







Idiomas
Páginas
Jurídico


DETERMINACIÓN DE LEISHMANIAVIRUS EN BIOPSIAS Y AISLAMIENTOS DE
PACIENTES CON DIAGNOSTICO CONFIRMADO DE LEISHMANIASIS MUCOSA Y
DE PACIENTES CON DIAGNÓSTICO DE LEISHMANIASIS CUTÁNEA EN FALLA
TERAPÉUTICA
Diana Marcela Parra Muñoz
Universidad Nacional de Colombia
Facultad de Medicina, Departamento de Salud Pública
Bogotá, D.C., Colombia
2017

DETERMINACIÓN DE LEISHMANIAVIRUS EN BIOPSIAS Y AISLAMIENTOS DE
PACIENTES CON DIAGNOSTICO CONFIRMADO DE LEISHMANIASIS MUCOSA Y
DE PACIENTES CON DIAGNÓSTICO DE LEISHMANIASIS CUTÁNEA EN FALLA
TERAPÉUTICA
Diana Marcela Parra Muñoz
Tesis presentada como requisito para optar al título de:
Magister en Infecciones y Salud en el Trópico
Director:
María Clara Echeverry G. MD-Ph.D
Laboratorio de Parásitología Universidad Nacional de Colombia
Codirector:
Clemencia Elena Ovale B. Ph.D
Centro Dermatológico- Federico Lleras Acosta- Bogotá-Colombia
Universidad Nacional de Colombia
Facultad de Medicina, Departamento de Salud Pública
Bogotá, D.C., Colombia
2017

4
«No dejaremos de explorar y el fin de nuestra
exploración sera encontrar el punto de partida y
conocer el lugar por primera vez»
Thomas Stearns Eliot

5
Agradecimientos
Dra. María Clara Echeverry, por la dirección de este trabajo, su respaldo y
acompañamiento.
Dra. Clemencia Ovalle, por la co-dirección de este trabajo y haberme dado la
oportunidad de iniciarme en la investigación.
Diana Alexandra Londoño M.Sc. y Carlos Esteban Franco M.Sc. por su apoyo en
la búsqueda de las biopsias de LM. Por sus observaciones técnicas, su amistad y
paciencia.
Dra. Myriam Consuelo López por su apoyo en el tratamiento de los modelos
murinos y su disposición para compartir sus valiosos conocimentos.
Alejandro Contreras y Catherine Aguilar B.Sc., por su apoyo invaluable en el
laboratorio. Por su actitud proactiva y generosa.
Juliana Laverde M. Sc. Y Samanda Aponte M.Sc., por sus observaciones técnicas
y su apoyo durante el estudio.
Grupo de jóvenes investigadores del Hospital Centro Dermatológico Federico
Lleras Acosta, por su disposición para colaborar con este estudio. A Jussep
Salgado por su apoyo en la tipificación de las biopsias.
Carlos Murcia, Angie Sanchez y Aura Sotelo por su compañía durante este proceso
de aprendizaje, su amistad y sus críticas siempre constructivas.
Mi familia, a Andres González y a Stefany Torres por su apoyo incondicional y
paciencia.
Dios por permitirme llevar a cabo este objetivo personal.
A las instituciones,
Laboratorio de Parasitología de la Facultad de Medicina de la Universidad Nacional
de Colombia, Sede Bogotá, en donde se ejecuto este estudio y de cuyo banco
provinieron las muestras clínicas de pacientes en falla terapéutica.
Programa Jovenes investigadores Colciencias, 2016 por mi financiación.
Hospital Centro Dermatológico Federico Lleras Acosta, por proveer las muestras
clínicas de pacientes con LM y poner a disposición su infrastructura y potencial
humano para la ejecución de este estudio.
Laboratorio de equipos comunes de la Facultad de Medicina de la Universidad
Nacional de Colombia, cede Bogotá en donde se ejecuto la mayor parte del estudio.

6
TABLA DE CONTENIDO
1 RESUMEN 10
2 ABSTRACT 12
3 INTRODUCCIÓN 14
4 MARCO TEORICO 16
4.1 GENERALIDADES DE LAS LEISHMANIASIS 16
4.1.1 DEFINICIÓN DE LA ENFERMEDAD. 16 4.1.2 AGENTE CAUSAL. 16 4.1.3 CICLO BIOLÓGICO. 17 4.1.4 TAXONOMÍA. 19 4.1.5 PRESENTACIONES CLÍNICAS. 20 4.1.6 EPIDEMIOLOGÍA. 21 4.2 LEISHMANIASIS CUTANEA. 22
4.2.1 RESPUESTA INMUNOLÓGICA DE LA LEISHMANIASIS CUTÁNEA. 22 4.2.2 TRATAMIENTO Y FALLA TERAPÉUTICA 26 4.3 LEISHMANIASIS MUCOSA. 29
4.3.1 RESPUESTA INMUNOLÓGICA DE LA LEISHMANIASIS MUCOSA. 30 4.3.2 OTROS FACTORES ASOCIADOS AL DESARROLLO DE LEISHMANIASIS MUCOSA. 31 4.3.3 TRATAMIENTO DE LA LEISHMANIASIS MUCOSA 33 4.4 LEISHMANIAVIRUS. 33
4.4.1 GENERALIDADES. 33 4.4.2 REPLICACIÓN Y TRADUCCIÓN. 36 4.4.3 EFECTO DE LRV EN EL HOSPEDERO HUMANO. 38 4.4.4 DETECCIÓN DE LRV EN MUESTRAS CLÍNICAS. 42
5 OBJETIVOS 47
5.1 OBJETIVO GENERAL 47
5.2 OBJETIVOS ESPECÍFICOS 47
6 METODOLOGÍA 48
6.1 TIPO DE ESTUDIO 48
6.2 CONSIDERACIONES ÉTICA 48
6.2.1 ASPECTOS AMBIENTALES. 49 6.2.2 BIOSEGURIDAD. 49 6.3 SELECCIÓN DE LAS MUESTRAS 49
6.4 CULTIVO DE AISLAMIENTOS PARASITARIOS 50
6.5 BIOPSIAS 50
6.6 PRODUCCIÓN DEL CONTROL POSITIVO PARA LA DETECCIÓN DEL VIRUS EN BIOPSIAS 50
6.7 EXTRACCIÓN DE RNA 51
6.8 RETROTRANSCRIPCIÓN 51
6.9 CONFIRMACIÓN DE LA CALIDAD DEL CDNA 51

7
6.10 IDENTIFICACIÓN DEL VIRUS EN AISLAMIENTOS PARASITARIOS 52
6.11 IDENTIFICACIÓN DEL VIRUS EN BIOPSIAS 53
6.12 IDENTIFICACIÓN DE ESPECIE EN AISLAMIENTOS PARASITARIOS Y BIOPSIAS 54
6.13 ANÁLISIS ESTADÍSTICO 54
6.14 ELABORACIÓN DEL MAPA DE DISTRIBUCIÓN DE ESPECIES 54
7 RESULTADOS 55
7.1 ESTANDARIZACIÓN DE LA DETECCIÓN DEL VIRUS EN EL LA CEPA DE REFERENCIA M4147.
55
7.1.1 DETECCION DEL VIRUS EN LA CEPA DE REFERENCIA M4147 POR PCR
CONVENCIONAL. 55 7.1.2 ESTANDARIZACIÓN DE LAS CONDICIONES DE DETECCIÓN DEL CDNA VIRAL POR QRT-
PCR. 56 7.2 DETECCIÓN DEL VIRUS EN AISLAMIENTOS CLINICOS DE PACIENTES CON LM Y LC EN FALLA TERAPÉUTICA. 58
7.3 ESTANDARIZACIÓN DE LA DETECCIÓN DEL VIRUS EN BIOPSIAS. 60
7.4 ALGORTIMO DE DETECCIÓN DEL VIRUS EN MUESTRAS CLINICAS DE PACIENTES CON LM Ó
LC EN FALLA TERAPÉUTICA. 63
7.5 FRECUENCIA DEL VIRUS 64
7.5.1 FRECUENCIA EN AISLAMIENTOS PARASITARIOS Y BIOPSIAS. 64 7.6 TIPIFICACIÓN DE ESPECIES 65
7.7 ANÁLISIS DE OTRAS VARIABLES. 66
7.7.1 ANÁLISIS ESTADÍSTICO. 66 7.7.2 ANÁLISIS GEOGRÁFICO. 67
8 DISCUSIÓN 68
9 CONCLUSIONES 74
10 BIBLIOGRAFÍA 75
11 ANEXOS 84
11.1 GLOSARIO 84
11.1.1 EFICIENCIA 84 11.1.2 COEFICIENTE DE DETERMINACIÓN (R2) 84 11.1.3 BASELINE 84 11.1.4 THERSHOLD CYCLE (CT) 84 11.1.5 THERSHOLD 85 11.2 PREPARACIÓN MEDIO DE CULTIVO SENEKJIE 85
11.3 PREPARACIÓN DE MEDIO SCHNEIDER 86
11.4 PREPARACIÓN DEL DE AGAROSA 87
11.5 PREPARACIÓN TAE 50X 88

8
Lista de Tablas
Tabla 1 Estadíos clínicos en LM Tabla 2 Cuadro resumen de ensayos de identificación de LRV en muestras clínicas. Tabla 3 Ensayo de titulación de cDNA para la estandarización de la qRT-PCR
usando como blanco molecular la region 5´UTR. Tabla 4 Cts promerdio y Tm° promedio obtenidos para la curva parasitaria en base
10 para la detección de LRV con los primers de Masayuki y colaboradores Tabla 5 Cts promerdio y Tm° promedio obtenidos para la curva parasitaria en base
10 para la detección de al cambiar condiciones del perfil térmico y utilizando dos concentraciones finales de primers LRV de Masayuki y colaboradoes.
Tabla 6 Cts promerdio y Tm° promedio obtenidos para los aislamientos de LM y LC en falla terapéutica negativos en la RT-PCR.
Tabla 7 Cts promerdio y Tm° promedio obtenidos para la curva del gen 18S usando cDNA proveniente de el modelo infectado con la cepa L. (V.) guyanensis M4147.
Tabla 8 Datos obtenidos aplicando el algoritimo para la detección de LRV en aislamientos y biopsias de LM ó LC en falla terapéutica.
Tabla 9 Datos de % de infectividad in vitro, IC50 para Glucantime y presencia o ausencia del LRV en aislamientos de LC en falla terapéutica.
Tabla 10 Datos de media, mediana, SD e IQR para la varible IC50. Tabla 11 Datos de media, mediana, SD e IQR para la varible % de infectividad.

9
Lista de Figuras
Figura 1 Esquema del aspecto de las formas amastigote y promastigote de Leishmania sp. al microscopio óptico.
Figura 2 Ciclo de vida de Leishmania sp Figura 3 Metaciclogénesis de Leishmania sp. en el intestino del vector. Figura 4 Espectro inmunológico de las Leishmaniasis en el huésped humano Figura 5 Mecanismo probable de acción de Sb+5. Figura 6 Ubicación en el genoma de sondas usadas en ensayo de Northern blot
para la identificación de LRV en aislamientos de Leishmania del Nuevo y Viejo mundo
Figura 7 Ubicación del sitio de clivaje en el extremo 5´UTR para autorregulación de la replicación y persistencia de la infección.
Figura 8 Ubicación de la unión de los ORF2 y 3 en el genoma viral. Figura 9 Cascada de señalización celular en respuesta a la liberación de dsRNA de
Leishmaniavirus en macrófagos infectados con L. (V.) guyanensis Figura 10 Diagrama del genoma viral de LRV1 con la ubicación de los primers
utilizados en el algoritmo de detección para aislamientos parasitarios. Figura 11 Amplificación del fragmento de 488pb correspondiente a la region de la
cápside mediante qRT-PCR y RT-PCR. Figura 12 Titulación de la cantidad de cDNA en la RT-PCR. Figura 13 Plot de amplificación en RT-qPCR para la curva parasitaria anteriomente
mencionada. Figura 14 Ejemplo de resultados obtenidos con los 4 aislamientos negativos, plot de
amplificación, curva melting y gel del aislamiento 707 de LM. Figura 15 Ejemplar murino infectado con lesion en hocico y verificación
parasitológica. Figura 16 Titulacion de la cantidad de cDNA de biopsia infectada con L. (V.)
guyanensis M4147 para la verifficacion de la calidad del mismo en la RT-PCR 18S.
Figura 17 Primer round de la RT-PCR anidada usando diferentes concentraciones de cDNA procedente de biopsia del modelo murino infectado con L. (V.) guyanensis M4147.
Figura 18 Segundo round de la RT-PCR anidada de los productos del primer round de las concentraciones 6.6ng, 19.98ng y 33.3ng.
Figura 19 Algortimo de detección de LRV en aislamientos y biopsias. Figura 20 Gráfica de biogotes obtenida del análisis de la prueba U de Mann-Whitney
para los grupos “LRV positivo” (1) y “LRV negativo” (0) respecto a las variables % de infectividad e IC50.
Figura 21 Mapa de distribución geográfica de los aislamientos y biopsias negativos y positivos paral LRV.

