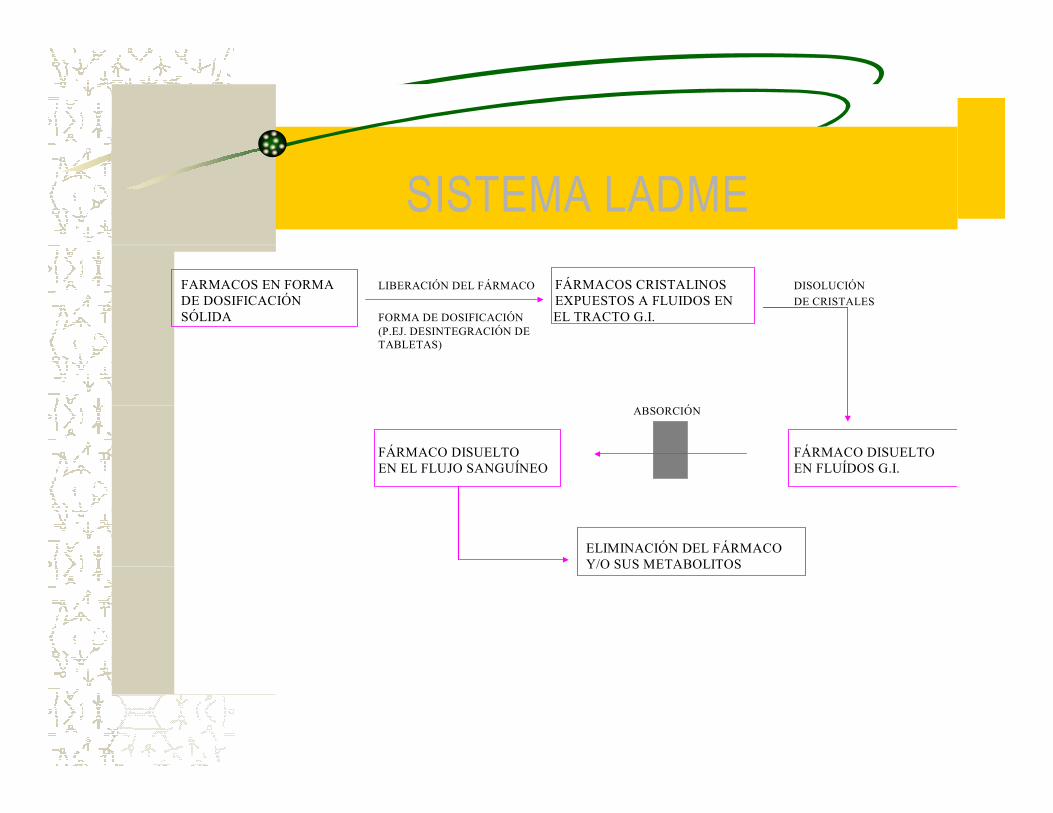
SISTEMA LADME FARMACOS EN FORMA LIBERACIÓN DEL FÁRMACO FÁRMACOS CRISTALINOS DISOLUCIÓN DE DOSIFICACIÓN EXPUESTOS A FLUIDOS EN DE CRISTALES SÓLIDA FORMA DE DOSIFICACIÓN EL TRACTO G.I. (P.EJ. DESINTEGRACIÓN DE TABLETAS) ABSORCIÓN
FÁRMACO DISUELTO FÁRMACO DISUELTO EN EL FLUJO SANGUÍNEO EN FLUÍDOS G.I. ELIMINACIÓN DEL FÁRMACO Y/O SUS METABOLITOS

Procesos
Factores limitativos. Los procesos son interdependientes y consecutivos. Si la velocidad de liberación del fármaco <<<< velocidad de absorción La liberación del fármaco es limitativa

En los estudios siempre existen riesgos, es necesario enfrentarlos de una manera sensible y con responsabilidad.


Estudios
En animales de experimentación (rata, perro, puerco enano, ). Toma de muestras de todos los tejidos (distribución del fármaco)En humanos, sangre y orina (datos plasmáticos y cantidades excretadas en orina de fármaco inalterado). Ideal.– Curvas de nivel plasmático– Curvas de excreción urinaria


Análisis compartimental

Modelos compartimentales
Compartimento, representa fracción de material biológico en el que el fármaco se supone distribuye uniformemente y presenta las mismas propiedades cinéticas (concepto cinético). – Puede ser un sector acuoso que ocupa un volumen
determinado (V), y contiene una cantidad de fármaco (Q)
– Concentración de fármaco (C), C = Q/V
Q VC k


Modelo compartimental
Simplificación: se considera al organismo por el mínimo número de compartimentos, siempre que se pueda interpretar una descripción farmacocinética
Lineal: Los procesos cinéticos corresponden a una cinética de primer orden (proporcionalidad directa entre concentración y velocidad o velocidad y cantidad y no hay variación de volumen), los parámetros farmacocinéticos no cambian al cambiar la dosis

Modelo compartimental
No lineal: Al variar la dosis de un fármaco dado, el valor de uno o más parámetros farmacocinéticos cambia, y la conc. A un tiempo dado no es directamente proporcional a la variación de la dosis.