10
1 RESUMEN
La leishmaniasis es una enfermedad de transmisión vectorial, endémica en regiones
tropicales y subtropicales, que puede tener compromiso en piel, mucosa y vísceras
dependiendo de la especie infectante y la condición inmunológica del huésped. Es una
enfermedad de baja mortalidad y alta morbilidad con cerca de 2.4 millones de años de vida
ajustados por discapacidad.
En Colombia la forma cutánea aporta el 95% de los casos reportados anualmente y aunque
su pronóstico es más favorable se presentan como principales complicaciones la falla
terapéutica o no respuesta al tratamiento y la producción de cuadros con compromiso
mucoso.
La falla terapéutica o no respuesta al tratamiento se registra en alrededor del 25% de los
casos tratados con antimoniato de meglumina, primeria línea de tratamiento en el país. La
respuesta al tratamiento está asociada a múltiples factores que incluyen la especie del
parásito, la edad y estatus inmunológico del huésped, la localización de las lesiones y la
vía de administración y duración del tratamiento.
Del 3-10% de las personas que padecen Leishmaniasis cutánea (LC) pueden desarrollar
leishmaniasis mucosa (LM) de manera concomitante o tardía. Esta forma complicada de
la LC que se caracteriza por afectar principalmente la mucosa nasofaríngea, es producto
de una diseminación linfo-hematógena de los parásitos de Leishmania spp. a partir de una
lesión cutánea distante o del inoculo del parásito en proximidad de las regiones nasal u
oral. En Colombia representa el 1.3% de los casos de Leishmaniasis según informó el
Sistema Nacional de Vigilancia en Salud Pública (SIVIGILA) para el año 2015. Esta
complicación es de gran impacto en la población por ser altamente deformante e
incapacitante. La presentación de la complicación LM es considerada de origen
multifactorial, involucrando componentes asociados tanto al huésped como al parásito.
Actualmente una hipótesis que ha tomado fuerza para explicar las complicaciones de la
LC, ya sea la falla terapéutica y/o el compromiso a nivel mucoso, es la infección del parásito
infectante por un virus de familia Totiviridae y género Leishmaniavirus. Este virus podría

11
estar involucrado en una exacerbación de la respuesta inmune y la persistencia del
parásito en los macrófagos.
Sin embargo las frecuencias reportadas del virus varian entre 0 y e 87% en los pacientes
con LM o LC en falla terapéutica, hecho que refleja una alta variablidad en la presencia del
virus que puede deberse a multiples factores incluyendo componenetes técnicos
asociados a la muestra y el método usado para la detección viral.
El presente estudio pretendió determinar la frecuencia con la que se detecta el virus en
una serie de biopsias y aislamientos parasitarios provenientes de pacientes con LM o LC
en falla terapéutica. Lo anterior fue posible mediante la combinación de técnicas de RT-
PCR y RT-qPCR que han sido descritas de manera individual en la detección de los virus
pertenecientes a este género. Se describen los hallazgos obtenidos al analizar un total de
31 muestras clinicas: 10 biopsias de pacientes con LM, 6 aislamientos parasitarios a partir
de lesiones mucosas y 15 aislamientos parasitarios a partir de pacientes que presentaron
falla terapéutica o recidiva al tratamiento con Glucantime®.
Se construyó un algoritmo diagnóstico que permitió detectar el virus en el 56.67% del total
de las muestras analizadas, las cuales estaban infectadas por parásitos de la especie L.
(V.) braziliensis, L. (V.) panamensis y Leishmania sp. y no se detectó un patrón de
distribucion geográfico predominante. En los aislamientos parasitarios asociados a la falla
terapéutica o recidiva en las lesiones cutáneas tratadas con Glucantime®, la presencia
viral no se relacionó con el porcentaje de infectividad ni con la sensibilidad “in vitro” de los
amastigotes intracelulares al Glucantime®.

12
2 ABSTRACT
Parasites from leishmania genus are associated with pleiomorphic human disease with
visceral or cutaneous manifestations. The clinical behaviour of the disease depends on
host’s factors and on parasites species. Leishmania genus has a worldwide distribution on
tropical regions, however the species circulating at the Old world have differences with
those found at the New world, particularly with species grouped into the Leishmania Viannia
subgenus. Those last are predominant in South America where they generate complicated
cutaneous forms of the diseases as mucosal (ML) form and non-responding cutaneous
leishmaniasis (NRCL) without parasite resistance.
The occurrence of the above-mentioned complications could be associated with the
presence of a cytoplasmic virus in the infecting parasite, known as Leishmania RNA virus
(LRV1). However the frequency of the virus in clinical samples ranges from 0 to 87% without
regular data that strongly support association. Those apparent inconsistent results could
obey to: the use of a diverse sort of experimental approaches, overlooking the parasite
species factor and the type of clinical samples that had been analysed.
In order to avoid detection bias when looking for LRV1 in clinical samples, the present study
used complementary approaches to assurance the reliability in the LRV1 frequency
informed. Taking into the account the parasites species and looking for differences in the
result when the source sample corresponds with isolates parasite of with lesion biopsies.
Also to determine the impact of the LRV-1-infection on the amastigote fitness, the
percentage of infections on human macrophages has been assessed as their in vitro
susceptibility to the first choice drug in South America for this parasite, Glucantime®.
31 samples were analysed from different clinical background 16 samples from patients with
ML (10 biopsies and 6 isolates), 15 from patients with NRCL and 1 reference isolate.
Analysing data by type of approach LRV1 observed frequencies were in RT-PCR and RT-
qPCR: 42.86%. Nested PCR: 88.8%. By clinical condition ML: 73.3%, NRCL: 40%. By type
of sample: biopsies: 88.8% and isolates: 42.86%, by parasite species: L. (V.) braziliensis:
76,4%, L. (V.) panamensis: 11,76% y Leishmania sp.: 11,76%.

13
Using One-way ANOVA on ranks, nor differences in Infectivity to the macrophages
(p=0.523) neither in Glucantime® susceptibility (p=0.728) were observed between L.
braziliensis-LRV1 positive (median value of infectivity 47.7% and of IC50, 120.7 ug/ml) and
L. braziliensis-LRV1 negative (median value of infectivity 44.9% and of IC50, 121.6 ug/ml).
Suggesting that the presence of the virus does not affect intracellular parasite fitness at
least at those levels.
Findings from the present study indicate that LRV-1 detection is highly dependent on the
type of sample, the parasite load and the experimental approach. They also suggest that
LRV1 is associated to L. (V.) braziliensis linked to ML in a higher frequency than to parasites
from same species related to NRCL. Also there is a higher detection of the virus in biopsies
than in isolates and this was always found in L. (V.) braziliensis but not L. (V.) panamensis.

14
3 INTRODUCCIÓN
La leishmaniasis es una parásitosis con una amplia distribución geográfica producida por
un protozoo del género Leishmania y trasmitida por un vector de la subfamilia
Phlebotominae cuyo género varía en el Nuevo y Viejo mundo. Son consideradas
endémicas en aproximadamente 98 países, afectando a 12 millones de personas y
poniendo en riesgo a 350 más [12, 13].
Estas enfermedades se caracterizan por presentar compromiso tegumentario y visceral.
Dentro de la forma tegumentaria se reconocen varios tipos, en donde la LC localizada
resulta ser la más frecuente, a diferencia de la LM o LC difusa, las cuales se consideran
complicaciones [14].
La LC localizada es causada por la picadura directa del vector usualmente en zonas
expuestas del cuerpo como extremidades superiores, inferiores y cara. La lesión aparece
usualmente entre 3-6 semanas posteriores a la picadura y tiene una morfología
característica. En las Américas, las especies de parásitos comúnmente incriminadas a esta
forma de la enfermedad son L. (V.) panamensis, L. (V.) braziliensis, L. (V.) guyanensis, L.
(V.) peruviensis y el complejo mexicana [15, 16]. La ocurrencia de LC está asociada en su
mayoría a la intromisión del hombre en zonas selváticas por factores socioeconómicos,
hipótesis coherente con los registros de la enfermedad en los que predominan como lugar
de procedencia regiones selváticas y pacientes del sexo masculino en edad productiva
[13].
La LM puede presentarse en el 3-10% de pacientes con previa LC localizada. Esta forma
es de desarrollo tardío y es considerada producto de la diseminación de los parásitos por
vía linfática o hematógena a las mucosas. También puede darse por la picadura del vector
en zonas cercanas a la mucosa oro-faríngea como es el caso de las lesiones faciales [17,
18]. La LM está limitada a las especies de Leishmania del subgénero Viannia,
principalmente al complejo braziliensis [18-20] y afecta la mucosa nasal y oral ocasionando
lesiones deformantes que dificultan la respiración, fonación y deglución [17].
En Colombia según el ultimo informe SIVIGILA del 2015 se presentaron 5113 casos de LC
y 67 de LM; la mayor tasa por cada 100.000 habitantes provino del departamento de

15
Guaviare para ambas formas de la enfermedad y se evidenció mayor impacto en la
población masculina en un amplio rango de edades [13].
Varios autores refieren que la respuesta al tratamiento en LC y el desarrollo de
complicaciones de la misma como la LM, responden a múltiples factores como la edad y
el sexo del huésped, estado inmunológico, enfermedades concomitantes, malnutrición, co-
infecciones bacterianas de las lesiones, la especie infectante y sus polimorfismos. Una
hipótesis que ha tomado fuerza es la presencia de un virus tipo RNA en el parásito
infectante conocido com Leishmania RNA virus (LRV) [21, 22].
Este virus perteneciente a la familia Totiviridae y al género Leishmaniavirus se ha asociado
con la exacerbación de la respuesta inmunológica del huésped, induciendo la
supervivencia del parásito en los macrófagos y por tanto fomentando su persistencia en el
individuo, lo que incrementa la probabilidad del desarrollo metastásico de la enfermedad
LC localizada [4, 11, 21, 23].
Sin embargo la serie de casos estudiados a la fecha corresponde a un número restringido
y no se ha logrado identificar la presencia del virus en todas las muestras clínicas
pertenecientes pacientes con cuadros mucosos [24-27] o que no hayan presentado
respuesta al tratamiento [27-30]. Por tanto el presente estudio quiso determinar la
frecuencia con la que se detecta el virus en una serie de muestras clínicas y aislamientos
parasitarios provenientes de pacientes colombianos que presentaron complicaciones a
partir de un cuadro cutáneo ya sea por la presencia de LM o por la respuesta fallida al
tratamiento con Glucantime®.
Mediante la construcción de un algoritmo diagnóstico que combina las técnicas RT-PCR y
RT-qPCR, el presente trabajo optimizó la detección del virus en diferentes tipos de
muestras clínicas, determinando la frecuencia de la presencia viral en muestras de
pacientes con LM y LC en falla terapéutica. En los aislamientos parasitarios asociados a
la falla terapéutica o recidiva se determinó el compromiso del fitness parasitario por la
infección viral mediante la evaluación de parametros como la infectividad parasitaria a
macrófagos y la susceptibilidad “in vitro” al Glucantime®. Finalmente se describe la
distribución geográfica en Colombia de L. (V.) braziliensis infectada con LVR1, en el grupo
de muestras estudiadas.

16
4 MARCO TEORICO
4.1 GENERALIDADES DE LAS LEISHMANIASIS
4.1.1 Definición de la enfermedad.
La Leishmaniasis es una enfermedad infecciosa causada por protozoarios del género
Leishmania, los cuales parasitan los macrófagos del huésped mamífero, generando una
sintomatología característica en vísceras o en piel y/o en mucosas. Se considera una
zoonosis debido a que las especies involucradas infectan, además del hombre, a
mamíferos como perros, osos perezosos, zarigüeyas y otros roedores, que actúan como
reservorios. Su transmisión se da por la presencia en el ciclo biológico de un vector hembra
del género Lutzomyia (Nuevo Mundo) y Phlebotumus (Viejo Mundo), que en la picadura
inyecta los parásitos al huésped [3, 17, 31-33].
4.1.2 Agente causal.
En el hospedero vertebrado la replicación es predominantemente asexual [3, 33], mientras
que en el vector además de la reproducción asexual [34], hay hallazgos que señalan una
recombinación genética por vía sexual [35, 36].
Dentro de su ciclo de vida se presentan dos
estadios: amastigote y promastigote,
haciéndolo digenético. El amastigote es la
forma intracelular e inmóvil del parásito, es
esférico u ovalado, con un diámetro que oscila
entre 1-5 µm, presenta núcleo grande,
kinetoplasto, axonema y blefaroplasto. Es el
estadio infectante para el vector y se
considera la forma diagnóstica en el huésped
mamífero [7, 37].
Por otro lado, el promastigote es la forma
extracelular y móvil, es alargado y delgado, su
longitud oscila entre los 15-20 µm, presenta
núcleo central grande, kinetoplasto próximo al
Fig. 1. Esquema del aspecto de las formas amastigote y promastigote de Leishmania sp. al microscopio óptico [7].

17
extremo anterior, axonema, blefaroplasto y un flagelo largo que puede alcanzar la longitud
total del parásito. En su forma metacíclica, es el estadío infectante para el huésped
mamífero (Fig. 1) [3, 7, 33, 37].
El kinetoplasto corresponde a DNA mitocondrial (kDNA) que debido a su alto nivel de
condensación incorpora los componentes básicos de coloraciones diferenciales, dando un
aspecto único de DNA extra-nuclear y que da nombre al orden Kinetoplastida. El
kinetoplasto se encuentra organizado en una condensada red de maxi (aprox. 40-50) y
mini-círculos (aprox. 5000-10000) [38] y ha sido comúnmente usado como blanco
molecular para la identificación de especies de Leishmania [39-42].
4.1.3 Ciclo biológico.
En el ciclo biológico, el vector infectado inocula al mamífero aproximadamente 10-1000
promastigotes metacíclicos durante la picadura [3]. Éstos son fagocitados por los
macrófagos y otras células fagociticas como células dendríticas y neutrófilos, en donde se
transforman en amastigotes y su replicación constante en la vacuola parasitófora genera
una lisis celular que resulta en la infección de otro gran número de macrófagos ya sea por
continuidad o por un fenómeno conocido como el caballo de Troya (Fig. 2) [15, 43].
Fig. 2. Ciclo de vida de Leishmania sp. [2, 3].

18
Cuando el vector se alimenta del huésped infectado, ingiere macrófagos con amastigotes,
que en su intestino se convierten en promastigotes procíclicos en un lapso de 24-48 horas
después de la ingestión. Una vez los parásitos han alcanzado cierta densidad poblacional,
se lleva a cabo un proceso conocido como metaciclogénesis (Fig. 3) [2, 44, 45].
La metaciclogénesis prepara al parásito para soportar importantes cambios de temperatura
y pH en el huésped mamífero. Este proceso consiste en transformar promastigotes
procíclicos, forma no infectiva caracterizada por adherirse al intestino del vector y
replicarse en él, en promastigotes metacíclicos, forma altamente infectiva e incapaz de
adherirse a la pared del intestino del vector ni de reproducirse. Esta última forma resulta
ser más pequeña y con gran movilidad, lo que facilita su migración a la probóscide del
vector; además presenta modificaciones bioquímicas que le confieren alta resistencia a las
barreras inmunológicas impuestas por el huésped mamífero (Fig. 3) [32, 46].
El proceso que se lleva acabo en el intestino del vector inicia posterior a la ingesta, cuando
la sangre infectada es cubierta en el intestino medio posterior por una matriz peritrófica
que protege los amastigotes de las proteasas intestinales y en donde ocurre la conversión
a promastigotes procíclicos, formas móviles que se replican abundantemente previa
transformación a formas más delgadas y alargadas denominadas nectomonas (48-72
horas) [5, 32, 45, 47-50].
Debido a la degradación de la matriz peritrófica se escapan nectomonas, se ubican en el
lumen del intestino medio, inundan la parte anterior del mismo y se incrustan por medio de
su flagelo a las microvellosidades migrando solo después de la excreción de la sangre
metabolizada al intestino medio torácico en donde se transforman a formas pequeñas
conocidas como leptomonas (4-7 días). Las leptomonas producen una sustancia
gelatinosa denominada PSG (promastigote secretory gel) que las recubre. Una vez
empiezan a invadir la válvula estomodeal, las leptomonas se convierten en haptomonas y
finalmente en promastigotes metacíclicos (5-7 días). El taponamiento de la válvula
estomodeal debido a la alta desidad parasitaria hace que la sangre succionada por el
vector no pueda ingresar al intestino y se genere un reflujo que devuelve al sitio de
picadura, parásitos infectantes para el huésped mamífero [5, 32, 45, 47-50].

19
Todas las formas promastigotes metaciclogénicas, a diferencia de los amastigotes, se
caracterizan por tener altas concentraciones en su superficie de Lipofosfoglicano (LPG) y
una metaloproteasa de zinc que confiere propiedades importantes de virulencia (GP63),
mientras que los únicos cambios bioquímicos dentro de las formas promastigotes en el
proceso de la metaciclogénesis, hacen referencia al grosor de LPG y los carbohidratos que
conforman algunos de sus dominios [32, 44].
4.1.4 Taxonomía.
La Leishmania es un parásito protozoario de la familia Trypanosomatidae, orden
Kinetoplastida y género Leishmania, el cual agrupa un gran número de especies de las
cuales aproximadamente 20 resultan patógenas para el hombre. Estas especies, aunque
morfológicamente son similares, presentan diferencias a nivel eco-biológico, bioquímico y
molecular, siendo estas dos últimas características las utilizadas para la actual clasificación
taxonómica que incluye dos grandes subgéneros: Leishmania y Viannia [51, 52].
Fig. 3. Metaciclogénesis de Leishmania sp. en el intestino del vector. 1) Amastigote; 2) Promastigote procíclico; 3) Nectomonas; 4) Leptomonas; 5) Haptomonas; 6) Promastigotes
metacíclicos [5, 8].

20
El subgénero Leishmania abarca las especies de parásitos que tienen afinidad para
replicarse en el intestino medio y anterior (región suprapilaria) del vector como son los
complejos de especies: L. donovani, L. tropica, L. major, L. aethiopica y L. mexicana.
Mientras que los parásitos incluidos en el subgénero Viannia tienen afinidad para replicarse
en el intestino posterior (región peripilaria) e incluyen las especies: L. (V.) braziliensis, L.
(V.) guyanensis, L. (V.) panamensis, L. (V.) naiffi y L. (V.) lainsoni (Fig. 3) [20, 51].
En el Nuevo Mundo se cuenta con una amplia distribución de especies tanto del subgénero
L. Leishmania como del subgénero L. Viannia, siendo este último restringido para el Nuevo
Mundo. Las formas y complicaciones de la enfermedad se pueden asociar a la especie
infectante, razón de peso que ha dado en los últimos tiempos una gran importancia a la
identificación de especies como factor pronóstico de la enfermedad y de respuesta al
tratamiento. [53-55].
4.1.5 Presentaciones Clínicas.
Existen tres formas clínicas de las leishmaniasis: cutánea, mucosa y visceral, cuyo
desarrollo está influenciado por la especie infectante y en gran parte por el estado
inmunológico del huésped [17]. Otros autores refieren solo dos formas clínicas: cutánea y
visceral, entendiendo que la primera puede manifestarse como cutánea localizada,
cutánea difusa, mucosa y mucocutánea [14].
La LC localizada se produce en el sitio de picadura del vector infectado con el parásito.
Consiste generalmente en el desarrollo de una úlcera redondeada, con fondo limpio y
bordes levantados, que resulta indolora y presenta curación espontanea, sin embargo,
también puede darse como lesiones verrugosas, maculares y papulomatosas sin
perforación. La prueba más común para el diagnóstico es la visualización de las formas
amastigotes del parásito en el examen directo de la lesión [17, 44].
La LM, es considerada producto de una diseminación linfohematógena de las formas
parasitarias, sin embargo, también puede darse por la picadura cercana del vector a una
mucosa. Consiste en lesiones deformantes y mutilantes de las vías aéreas superiores,
siendo la forma más común la perforación del tabique nasal, que está precedida de
síntomas como congestión, obstrucción, prurito y epistaxis. Su diagnóstico definitivo se

21
logra al evidenciar la presencia del parásito en el análisis histopatológico, sin embargo las
formas amastigotes son escasas en las lesiones lo que hace necesario utilizar un algoritmo
que incluya información epidemiológica, pruebas serológicas y análisis de la historia clínica
[17, 44, 56].
La leishmaniasis visceral (LV) es de progresión lenta y puede resultar fatal. Consiste en la
diseminación de los parásitos a médula ósea, hígado y bazo, lo que conlleva a síntomas
como organomegalia, anemia producto de fuertes hemorragias, pérdida de peso y
adenopatías. Afecta principalmente a huéspedes con inmunosupresión, su diagnóstico al
igual que en las otras formas incluye la visualización del parásito en muestras de biopsia
de medula ósea o bazo y el apoyo en técnicas serológicas [17, 44].
4.1.6 Epidemiología.
La forma de la enfermedad con mayor prevalencia en el mundo es la leishmaniasis cutánea
siendo Afganistán, Argelia, Brasil, Colombia, Irán, Perú, Siria y Turkmenistán los países
que aportan el mayor número de casos según el último informe de la Organización Mundial
de la Salud (OMS) [57].
En Colombia, el último informe del SIVIGILA del 2015 reportó 5113 casos de leishmaniasis,
en donde el 98.4% correspondieron a la forma cutánea, el 1.3% a la mucosa y el 0.3% a
la visceral. Este patrón de distribución es constante año a año, sin embargo, es importante
resaltar que hubo una reducción del 39.5% en los casos de LC respecto al año anterior
[13].
Los departamentos que presentaron mayores tasas por 100.000 habitantes de la forma
cutánea fueron Vaupés, Vichada y Guaviare, concentrando el 79.5% de casos en la
población masculina principalmente entre los 15-34 años, esto probablemente está
asociado a una contundente transmisión zoonótica producto de la irrupción del hombre a
zonas selváticas por motivos socioeconómicos. Las lesiones se ubicaron principalmente
en extremidades superiores e inferiores, un 20.4% en cara y un 16.7% tronco. [13].
Estudios de distribución de especies en el país, asocian a esta forma de la enfermedad
principalmente a L. (V.) panamensis y L. (V.) braziliensis [16, 58, 59].