Modelo monocompartimental
Es el más sencillo
Un solo compartimento donde existe una distribución instantánea
Propuesto por Widmark y Tanberg en 1924; Dost lo desarrolló a aplicaciones clínicas.
Compartimento único de carácter acuoso.

Considerando 12 h

Modelo monocompartimental
Esquema: Dosis iv, rápida
Div
Q Vd QelC kel
Ecuación de desaparición (eliminación) de fármaco:dQ/dt = -kel*Q, entre Vd:dC/dt = -kel*C

Modelo monocompartimental
Cuando la administración no es intravenosa rápida, sino extravasal, existe la fase de absorción. Esquema:
Qaka Q Vd Qel
C kel
Ecuación: dQ/dt = ka*Qa – kel*Q

Modelo monocomopartimental
Bolo intravenoso– Distribución instantánea y
uniforme (ideal)– Se facilita el concepto,
parámetros y constantes farmacocinéticas para explicar el tránsito del fármaco en el organismo
– El modelaje es útil para la estimación de dosis y pautas de dosificación mas adecuadas para cada medicamento
Cantidad de fármaco Q, que varía a lo largo del tiempo en el compartimento y estáregido por el volumen acuoso Vd (constante) y la eliminación del organismo kel (constante de velocidad de primer orden):
dQ/dt = -kel * QIntegrando: ln Q –lnQo = -kel
*(t-0)Q = Qo * e-kel*t
Q = D * e -kel*t, o sea C = D/Vd * e -kel*t C = Co * e -kel*t

Parámetros farmacocinéticos
Constante de eliminación, kel.– Proceso de primer orden– Valor absoluto de la inclinación
de la recta en la ecuación con ln:
lnC = -kel * t + lnCoEn base log decimal:Log C = - kel/2.303 * t + log Co- Unidades kel = dQ/dt/Q - Kel = (mg/min)/mg = 1/min =
min-1
Kel = velocidad de eliminación del fármaco/cantidad de fármaco remanente en el organismo


Gráficas en papel milimétrico y semilog.

Parámetros farmacocinéticos
Vida media biológica de eliminación, t1/2 :– Información acerca de la
permanencia o fugacidad del fármaco en el organismo
– Tiempo en que una determinada concentración del fármaco se reduce a la mitad de su valor
– Proceso cinético de orden uno.– Valor constante e
independiente de la dosis administrada y de la concentración inicial considerada.

Parámetros farmacocinéticos
Significado de e-kel*t
Si tenemos Q/D = e-kel*t
Q/D, fracción de dosis remanente en el organismo.
Si t = n * t1/2T = n * (0.693/kel)Q/D = e-0.693n
e-0.693n= ½Fracción de dosis remanente =
(1/2)n
Ecuación anterior indica:– Cuando ha transcurrido
una vida media (n=1) se ha eliminado el 50% de la dosis
– Si n=2, solo queda el 25% de dosis, se ha eliminado el 75% de la misma.


Parámetros farmacocinéticos
Volumen de distribución Vd:– Teóricamente representa el
volumen acuoso del organismo en el cual se distribuye una cantidad dterminada de fármaco de acuerdo con sus características fisicoquímicas.
– En este modelo el volumen es en el que instantáneamente se distribuye el fármaco, la conc. plasmática equivale a la conc. en el organismo, ya que el plasma es una parte alícuota del volumen.
Vd = Q/C
Vd = D/Co, mg /mg/L
Unidades, L ó L/kg (D=mg/kg)
Unicamente se calcula el Vd tras la administración iv. (cuando se conoce la cantidad de fármaco remanente en el organismo D a tiempo cero y Co)


Volumenes acuosos de los diferentes compartimentos corporales
Compartimento acuoso
% del peso corporal
% de agua total
Plasma
Agua extracelular total
Agua intracelular total
Agua corporal total
4.5
27
33
60
7.5
45
55
100

Valores de volumen de distrobución aparente de diferentes fármacos
Fármaco Vd (L) (70 kg) Vd (L/kg)
Warfarina
Ac. Salicílico
Gentamicina
Digitoxina
Diazepam
Propanolol
Digoxina
Imipramina
Cloroquina
Quinacrina
7
12
18
36
77
270
560
1050
18450
43400
0.11
0.17
0.25
0.51
1.1
3.9
8
15
235
620