22
En cuanto a la forma mucosa, el departamento con una tasa de incidencia por 100.000
habitantes relevante fue Guaviare. El 89.6% de los casos correspondió a hombres
principalmente entre 15-44 años. Los signos más comunes fueron rinorrea y epistaxis y en
menor porcentaje hiperemia, obstrucción nasal, disfonía y disfagia, sin embargo, se
reportaron 13 casos con perforación y/o destrucción del tabique [13]. Aunque en las
Américas la principal especie incriminada en esta forma de la enfermedad es L. (V.)
braziliensis, el país hay reportes de casos por L. (V.) panamensis y L. (V.) guyanensis [18,
19, 60].
La forma visceral se confirmó en municipios pertenecientes a los departamentos de Sucre,
Bolívar y Córdoba. El 57.1% de los casos correspondieron a mujeres. Los síntomas
reportados en la mayoría fueron fiebre, organomegalia y anemia [13].
4.2 LEISHMANIASIS CUTANEA.
La LC localizada consiste en una lesión en el sitio de picadura del vector, siendo ubicación
frecuente los sitos expuestos por el huésped como extremidades y rostro. El periodo de
incubación oscila entre 3 semanas incluso hasta 6 meses, en donde posterior a la
inoculación parasitaria aparece una lesión indolora con posterior presentacion ulcerativa,
nodular, macular o verrucosa. Estas lesiones pueden llegar a curarse espontáneamente
sin ninguna intervención farmacéutica, lo cual se ha visto más comúnmente en las formas
de la enfermedad producida por parásitos del subgénero L. Leishmania [20, 33].
La lesión mas frecuente es la forma ulcerada que se caracteriza por ser redondeada, con
bordes elevados y fondo limpio, a excepción de cuando se sobreinfecta con bacterias
llegando incluso a ser dolorosa. El diagnóstico diferencial deberá incluir enfermedades
micóticas y bacterianas, tales como esporotricosis, cromomicosis, pioderma gangrenoso y
tuberculosis cutánea entre otras [17].
4.2.1 Respuesta inmunológica de la leishmaniasis cutánea.
En LC la respuesta inmunológica, tanto innata como adaptativa, es principalmente de tipo
celular, asociando incluso el predominio de la respuesta humoral a una ineficiente
eliminación del parásito que conlleva a la cronicidad de la enfermedad [61-63].

23
Algunos autores afirmar que durante la picadura al huésped mamífero, el vector infectado
regurgita promastigotes metacíclicos que son inoculados junto con componentes de la
saliva como el maxadilan, una péptido vasodilatador que tiene propiedades inhibitorias de
la respuesta de hipersensibilidad tardía, lo que favorece la infección parasitaria [3, 64, 65].
Otros componentes salivares que también son inoculados son 5´-nucleotidasa, adenosin
deaminasa, aspirasa y hialuronidasa, enzimas con funciones hemostáticas y anestésicas
[33].
Debido a la laceración de la piel son atraídas células de respuesta inmune innata como
monocitos, neutrófilos, macrófagos dérmicos y células dendríticas (células de
Langerhans), todas con capacidad fagocitica y las dos últimas reconocidas como
importantes células presentadoras de antígeno (CPA). Por otro lado, los queratinocitos,
células predominantes en la epidermis, se han asociado con la secreción de sustancias
inmunomodulatorias contra el parásito en la etapa temprana de la infección [61, 65, 66].
Los promastigotes metacíclicos estan recubiertos de la proteína de superficie GP63 y
glicolipidos como el LPG y el glicosilfosfatidilinositol (GIP) que le sirven como patrones
moleculares asociados a patógenos (PAMPs) para ser reconocidos por los receptores de
las células fagociticas del huésped facilitando su internalización en ellas. En el caso de la
GP63, su acción favorece la conversión de C3b en su forma inactiva C3bi que funciona
como opsonina y facilita el reconocimiento al receptor del complemento tipo 3 (CR3) de
los macrófagos. Otros receptores implicados en el reconocimiento de las proteínas de
superficie parasitarias incluyen el receptor del complemento tipo 1 (CR1) y el receptor
fucosa-manosa (MFR) [62, 63, 67].
La primera línea de defensa son los neutrófilos, células con una vida media corta que
presentan un núcleo segmentado y gránulos citoplasmáticos primarios, secundarios y
terciarios que contienen enzimas altamente proteolíticas. Una vez llegan al sitio de la
lesión, son capaces de fagocitar los parásitos opsonizados en donde su destrucción es
posible mediante la formación de un fagolisosoma en el cual se lleva a cabo el estallido
respiratorio, proceso en el que se originan especies reactivas de oxigeno por activación de
la enzima Nicotinamida adenina dinucleótido fosfato (NADPH) tales como los singletes de
oxígeno, aniones superóxido, peróxido de hidrogeno entre otros. Otro mecanismo de
aniquilamiento presente en los neutrófilos e independiente de oxígeno incluye la

24
degranulación de compartimientos citoplasmáticos en el fagolisosoma que liberan enzimas
liticas contra el patógeno intracelular [43, 67-69].
Como proceso natural, los neutrófilos entran en apoptosis y estimulan mediante la
secreción de las proteínas inflamatorias de macrófagos MIP-1α y MIP-1β la llegada de
macrófagos que fagocitaran los neutrófilos muertos e infectados. Dicho proceso puede
funcionar como un mecanismo de “caballo de Troja” en la infección si hay un
funcionamiento inadecuado de los polimorfonucleares asociado a las condiciones
genéticas del huésped humano que faciliten mecanismos alternos como la fagocitosis de
parásitos no opsonizados que no activan los mecanismos de destrucción intracelular y/o
el silenciamiento de los mismos [43].
Otro mecanismo de destrucción descrito en los neutrófilos ha sido la formación de trampas
extracelulares de los neutrófilos (NETs) que consisten en la liberación extracelular de
cromatina, histonas y gránulos con el fin de atrapar y eliminar los patógenos circundantes,
lo que finalmente promueve la muerte celular por NETosis [65, 69, 70]. Este mecanismo
ha sido descrito en promastigotes y amastigotes de L. (L.) mexicana mostrando
experimentalmente una actividad leishmanicida, sin embargo faltan más estudios que
muestren la influencia de las NETs en la supervivencia de los parásitos de Leishmania [71].
Los macrófagos aunque llegan tardíamente al sitio de la lesión, asumen funciones de
activación de la respuesta inmune adaptativa y fagocitosis, este último proceso funciona
como se mencionó anteriormente en los neutrófilos. Si la replicación es exitosa en los
macrófagos estos se lisan por la gran cantidad de parásitos, liberando en ese momento
amastigotes que infectan otros macrófagos circulantes. El estallido respiratorio que se lleva
a cabo en estas células es eficiente solo en caso de que hayan sido previamente activadas
por el Interferón gamma (IFNγ) [61, 63].
Tanto los macrófagos como las células dendríticas funcionan como CPAs en los nódulos
linfáticos vecinos. La presentación de antígenos se hace a los linfocitos T
predominantemente CD4+ quienes se diferencian en dos subpoblaciones celulares, Th1 y
Th2, iniciando así la respuesta inmune adaptativa. El espectro de la enfermedad en cada
huésped dependerá de la relación entre ambas subpoblaciones [3, 61].

25
Paralelamente, durante la fagocitosis hay el reconocimiento de PAMPs parasitarios como
el LPG y GIP por receptores tipo toll (TLR) ubicados en la superficie celular y a nivel
endosomal de macrófagos, células dendríticas y células NK. Los TLRs funcionan como
receptores de reconocimiento de patrones (PRRs) desencadenando vías de señalización
celular que promueven mediante el factor nuclear potenciador de las cadenas ligeras
kappa de las células B activadas (NFκβ), la trascripción de genes de interleucinas pro-
inflamatorias quienes también estimularan el desarrollo de una respuesta inmune
adaptativa [72, 73].
Los TLRs involucrados en el control de los parásitos de Leishmania spp. han sido TLR2
[74, 75], TLR4 [75, 76] y TLR9 [77, 78] y algunos autores sugieren actividad de TLR3 en
casos donde el parásito infectante esta a su vez infectado con un tipo de virus RNA de
doble cadena cononcido con Leishmaniavirus. Esto debido a que TLR3 es un receptor
intracelular que tiene la capacidad de reconocer RNA de doble cadena [72, 73, 79].
Para el control de la Leishmaniasis es indispensable la participación de linfocitos Th1
(LTh1), células capaces de estimular la producción de interleucinas 2, 12, IFNγ y el Factor
de necrosis tumoral alfa (TNF-α). La IL-12 juega un papel importante ya que estimula los
efectos tóxicos, la producción de IFNγ y la proliferación de más LTh1, mientras que suprime
la producción de IL-4 [80, 81]. El IFNγ y TNF-α trabajan sinérgicamente para la eliminación
de los parásitos mediante la activación de macrófagos en los que induce la producción de
óxido nítrico [33, 61, 81].
Por otra parte la participación de linfocitos Th2 (LTh2) no tiene una eficiente actividad
leishmanicida y su predominio está asociado a una mal pronóstico de la enfermedad. Esta
afirmación se soporta en las observaciones hechas en pacientes con LC difusa y en
experimentos con modelos murinos en los que se presenta una clara dicotomía entre
resistencia y susceptibilidad a la infección ante respuesta Th1 y Th2 respectivamente. Los
LTh2 estimulan las interleucinas 4-6, 9-10 y 13, siendo reconocidos los efectos de la IL-4
como potente activador de linfocitos B y de la IL-10 como supresora de la respuesta celular,
lo que facilitaria la persistencia del parásito [63, 67, 68]. El factor de crecimiento
transformante β (TGFβ) es una interleucina producida por los macrófagos que también se
ha asociado con un inhibición de los efectos tóxicos en ellos y la supresión de células NK
[3, 62].

26
Un respuesta Th1 regulada pero predominante respecto a Th2 se refleja en un cuadro
clínico moderado de la enfermedad que puede incluso llegar a la curación espontanea en
el 90% de los casos. El tiempo en el que se logra la curación dependerá, en un escenario
inmunológico adecuado, de la especie infectante (Fig. 4) [3, 61].
4.2.2 Tratamiento y falla terapéutica
El medicamento de primera línea para el tratamiento de LC son las sales de antimonio
pentavalente (Sb+5) que incluyen el estibogluconato de sodio (Pentostam) y el antimoniato
de meglumia (Glucantime®). En el país se administra gratuitamente el Glucantime de
acuerdo a lo indicado por la “Guía de atención de la leishmaniasis”, dentro del marco de la
resolución 00412 del 2000 que reglamenta las normas técnicas y guías de atención para
enfermedades de interés en salud pública [31, 82]. El esquema es únicamente
suministrado cuando se cuenta con confirmación parásitológica de la enfermedad. La dosis
Fig. 4. Espectro inmunológico de las Leishmaniasis en el
huésped humano [4, 5]

27
indicada es de 20 mg/Kg de peso/día durante 20 días, la cual es suministrada vía
intramuscular [17].
El porcentaje de curación con este medicamento está alrededor del 76% si el esquema se
aplica completamente, pues interrupciones en el mismo han sido relacionadas con
recidivas [83, 84]. El Glucantime® se distribuye en plasma, hígado y bazo y es eliminado
por la orina hasta en promedio 76 horas después de la administración [33, 85], factores
que hacen necesaria la realización de una valoración cínica y paraclínica del sistema
cardiaco, hepático y renal antes de iniciar el esquema [17].
Para su activiación se requiere la reducción del antimonio pentavalente, considerado como
una prodroga, en una forma trivalente con mayor poder leishmanicida. En esta reducción
podrían estar implicados tioles del huésped como glutatión, cisteína y cisteína-glicina y del
parásito como tripanotion, compuesto conformado por glutatión (GSH) y espermidina. Por
otro lado, se sugiere la participación de enzimas parasitarias como la Reductasa
dependiente de tiol (TDR1) y la Antimoniato reductasa (ACR2). La reducción se ve
favorecida por condiciones ambientales de acidez y elevada temperatura y aunque no es
claro si se lleva a cabo en los amastigotes y/o en el macrófago, estudios señalan un mayor
concentración de antimoniato trivalente en amastigotes [86-88].
La internalización del medicamento está asociada a proteínas transmembranales de la
familia de proteínas integrales de membrana denominas Acuaporinas tipo 1 (AQP1s) [81].
El efecto leishmanicida se refleja en la inhibición de compuestos enzimáticos parasitarios
y de síntesis de material genético [3], estudios han mostrado que la acumulación de
antimonio trivalente estimula la apoptosis de amastigotes al fragmentar el DNA y
externalizar la fosfatidlserina. Sin embargo no se ha encontrado una clara actividad de
caspasas, proteínas directamente asociadas a la muerte por apoptosis [89].
En caso de que la reducción ocurra total o parcialmente en el macrófago, el Sb+3 tendría
que ser transportado al parásito en donde las AQP1 tendrían un rol fundamental, pues este
canal de membrana se ha asociado al ingreso del Sb+3 en los parásitos. La activación del
medicamento, traducida en la reducción del mismo, podría ser de manera no enzimática
producto de los tioles del parásito como el tripanotion y los tioles del macrófago como la
glicil-cisteina. Dichas moléculas tiene una participación importante en el mantenimiento de

28
potencial redox intracelular. Otros estudios han sugerido que TDR1 usaría el GSH como
agente reductor y que la ACR2 mostró tener capacidad de reducción de Sb+5 usando como
cofactores GSH y glutaredoxin para la actividad enzimática (Fig. 5) [81].
La falla terapéutica se puede dar de manera temprana o tardia y puede asociarse a
multiples factores como: a) condiciones intrínsecas del parásito infectante, b) carga
parasitaria, c) adherencia al tratamiento, d) metabolismo del medicamento y e) respuesta
inmune del huésped [81, 87, 88, 90].
La especie infectante juega un rol fundamental en el fenotipo de la enfermedad y en su
respuesta al tratamiento [91] por lo que en la actual guía basada en la evidencia de la
Organización Panamericana de Salud (PAHO) se hacen recomendaciones terapéuticas de
acuerdo a la especie [92]. Mecanismos como la amplificación de genes de resistencia
natural, la modulación de la entrada del medicamento y su inactivación son otros
potenciales factores asociados a la falla terapéutica [87].
Fig. 5. Mecanismo probable de acción de Sb+5. Cys (cisteína) γGCS (γ-glutamilcisteina
sintetasa), GSH (glutanion), Orn (orinitina), ODC (ornitina decarboxilasa) TSH (tripanotion),
TPx (tripanotion peroxidasa, TR (tripanotion reductasa), MRPA (proteína asociada a la
resistencia multidroga).

29
Otro factor que influye en la resistencia asociada a la especie del parásito son los ciclos
de transmisión; en ciclo zoonótico en donde la trasmisión es a partir de un reservorio animal
hay menor probabilidad de infectarse con una especie del parásito que previamente haya
sido sometida a la presión del medicamento, caso contrario a la transmisión
antropoonótica. Esto sustenta el hecho de que la falla terapéutica resulta ser frecuente en
áreas endémicas [81].
En cuanto al huésped humano, la acción del medicamento parece depender
significativamente del estatus inmunológico del paciente pues la droga tendría la capacidad
de activar en los macrofagos la generación de radicales libres, capaces de eliminar el
parásito [81]. En el estudio de Saldanha en 2012 se mostró que al séptimo dia de
tratamiento antimonial, monocitos humanos infectados incrementaron la fagocitosis y la
producción de TNF-α, ambas actividades tienen efectos leishmanicidas importantes [93].
Lo anterior sugiere que ante una inadecuada implantación de la respuesta inmunológica
los efectos del medicamento no tendrían los mismos alcances.
Este factor sumado a otros mecanismos del parásito como la disminución de la reducción
del Sb+5, la disminución del aflujo o el incremento del eflujo del medicamento y el
incremento de niveles de tripanotion para restaurar el potencial redox se han traducido en
la falla terapéutica de los pacientes que incluye no solo la no respuesta al mismo sino
también la recaída después de una cura inicial [81].
4.3 LEISHMANIASIS MUCOSA.
La LM o espundia es una complicación de la LC producida por la diseminación hematógena
o linfática de las formas amastigotes del parásito a las mucosas [18], o por picadura del
vector cercana a ellas. Es posible evidenciar su sintomatología tiempo después de la cura
de una lesión cutánea o coexistiendo con ella, tomando en ese caso el nombre de
mucocutánea. Las lesiones se ubican en la mucosa nasal y oral afectando nariz, laringe,
faringe, paladar y labios, y por tanto la deglución, respiración y tono de la voz de los
pacientes afectados [18, 60].
Esta es una enfermedad con un alto costo social y económico debido a las deformaciones
faciales que aíslan e incapacitan el paciente. La baja respuesta al tratamiento, la

30
importante tasa se reincidencia, la sobreinfección bacteriana y el diagnóstico tardío,
convierten el cuadro de LM en una enfermedad de mal pronóstico [94, 95].
Debido a la frecuencia con la que se ve involucrado el tejido de la mucosa nasal, Lessa y
colaboradores sugirieron una clasificación de los estadíos de la enfermedad de acuerdo al
compromiso de éste tejido. La clasificación se hizo con base en los hallazgos clínicos en
50 pacientes diagnosticados con LM en Corte de Pedra, población ubicada en el suroeste
de Brasil, estableciendo 5 estadios: I, II, III, IV y V. Cabe aclarar que esta clasificación no
incluyó pacientes con lesiones a nivel oral [1]:
4.3.1 Respuesta inmunológica de la leishmaniasis mucosa.
La LM está asociada a una respuesta inmunológica exacerbada, presenta un alto
porcentaje de recurrencia y muestra mayor falla terapéutica en el tratamiento con derivados
de Sb+5 que las otras leishmaniasis [11, 22, 96, 97].
Se ha señalado en la LM la presencia hiperactividad de los linfocitos T, acompañada de
una respuesta inflamatoria progresiva producto de altos niveles de interleucinas pro-
Tabla 1. Estadíos clínicos en LM [1].

31
inflamatorias como IFNγ y TNF-α, al igual que una respuesta anti-inflamatoria disminuida
por baja eficiencia de la IL-10 [11].
En caracterizaciones histológicas de lesiones de LM antes y despues del tratamiento se
observa una disminución de macrófagos y linfocitos T CD4+ después del tratamiento,
mientras que los niveles de células de Langerhans, Linfocitos B, linfocitos T CD8+ y células
NK permanecieron sin cambios significativos. En cuanto a las interleucinas hubo una
disminución post-tratamiento de INFγ, TNF-α e IL-4, en tanto que la IL-10 no tuvo cambios
relevantes como se esperaba [96].
Aproximaciones similares en pacientes que presentaron tanto cura como recaída,
mostraron que los conteos de linfocitos T CD4+, macrófagos, IFNγ, TNF-α, IL-4 y IL-10
disminuyeron posterior al tratamiento de manera similar en las dos poblaciones, sin
embargo la IL-10 disminuyó menos en los pacientes en cura. Los linfocitos T CD8+ aunque
se encontraron aumentados en ambas condiciones, lo estuvieron más en los pacientes en
recurrencia [97].
La LM se encuentra en un extremo del espectro inmunológico de la enfermedad, por lo que
hay una respuesta incontrolada de tipo Th1 caracterizada por la estimulación de factores
pro-inflamatorios que se asocian a una destrucción del tejido (Fig. 4) [98]. El hallazgo de
antígeno en lamina propia en el 40% los pacientes post-tratamiento, así como la
persistencia de células como células de Langerhans, linfocitos B, células NK [96] y
linfocitos T CD8+ [97], podría estar relacionado con la persistencia de la enfermedad.
Respecto a la IL-10, dadas las funciones inhibitorias atribuidas a ella, se ha sugerido una
probable disminución en los casos de LM, sin embargo su rol aun no es claro.
4.3.2 Otros factores asociados al desarrollo de leishmaniasis mucosa.
4.3.2.1 Huésped.
El desarrollo a la LM ha sido asociado a diferentes factores como edad, ocupación, estado
nutricional, co-infección de las lesiones y defectos en el establecimiento de la respuesta
inmune ya sea por genética o por la co-morbilidad con HIV [99].

32
En algunos estudios la LM ha sido encontrada predominantemente en la población
masculina y comprometiendo edades avanzadas [22, 100, 101], quizá asociado al
detrimento de la función inmunológica [100]. El desarrollo de una LC previa y de lesiones
cutáneas en cualquiera área por encima de la cintura también fueron factores de riesgo
asociados [101].
En cuanto al estado nutricional, un déficit podría comprometer la resolución de la
enfermedad [102]. La asociación LM y el bajo peso [103], puede ser circunstancial dadas
las manifestaciones clínicas propias de la enfermedad que limitarían una correcta ingesta
de los alimentos.
Otros factores que se han descrito como de riesgo en el desarrollo metastásico de la
enfermedad son la presencia de inmunodeficiencias de base o enfermedades
concomitantes [21, 99, 104, 105]. Referente a la comorbilidad con HIV, los pacientes con
esta condición tienden a desarrollan lesiones agresivas y atípicas de la leishmaniasis [106-
108].
4.3.2.2 Parásito.
En principio el desarrollo de la LM estaba asociado únicamente a L. (V.) braziliensis, sin
embargo, varios estudios indican que esta forma de la enfermedad también se asocia a L.
(V.) panamensis y L. (V.) guyanensis. [16, 60, 109, 110]. La LM se encuentra restringida a
las especies del subgénero Viannia [111].
Dentro de la misma especie hay una gran variabilidad genética lo que podría asociarse a
diferentes patrones de la infección [112]. Sumado a esto, se han encontrado genes
asociados a mayor virulencia en L. (V.) braziliensis [113, 114].
Actualmente se ha descrito presencia de un virus de familia Totiviridae dentro de los
parásitos de algunas especies de Leishmania que podría funcionar como factor de
virulencia, situación descrita en otros parásitos como Trichomona, Cryptosporidium,
Giardia y Acantamoeba con virus de la misma familia [115]. La presencia del virus podría
promover interleucinas pro-inflamatorias como TNF-α, IL-6 y quimioquinas como CCL5 y
CXCL10 [116].

33
4.3.3 Tratamiento de la leishmaniasis mucosa
El medicamento de primera línea, al igual que en LC, son los Sb+5 cuya dosis indicada es
de 20 mg/Kg de peso/día durante 28 días [17]. El porcentaje de curación con este
medicamento está dentro del 30-90%. Otras opciones terapéuticas incluyen Anfotericina B
y Pentamidina [117].
Al finalizar el esquema se evaluará al paciente, luego a los 45 días, a los 6 meses y
posteriormente cada 6 meses durante 2 años [31]. Los criterios de curación incluyen la
mejora de las lesiones en las mucosas comprometidas, deseable disminución de los títulos
de inmunofluorescencia y ausencia de parásitos y patrones de inflamación en la biopsia
[17].
4.4 LEISHMANIAVIRUS.
4.4.1 Generalidades.
En el año 1974, tras un análisis de ultra-estructura celular por microscopio electrónico,
fueron vistas inclusiones citoplasmáticas en promastigotes en cultivo de L. (L.) hertigi,
especie de leishmania originaria de Panamá que fue encontrada por Herrer en 1971 en un
Coendou rothschildi, roedor de la familia Erethizontidae endémico en ese país. Estas
inclusiones que fueron denominadas partículas similares a virus (VPLs) estaban
distribuidas ampliamente por todo el citoplasma y en cultivo no afectaron la división celular
de los parásitos ni dañaron la estructura de los mismos [118]. Años antes ya se habían
descrito VPLs en otros protozoos como Entamoeba hystolitica, Plasmodium sp y Naegleria
grubei [119].
En 1988 Tarr y colaboradores detectaron un ácido nucleico tipo RNA de aproximadamente
6000 nucleótidos localizado de manera exclusiva en el citoplasma de promastigotes de L.
(V.) guyanensis CUMC1-1A en donde se denominó LR1 y de L. (V.) braziliensis CUMC3
en donde se denominó LR2, ambos parásitos aislados de huéspedes humanos. No fue
encontrado en los demás stocks de parásitos analizados que incluían especies como L.
(V.) guyanensis, L. (V.) braziliensis, L. (V.) panamensis, L. (L.) chagasi, L. (L.)
amazonensis, L. (L.) mexicana y L. (L.) major, Trypanosoma brucei y Trypanosoma cruzi.
En este estudio se sugirió que el RNA de LR1 era de cadena sencilla (ssRNA) y que este
material genético se encontraba contenido en partículas de 32 nm. El aislamiento que
contenía el LR1 provenía de un paciente que visitó Surinam, quien contrajo una LC que le