Parámetros farmacocinéticos
Area bajo la curva de
niveles plasmáticos
(ABC)
– La mayoría de estudios farmacocinéticos se llevan a cabo utilizando curvas de niveles plasmáticos contra tiempo. (Papel milimetrado)
Cálculo mediante integración numérica:– Se basa en calcular el
área entre dos puntos experimentales consecutivos de la curva de niveles plasmáticos (METODO DE LOS TRAPEZOIDES):
– Area = ∆t * (C1+C2)/2, ∆t = t2-t1


Parámetros farmacocinéticos
Area bajo la curva de niveles plasmáticos(ABC) desde tiempo cero a tiempo t:
– ABCo-t = ∑o-t ∆t * (Ct-1 + Ct)/2
– El área total bajo la curvaABCo- ∞ = ABCo-t + ABCt-∞
- Unidades mg/L*h, ó mg*h*L-1
El valor de ABCo-t se obtiene por el método de los trapezoides , el de ABCo- ∞ , se obtiene de Co/kel (fracción extrapolada mas allá del último tiempo de toma de muestras).– Co es la conc. Plasmática del
último valor experimental (Ct)

Parámetros farmacocinéticos
Consideraciones de ABC:– El cálculo del valor del área al
primer trapecio (de tiempo cero a la primera toma de muestras) presenta la dificultad de que se desconoce el valor de la conc plasmática a tiempo cero y se calcula por extrapolación.
Consideraciones de ABC:• El valor de la fracción del
área extrapolada no se basa en ningún valor obtenido experimentalmente por lo que se desconoce el tramo recto semilogarítmico, para evitar errores se recomienda que el valor del área extrapolada no sea superior al 20% del área total.

Método de los trapezoides

Método de los trapezoides

Método de los trapezoides

Parámetros farmacocinéticos
Relación entre ABC y
Vd
– Vd = D/Co, y
– ABCo- ∞ = Co/kel
– Despejando
Co = ABCo- ∞ *kel
Sustituyendo en Vd
Vd = D/ ABCo- ∞ * kel , ó
ABCo- ∞ = D/ Vd*kel
Aclaramiento plasmático (Clp)– Es el volumen de sangre
totalmente depurado de fármaco por unidad de tiempo, por parte del órgano (renal, hepático, etc) o por el organismo
– Relaciona la velocidad de eliminación del fármaco con su conc. Plasmática al mismo tiempo: Cl = dQ/dt / C
Donde: dQ/dt = Clp * C

Parámetros farmacocinéticos
Aclaramiento plasmático (Clp)
Si dQ/dt = kel * Q y Vd = Q/C:
Kel * Q = Clp * C yKel * Vd * C = Clp * C donde Clp = Vd * kel,El aclaramiento plasmático
representa la eliminación del fármaco en función del volumen de distribución.
Si t1/2 = 0.693/kel
Clp = Vd * 0.693/ t1/2, o bien
T1/2 = Vd * 0.693/ Clp
Si el Vd y el Clp de un fármaco A presenta valores elevados, puede presentar la misma vida media biológica que un fármaco B con valores bajos de aclaramiento y Vd.

Parámetros farmacocinéticos
Relación entre Clp, ABC y Vd
Si dQ/dt = kel * Q y Vd = Q/C:dQ/dt = kel * Vd * C, odQ/dt = Clp * C.Si dQ = Clp*C*dt yƒo- ∞ dQ = ƒo- ∞ Clp*C*dtClp es constanteƒo- ∞ dQ = Clp * ƒo- ∞ C*dt,Q ∞ – Qo = Clp * ABCo- ∞A tiempo cero Q es cero y en
administración iv, Q a tiempo ∞ es la dosis D
D = Clp * ABCo- ∞
Clp = D / ABCo- ∞∞∞∞
ABCo- ∞ = D / Clp,
Si Clp disminuye, ABC aumenta
Si Clp = Vd * kel
Vd =Clp / kel = D / ABCo- ∞ * kel
Si kel= 0.693/t12
Vd = D*t1/2 / ABCo- ∞ * 0.693 o
Vd = 1.44 * D*t1/2 / ABCo- ∞∞∞∞
El cálculo de Vd se aplica también para infusión iv.

Modelo monocompartimental
Considerando el comportamiento del fármaco lineal, con modelo monocompartimental en una admon iv. (figuras siguientes):– Al aumentar la dosis D
aumenta proporcionalmente el valor de Co, kel NO se modifica (rectas paralelas).

Modelo monocompartimental
– Las rectas semilog conc./tiempo presentan inclinaciones mas pronunciadas cuanto menor es el valor de la vida media (t1/2 es inversamente proporcional a kel)

Modelo monocompartimental
– Si Vd es constante, variaciones en el aclaramiento (Clp) se observan diferentes inclinaciones de la recta semilog, que representa el proceso de eliminación
Top Related