34
produjo lesiones satelitales y compromiso de nódulos linfáticos. Una vez aislado del
paciente e inoculado en un modelo murino, el parásito le produjo LM al animal [120].
En el siguiente año, Widmer y colaboradores buscaron indicios de replicación viral en una
serie de aislamientos de Leishmania spp. encontrando una actividad RNA polimerasa
dependiente de RNA (RDPR) en dos de los aislamientos: L. (V.) guyanensis M4147 y L.
(V.) guyanensis WR677. Su estudio también sugirió la posibilidad de que el RNA viral no
fuera de cadena sencilla sino de cadena doble (dsRNA) al observar una gran estabilidad
del material genético posterior extracción [121] y al ver en experimentos posteriores la
resistencia del mismo a la digestión con RNAsa en altas concentraciones de sal [122].
Esta inquietud fue finalmente resuelta por Weeks y colaboradores en 1992 quien obtuvo
dos clases de partículas de LR1 mediante el uso de gradientes de densidad como la
sucrosa y el cloruro de celsio (CsCl), las partículas densas contenían dsRNA mientras que
las menos densas contenían ssRNA. Ambos ácidos nucleicos se encontraron mucho más
abundantes en la fase estacionaria que en la fase logarítmica de la curva de crecimiento
de promastigotes de L. (V.) guyanensis CUMC1-1A [123].
En el mismo año Stuart y colaboradores describieron la secuencia completa del virus del
aislamiento L. (V.) guyanensis CUMC1-1A. La secuencia contiene 5284 nucleótidos
organizados en tres marcos abiertos de lectura (ORF) que se ubican en la hebra positiva
del dsRNA. Los ORF 2 y 3 codifican para la cubierta viral y la RPDR respectivamente.
Ambos ORFs se sobrelapan en 71 nucleótidos. El ORF1 corresponde al extremo 5´-UTR,
se encuentra altamente conservado en las especies virales y parece tener un rol importante
en la iniciación de la traducción y la estabilidad del RNA. Se sugiere un origen ancestral
común con el virus de levaduras ScV L-A dado la semejanza en la organización genómica,
el ciclo replicativo y la secuencia nucleotidica de la RPDR [124].
Paralelamente, Guilbride y colaboradores buscaron mediante un análisis de hibridización
identificar el virus en 71 aislamientos de Leishmania provenientes tanto del Nuevo como
del Viejo mundo, encontrando 12 de ellos positivos pertenecientes a las especies
parasitarias L. (V.) braziliensis y L: (V.) guyanensis. Se usaron 6 sondas dirigidas a
diferentes partes del genoma viral obtenidos del virus del aislamiento L. (V.) guyanensis
CUMC1-1A (Fig. 6). Todos los aislamientos positivos recibieron el nombre LRV1 y se les

35
asignó un sufijo numérico para diferenciarlos entre sí, dicha clasificación de especies de
Leishmaniavirus es la que se encuentra vigente. Es de resaltar que todos los aislamientos
hibridizaron con la sonda LP28 dirigida a la región 5´-UTR reafirmando que esta región se
encuentra altamente conservada, mientras que no todos los aislamientos hibridizaron con
la sonda LP29 correspondiente a la cápside, sugiriendo polimorfismos en esta región [6].
En 1994 Sheffter y colaboradores publican la secuencia completa del virus del aislamiento
L. (V.) guyanensis M4147 (LRV1-4) el cual comparte el 77% de identidad con LRV1-1. Se
reiteró que la región más conservada a nivel de genoma es la región 5’UTR mientras que
los ORF 2 y 3 son más conservados a nivel de proteína [125]. Cerca al extremo 5´, además
del ORF1, parece encontrarse un segundo marco de lectura denominado ORFx [126].
En experimentos posteriores se consiguió identificar el virus en el aislamiento parasitario
L. (L.) major de un paciente proveniente de Turkmenistán; el virus fue denominado LRV2-
1 debido a ser el primer virus identificado en parásitos del subgénero L. Leishmania.
Nuevamente Sheffter y colaboradores logran la secuencia completa del virus en 1995 y
señalan que esta secuencia difiere considerablemente de la reportada para LRV1-4
sugiriendo una transmisión vertical del virus previa divergencia de los parásitos del Nuevo
y Viejo mundo. Dentro de las principales diferencias se encuentra que los ORF 2 y 3 no se
sobrelapan como en el caso de LRV1-4, lo que tiene implicaciones importantes en el
proceso de traducción [127].
Dichas diferencias taxonómicas se reflejaron en análisis filogenéticos hechos por Widmer
y colaboradores quienes compararon dos fragmentos de secuencia pertenecientes al
ORF1 y ORF3 de siete tipos de virus (LRV1-2, LRV1-3, LRV1-4, LRV1-9, LRV1-13,
LARV1-14 y LRV2-1), encontrando entre LRV1 y LRV2 una baja similaridad en la
Fig. 6. Ubicación en el
genoma de sondas usadas
en ensayo de Northern blot
para la identificación de
LRV en aislamientos de
Leishmania del Nuevo y
Viejo mundo [5, 6].

36
secuencia nucleotidica del ORF1 que impidió obtener una alineamiento significativo y una
similaridad entre LRV1-4 y LRV2-1 del 50% en el ORF3. Entre los tipos de LRV1 la similitud
en el ORF3 oscilo entre 71-100%. A nivel de proteína, la similitud en la secuencia de
aminoácidos correspondiente al ORF3 entre LRV1-4 y LRV2-1 fue del 60%. Paralelamente
se estimaron las distancias genéticas entre cada aislamiento de Leishmania spp. que
contenía cada tipo viral. Al sobrelapar los arboles genéticos elaborados tanto para el
aislamiento del parásito como para su respectivo virus, se encontró un patrón similar de
asociación en ambos grupos consistente con la procedencia geográfica y probablemente
con un proceso de co-evolución, teoría que dice soportarse en la reproducción del parásito
predominantemente asexual y la ausencia demostrable de un ciclo de infección viral. [128].
Recientemente se detectó el virus en 4 aislamientos parasitarios de L. (L.) aethiopica, tres
de ellos se secuenciaron y al compararse con las secuencias reportadas de LRV1 y LRV2,
mostraron mayor similitud con LRV2 (65%), sin embargo en cuanto a la organización de
su genoma presenta mayor semejanza con LRV1, pues sus ORFs se sobrelapan, en este
caso, en 46 nucleótidos. [10].
4.4.2 Replicación y traducción.
Los virus RNA de Leishmania pertenecen al género Leishmaniavirus (LRV) y familia
Totiviridae. Presentan un diámetro entre 32-33 nm, una molécula lineal de ARN de doble
cadena y una cápside con morfología icosaédrica compuesta por 12 dímeros pentaméricos
[129]. En su genoma el ORF2 codifica para la proteína de la cápside caracterizada por un
peso de 82 KDa y el ORF3 codifica para la RPDR, enzima de 92 KDa, que tendrá actividad
tanto en la replicación como en la traducción [4, 124, 130, 131].
La replicación es llevada a cabo dentro de la cápside viral y está a cargo de la RPDR
usando como molde tanto la hebra positiva como la negativa, sugiriendo un proceso
semiconservativo. Macbeth y colaboradores reportan una persistencia natural de la
infección por LRV al autorregularse usando la actividad endonucleasa de la cápside que
impide una adecuada replicación y traducción de los ORFs. El sito de clivaje se encuentra
ubicado en la región 5’UTR de la hebra positiva en una secuencia consenso 5´-
GNUC*CGNA-3´ obtenida de las secuencias de los virus LRV1-1, LRV1-4 y LRV2-1. El

37
clivaje origina pequeños fragmentos que oscilan entre los 284-320 nucleótidos (Fig. 7). [9,
132].
La replicación viral parece estar asociada al ciclo de vida del parásito hospedero. Chung y
colaboradores midieron el número de genomas virales por célula infectada desde su fase
logarítmica hasta su fase estacionaria, encontrando las mayores cantidades de RNA viral
en la fase estacionaria producto de un incremento de la actividad de la polimerasa viral.
En el momento en que hay mayor carga viral ocurre el clivaje del extremo 5´, proceso que
disminuye en la fase estacionaria tardía del parásito. Lo anterior se traduce en menor
número transcritos disponibles para la traducción y encapsulación y en disminución de la
replicación viral [133]. Durante la fase logarítmica en donde se encontrarían bajas
densidades virales una porción del genoma viral evitaría el clivaje y sería traducido y
posteriormente encapsulado mientras que cuando la densidad viral aumenta en la fase
estacionaria del parásito, la mayor parte de transcritos virales podrían ser clivados [134].
Widmer había reportado previamente una correlación negativa entre los productos virales
de cadena sencilla y doble y la densidad parasitaria [135].
Posterior a la replicación, la hebra positiva del RNA viral es extrudida de la cápside y asume
el papel de RNA mensajero [9], indicando una transcripción conservativa [135]. Al no haber
una estructura que facilite la traducción como es el caso de cap-5´ en organismos
eucariotas, se presume una secuencia en el genoma viral que sirve como sitio interno de
entrada al ribosoma (IRES) que permitirá el acoplamiento del ribosoma para el inicio de la
traducción [124, 125, 136].
El sobrelapamiento de los ORFs en las secuencias virales descritas a excepción de LRV2-
1 sugiere la producción de una proteína fusión tipo gag-pol producto de un fenómeno en
Fig. 7. Ubicación del sitio de clivaje en el extremo 5´UTR para autorregulación de la
replicación y persistencia de la infección. *Lugar donde se origina el clivaje [8, 9].

38
la traducción denominado “Frameshift” o cambio de marco, el cual consiste en el
deslizamiento del ribosoma a una base ya sea en dirección 5´ (-1) o 3´ (+1) [124]. En LRV1-
4 se describió la carencia de una potencial sitio de iniciación en el ORF3 sugiriendo la
producción de una proteína fusión de 189 kD, en la región del cambio de marco se detectó
un sitio resbaladizo “Slippery site” y una estructura “Pseudoknot” que facilitarían un cambio
de marco -1 [126]. La proteína fusión gag-pol seria clivada cerca de la unión de los
dominios funcionales por una proteína tipo cisteína perteneciente al parásito hospedero
[137] (Fig. 8).
En LRV2 detectado en L. (L.) aethiopica también presenta un sobrelapamiento de marcos
de lectura que requiere para la traducción un cambio de marco +1 [10]. Mientras que en el
LRV2 detectado en L. (L.) major no se sobrelapan los ORFs ya que se encuentran
separados por un codón de parada; en el extremo 3´ del gen de la cápside fue detectada
una estructura de “Pseudoknot” que sugiere un proceso de “Hopping” ribosomal para llevar
a acaba la traducción (Fig. 9) [127].
4.4.3 Efecto de LRV en el hospedero humano.
En el 2011, Ives y colaboradores encontraron la sobreexpresión de genes codificantes para
CCL5, CXCL10, TNF-α e IL-6 en macrófagos infectados con clones del aislamiento L. (V.)
guyanensis M5313 LRV+, respecto a macrófagos infectados con clones del mismo
Fig. 8. Ubicación de la unión de los ORF2 y 3 en el genoma viral. *Codón de parada. [10].

39
aislamiento pero LRV- y macrófagos infectados con el aislamiento LV39 de L. (L.) major el
cual no estaba infectado conel virus. En los modelos murinos, la infección con clones
portadores de virus indujo mayor cantidad de transcritos de IFN-β respecto a los infectados
con clones negativos para el virus. Lo anterior sugiere que la presencia de leishmaniavirus
en parásitos infectantes podría estar asociada con la exacerbación de la respuesta
inmunológica por parte del huésped mamífero desencadenando una presentación de la
enfermedad pro-inflamatoria como la LM [116].
Los autores también pudieron demostrar que la cascada de señalización que desencadena
el material genético viral estaría asociada al reconocimiento del mismo por el TLR3
utilizando el adaptador que contiene el dominio TIR e induce IFN-β (TRIF). Incluso,
modelos murinos deficientes en TLR3 infectados con parásitos LRV+ mostraron
disminución de las lesiones en las almohadillas plantares y de la carga parasitaria. Además
se logró establecer que la inducción de interleucinas dependiente de los clones infectados
con el virus requiere de la entrada del parásito dentro de la célula hospedera y el secuestro
del fagolisosoma, lo que tiene concordancia con el hecho de que los virus estan ubicados
en el citoplasma del parásito y solo podrían liberarse cuando estos mueren [116].
Dados estos hallazgos, Ronet y colaboradores sugieren un modelo de señalización celular
mediada por la liberación de LRV. Una vez los promastigotes de Leishmania son
fagocitados por el macrófago y cercados en el fagolisosoma, mecanismos de aniquilación
como el estallido respiratorio producen su muerte. La ruptura de la membrana celular
parasitaria permitiría la liberación de las partículas virales en el fagolisosoma, en donde la
cápside viral seria destruida dejando el dsRNA viral disponible para el reconocimiento del
receptor TLR3 (Fig. 9) [11] .
El TLR3 es un receptor endosomal transmembranal que reconocerá el dsRNA viral con la
región N-terminal mientras desencadena una cascada de señalización celular mediada por
el dominio C-terminal que va hacia el citoplasma, designado dominio TIR. En este dominio
citoplasmático se acoplará al dominio adaptador TIR que induce IFN-β (TRIF), el cual
promoverá la producción de IFN-β, interleucinas y citoquinas y la maduración de DC
mediante la activación de 4 factores de transcripción IRF-3, NFκβ, JNK y p38 [138, 139].
En el modelo propuesto por Ronet y colaboradores se menciona la activación de los
factores de transcripción IRF3 y NFκβ. El IFN-β liberado se uniría al receptor de interferón

40
tipo I (IFNAR) de los macrófagos fomentando la producción de sustancias antivirales (Fig.
10) [11].
Posteriormente las citoquinas pro-inflamatorias liberadas generarían un desbalance en la
respuesta inmune en el hospedero humano generando así una infección e inflamación
crónica que exacerbaría la enfermedad y probablemente estaría asociada con resistencia
a los medicamentos leishmanicidas y la persistencia del parásito en el individuo [140].
En el modelo murino infectado con L. (L.) aethiopica se encontraron resultados similares a
los obtenidos por Ives con el modelo infectado con L. (V.) guyanensis. La IL-6 y el TNF-α
estuvieron elevadas en los sobrenadantes de macrófagos infectados con los aislamientos
Fig. 9. Cascada de señalización celular en respuesta a la liberación de dsRNA
de Leishmaniavirus en macrófagos infectados con L. (V.) guyanensis [11].

41
con mayor carga viral, llamando la atención que uno de los aislamientos que se encontraba
positivo para el virus pero presentaba una baja carga viral no mostro dicho patrón
inmunológico. Lo anterior sugiere que la carga viral puede tener un rol fundamental en la
producción por lo menos de la IL-6 y el TNF-α [10].
Recientes estudios han sugerido que parásitos L. (V.) guyanensis positivos para el virus
logran regular positivamente la expresión de miR-155, un microRNA humano que regularía
la activación de la quinasa Akt reflejándose en la al persistencia del parásito por aumento
de la sobrevida del macrófago [23]
Dados estos hallazgos en modelos animales, algunas investigaciones clínicas han
estudiado la posibilidad de una asociación entre la presencia del virus y la LM o la falla al
tratamiento. Tal es el caso de Cantanhêde y colaboradores quienes en el 2015 reportaron
un riesgo relativo de 2.93 veces más en pacientes con compromiso mucoso positivos para
el virus. Además se encontró una mayor prevalencia del virus en pacientes con LM que en
pacientes con LC. Es de resaltar que en dicho estudio se reportaron como portadores del
virus además de L. (V.) braziliensis y L. (V.) guyanensis a las especies L. (L.) amazonensis
y L. (V.) lainsoni [27].
Massayuki y colaboradores reportaron una prevalencia del 70.3% del virus en pacientes
con LM, sin embargo no se lograron asociaciones significativas de la presencia o ausencia
del virus respecto al tiempo del comienzo de la lesión primaria, el tiempo de inicio de los
síntomas nasales ni la severidad de la LM según la clasificación de Lessa y colaboradores.
No obstante de los pacientes positivos para el virus, el 76.9% se encontraba en los estadios
más avanzados III, IV y V. El virus fue encontrado en LM producidas por L. (V.) braziliensis
y L. (V.) guyanensis [26].
Caso contrario reportan otros autores como Ramos y colaboradores, quienes solo
encontraron una prevalencia del 4,2% en pacientes con diferentes presentaciones de la
enfermedad, dicho porcentaje fue a expensas de los pacientes con LC. Dado que los
aislamientos positivos para el virus correspondían a L. (V.) guyanensis los autores sugieren
que la baja prevalencia puede ser producto de una menor eficiencia del sistema de
interferencia en esta cepa [25]. Por su parte, Alves y colaboradores al estudiar cambios
entre los parásitos L. (V.) braziliensis obtenidos de LC y LM del mismo paciente, no

42
encontraron la presencia del virus, descartando que en dicho caso el comportamiento
metastásico de la enfermedad fuera atribuido a la presencia de LRV [141].
En cuanto a la falla terapéutica, Adaui y colaboradores encontraron un Odds ratio (OR) de
3.99, indicando la presencia del virus como un factor de riesgo para desarrollar falla
terapéutica. En el análisis estadístico la falla terapéutica incluyo la no respuesta al
tratamiento y la recidiva. Se subraya que pacientes con LC mostraron un mayor riesgo de
falla terapéutica respecto a los que presentaban compromiso mucoso. En cuanto a la
posibilidad de que el virus confiriera una resistencia intrínseca a los parásitos frente al
medicamento, no se encontró una asociación significativa entre la presencia y ausencia
del virus y las líneas de macrófagos resistentes y sensibles a Sb+5. Lo anterior favorece la
hipótesis de que los cambios mediados por la presencia del virus en la respuesta al
tratamiento provendrían de la modificación de la respuesta inmune del huésped [28].
En otro estudio similar, Borreau y colaboradores reportaron falla terapéutica de 12
pacientes en una cohorte de 74 pacientes monitoreados; los 12 pacientes fueron
reportados como positivos para el LRV. La prevalencia general fue de 58.7% usando como
método de detección la qPCR y de 87% usando como método de detección la identificación
de dsRNA con el anticuerpo J2. Además, se informó una asociación significativa entre la
presencia del virus y niveles elevados de marcadores intralesionales de inflamación como
CCL5, IL-6, CXCL10 y TNF-α. Todos los casos analizados correspondían a la infección
por L. (V.) guyanensis [30].
4.4.4 Detección de LRV en muestras clínicas.
En aras de evaluar los mecanismos de identificación del virus en muestras clínicas,
Zangger y colaboradores en el 2013 probaron 7 técnicas para detectar el virus en
aislamientos de parásitos con diferentes cargas virales y en la biopsia de un modelo murino
infectado con una cepa positiva para el virus. Las técnicas utilizadas fueron la extracción
de dsRNA, qRT-PCR, western blot, Inmunofluorescencia, slot blot, dot blot y ensayo por
inmunoadsorción ligado a enzimas (ELISA). En las cepas con baja carga viral, el virus no
pudo ser detectado por ninguna técnica [142].

43
En este estudio, la única técnica que logro detectar el virus en la totalidad de los
aislamientos con alta carga viral y en la biopsia del modelo murino el virus fue el dot blot,
una técnica de hibridización en la que se detectó dsRNA mediante un anticuerpo
denominado J2 que reconoce dsRNA independientemente de la secuencia y la
composición nucleotidica del mismo. La siguiente técnica con mayor poder de detección
fue la extracción de dsRNA, sin embargo los autores señalan que el incoveniente de su
uso radica en la necesidad de disponer de por lo menos 108 parásitos [142].
Ginouves y colaboradores quienes buscaron el virus con RT-PCR en aislamientos,
discuten el hecho de las muestras usadas para la detección, pues se han visto mayores
frecuencias en estudios que utilizan aislamientos parasitarios respecto a los que usan
biopsias, en donde se sabe hay baja carga parasitaria y material genético tanto del
huésped humano como del parásito [143]. Esto se vio claramente en el estudio de
Bourreau quien obtuvo una prevalencia del virus de 87% usando aislamiento versus una
prevalencia de 58.7% en biopsias. Cabe resaltar que con los aislamientos se utilizó la
técnica de dot blot con el anticuerpo J2 mientras que con las biopsias se usó qRT-PCR
[30].
La tabla 2 relaciona en detalle los estudios a la fecha que han buscado detectar el virus
en muestras clínicas:
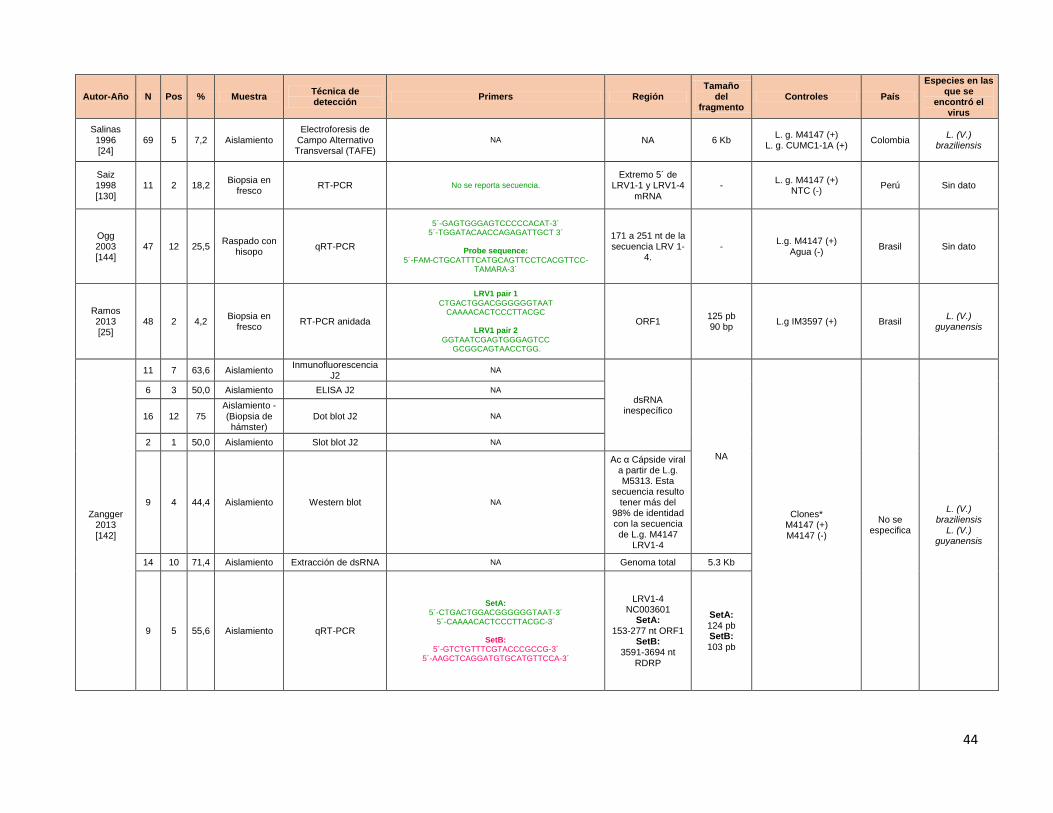
44
Autor-Año N Pos % Muestra Técnica de detección
Primers Región Tamaño
del fragmento
Controles País
Especies en las que se
encontró el virus
Salinas 1996 [24]
69 5 7,2 Aislamiento Electroforesis de
Campo Alternativo Transversal (TAFE)
NA NA 6 Kb L. g. M4147 (+)
L. g. CUMC1-1A (+) Colombia
L. (V.) braziliensis
Saiz 1998 [130]
11 2 18,2 Biopsia en
fresco RT-PCR No se reporta secuencia.
Extremo 5´ de LRV1-1 y LRV1-4
mRNA -
L. g. M4147 (+) NTC (-)
Perú Sin dato
Ogg 2003 [144]
47 12 25,5 Raspado con
hisopo qRT-PCR
5´-GAGTGGGAGTCCCCCACAT-3´ 5´-TGGATACAACCAGAGATTGCT 3´
Probe sequence:
5´-FAM-CTGCATTTCATGCAGTTCCTCACGTTCC-TAMARA-3´
171 a 251 nt de la secuencia LRV 1-
4. -
L.g. M4147 (+) Agua (-)
Brasil Sin dato
Ramos 2013 [25]
48 2 4,2 Biopsia en
fresco RT-PCR anidada
LRV1 pair 1
CTGACTGGACGGGGGGTAAT CAAAACACTCCCTTACGC
LRV1 pair 2
GGTAATCGAGTGGGAGTCC GCGGCAGTAACCTGG.
ORF1 125 pb 90 bp
L.g IM3597 (+) Brasil L. (V.)
guyanensis
Zangger 2013 [142]
11 7 63,6 Aislamiento Inmunofluorescencia
J2 NA
dsRNA inespecífico
NA
Clones* M4147 (+) M4147 (-)
No se especifica
L. (V.) braziliensis
L. (V.) guyanensis
6 3 50,0 Aislamiento ELISA J2 NA
16 12 75
Aislamiento -
(Biopsia de hámster)
Dot blot J2 NA
2 1 50,0 Aislamiento Slot blot J2 NA
9 4 44,4 Aislamiento Western blot NA
Ac α Cápside viral a partir de L.g. M5313. Esta
secuencia resulto tener más del
98% de identidad con la secuencia de L.g. M4147
LRV1-4
14 10 71,4 Aislamiento Extracción de dsRNA NA Genoma total 5.3 Kb
9 5 55,6 Aislamiento qRT-PCR
SetA: 5´-CTGACTGGACGGGGGGTAAT-3´
5´-CAAAACACTCCCTTACGC-3´
SetB: 5´-GTCTGTTTCGTACCCGCCG-3´
5´-AAGCTCAGGATGTGCATGTTCCA-3´
LRV1-4 NC003601
SetA: 153-277 nt ORF1
SetB: 3591-3694 nt
RDRP
SetA: 124 pb SetB: 103 pb

45
Zangger 2014 [10]
8 4 50,0 Aislamiento (Pacientes)
Dot blot J2 NA dsRNA
inespecífico NA
Clones M4147 (+) M4147 (-)
Etiopia
L. (L.) aethiopica 5 4 80,0
Aislamiento (Pacientes y
cepa de referencia)
Extracción de dsRNA NA Genoma total 5.3 Kb Etiopia
3 1 33,3 Aislamiento (Cepa de
referencia) RT-PCR
Primers Universales
UNIVf: 5´-TBRTWGCRCACAGTGAYGAAGG-3´ UNIVr: 5´-CWACCCARWACCABGGBGCCAT-3
Cápside viral 488 pb Etiopia
(Cepas de referencia)
Massayuki 2014 [26]
37 26 70,3
Muestra de la base de biopsia
obtenida con cepillos
cervicales
RT-PCR F: 5´ATGCCTAAGAGTTTGGATTCG-3´´ R: 5´ACAACCAGACGATTGCTGTG-3
ORF1 245 pb No se informa Brasil L. (V.)
braziliensis L. (V.) guyanensis
Cantanhede 2015 [27]
156 71 45,5
Muestra del borde interno de la lesión con cepillo
cervical estéril.
RT-PCR LRV1 F–5’-ATGCCTAAGAGTTTGGATTCG-3’ LRV1 R– 5’-ACAACCAGACGATTGCTGTG-3’
ORF1 240 pb L. g. M4147 (+) Brasil
L. (V.) braziliensis L. (V.) guyanensis
L. (L.) amazonensis L. (V.) lainsoni
Adaui 2015 [28]
97 32 33,0 Aislamiento RT-PCR Primers Universales
UNIVf: 5´-TBRTWGCRCACAGTGAYGAAGG-3´ UNIVr: 5´-CWACCCARWACCABGGBGCCAT-3
Cápside viral 488 pb
L. g M5313 (+) L.g. 17 sin LRV1 (-)
Buffer (-) Mock cDNA sin
retrotranscriptasa (-)
Perú Bolivia
L. (V.) braziliensis
Bourreau 2015 [30]
54 47 87,0 Aislamiento Dot blot J2 NA NA NA No se informa
Guyana francesa
L. (V.) guyanensis
75 44 58,7 Biopsia en
fresco qRT-PCR
LRV1-F 5′-GAGTGGGAGTCCCCCACAT-3′; LRV1-R, 5′-TGGATACAACCAGACGATTGCT-3′
LRV1 MGB probe: 5′-FAM -CATTTATGTAGTTCCT-3′.
ORF1 - L. g. M4147 (+)
L. p. LS94(-)
Alves 2015 [141]
4 0 0,0 Aislamiento RT-PCR Fr 5’-CGGTAGAGCATTAAGGGCTAGC-3’ Re 5’-CGGCAGTAACCTGGATACAACC-3’
ORF1 182 pb L. g. M4147 (+) Brasil L. (V.)
braziliensis
Macedo 2016 [29]
44 0 0,0 Aislamiento RT-PCR Primers Universales
UNIVf: 5´-TBRTWGCRCACAGTGAYGAAGG-3´ UNIVr: 5´-CWACCCARWACCABGGBGCCAT-3
Cápside viral 496 pb L. g. M4147 (+) L. b. M2903 (-)
Brasil L. (V.)
braziliensis
Ginouves 2016 [143]
129 96 74,4 Aislamiento RT-PCR LRV1-F1: 5'-CTG ACTGGACGGGGGGTAAT-3'
LRV1-R1: 5'-CAAAACACTCCCTTACGC-3' ORF1 124 pb
L. g. LBC6 (+) Agua (-)
Guyana francesa
L. (V.) braziliensis L. (V.) guyanensis

46
Hartley 2016 [145]
78 30 38,5 Aislamiento qRT-PCR Fwd: 5’-GAGTGGGAGTCCCCCACAT-3’;
Rev: 5’-TGGATACAACCAGACGATTGCT-3’; MGB probe:5’-FAMCATTTATGTAGTTCCT-3’
ORF1 - L. g. M4147 (+) L. p. LS94 (-)
Guyana francesa
L. (V.) guyanensis
Hajjaran 2016 [146]
50 2 4,0 Aislamiento RT-PCR semi anidada
LRV F-HR, 5´-4304TGTAACCCACATAAACAGTGTGC-3´ LRV R-HR, 5´-4809ATTTCATCCAGCTTGACTGGG-3´
LRV F2-HR 5´-4515AGGACAATCCAATAGGTCGTGT-3´
y LRV R-HR.
RDRP 526 pb 315 pb
Muestra LRV (-) NTC
Irán L. (L.) major
L. (L.) infantum
Tabla 2. Cuadro resumen de ensayos de identificación de LRV en muestras clínicas. *Cepas incluidas dentro de la identificación. Verde: primers
dirigidos a ORF1 del genoma vira, morado: primers dirigidos al ORF2 y fucsia: primers dirigidos a ORF3.

47
5 OBJETIVOS
5.1 OBJETIVO GENERAL
Determinar la frecuencia en que se detecta Leishmaniavirus en biopsias y aislamientos de
pacientes con diagnostico confirmado de LM y en pacientes con diagnóstico de LC en falla
terapéutica.
5.2 OBJETIVOS ESPECÍFICOS
Establecer un método de detección del virus mediante técnicas de RT-PCR y
RT-qPCR.
Determinar la frecuencia en la que se detecta el virus en aislamientos clínicos
de pacientes con LM y LC en falla terapéutica.
Determinar la frecuencia en la que se detecta el virus en biopsias de pacientes
con LM.
Determinar la especie infectante de Leishmania sp. asociadas a las muestras
clínicas analizados en el presente estudio.

48
6 METODOLOGÍA
6.1 TIPO DE ESTUDIO
Estudio de tipo descriptivo, con componente experimental, retrospectivo y de corte
transversal.
6.2 CONSIDERACIONES ÉTICA
El proyecto fue concebido de acuerdo a las consideraciones de la Declaración de Helsinki.
El diseño metodológico del estudio cumplió con los lineamientos de la normatividad
colombiana expuestos en la Resolución 008430 de 1993 del Ministerio de Salud para el
desarrollo de la actividad investigativa en salud. La investigación fue considerada sin riesgo
según lo define el artículo 11 de la mencionada resolución, pues no implico la intervención
o modificación de variables biológicas, fisiológicas, psicológicas o sociales de los
individuos que participaron en el estudio.
Los aislamientos parasitarios y las muestras clínicas incluidas hacen parte de los bancos
biológicos del Hospital Universitario Centro Dermatológico Federico Lleras Acosta
(CDFLLA) y del laboratorio de parásitología de la facultad de Medicina de la Universidad
Nacional de Colombia (UNAL). Todos cuentan con formato de preservación de muestras
en los mencionados bancos biológicos, el cual fue firmado por el paciente en el momento
de la toma de la muestra.
Se garantizó el anonimato y la confidencialidad de la información personal identificable y
de contacto (nombre, documento de identidad, dirección, teléfono) de los pacientes. Para
el caso de las muestras procedentes del CDFLLA, estas fueron solicitadas por el co-
director del presente trabajo, al coordinador del banco biológico del CDFLLA, quien hizo la
entrega. El co-director asumió la responsabilidad directa del material, garantizó el uso
adecuado de las muestras biológicas y se aseguró de que en el caso de existir excedentes
de las muestras utilizadas al final del proyecto, este material biológico fuera destruido.
Respecto a la experimentación con animales y con el fin de dar cumplimiento a los
principios de ética animal pactados en el Comité de Ética para el Uso de Animales
Experimentales de la Universidad y las leyes de la República de Colombia, se siguieron

49
las normas nacionales e internacionales para todos los procedimientos de alojamiento,
manipulación, sedación y eutanasia.
6.2.1 Aspectos ambientales.
El desarrollo de este proyecto implicó actividades que podían contaminar el medio
ambiente ya que incluían el manejo de microorganismos. Desde la perspectiva del riesgo
biológico, la manipulación de los microorganismos se realizó sistemáticamente en un
espacio cerrado y en cámara de flujo laminar clase II.
Se aplicaron los procedimientos de esterilización y manejo de residuos acorde con el
decreto 351 del año 2014, sobre el cual se reglamenta la gestión integral de los residuos
generados en la atención en salud y otras actividades. De igual manera los reactivos
empleados para las técnicas de biología molecular, se clasificaron y fueron etiquetados
con las convenciones requeridas para el manejo seguro.
6.2.2 Bioseguridad.
De acuerdo a la naturaleza de esta investigación, se asumió lo descrito en el capítulo I del
título IV de la Resolución 008430 de 1993 del Ministerio de Salud para el desarrollo de la
actividad investigativa en salud. Durante todo el proceso investigativo se tuvieron en cuenta
las normas de bioseguridad establecidas en el CDFLLA y en el manual de procedimientos
del laboratorio de parasitología de la Facultad de medicina de la UNAL. En cada ensayo
se hizo uso de todas las barreras de protección requeridas y se utilizaron las áreas
acondicionadas para dicho fin según los protocolos de uso.
6.3 SELECCIÓN DE LAS MUESTRAS
Se trabajó con un muestreo a conveniencia con un total de 31 muestras clínicas, de estas
16 estaban asociadas a lesiones de pacientes del CDFLLA que tenían diagnostico
confirmado de LM. Las muestras se distribuian: 6 aislamientos parasitarios de LM, 10
biopsias en fresco de LM y 15 aislamientos parasitarios del banco biológico del laboratorio
de parasitología de la Facultad de medicina de la UNAL provenientes de pacientes con LC
en falla terapéutica [147]. Todas las muestras tenían consentimiento informado.

50
6.4 CULTIVO DE AISLAMIENTOS PARASITARIOS
Los stocks de parásitos criopreservados fueron descongelados en medio Senekjie, el cual
fue rehidratado cada tercer día con 300 µl de medio Schneider (Schneider’s Drosophila
Insektenmedium, Genaxxon Bioscience) suplementado con 10% de suero fetal bovino y
gentamicina. Una vez se logró el crecimiento adecuado de los parásitos, estos fueron
amplificados hasta alcanzar la fase estacionaria en medio Schneider suplementado a una
temperatura de 26°C. Dicho procedimiento también se efectuó con las cepas de referencia
MHOM/BR/75/M4147 y MHOM/PA/71/LS94, usadas como controles positivo y negativo
respectivamente.
6.5 BIOPSIAS
Los fragmentos de biopsias se encontraban sin ningún persevante en viales estériles
congelados a -80°C y fueron manipuladas sobre hielo previa extracción de RNA. Todas las
biopsias contaban con confirmación parasitológica. El CDFLLA otorgó también un
fragmento de biopsia de un paciente con diagnóstico de lepra lepromatosa como control
negativo de los ensayos.
6.6 PRODUCCIÓN DEL CONTROL POSITIVO PARA LA DETECCIÓN DEL VIRUS EN BIOPSIAS
Para la estandarización de la detección del virus en las biopsias de los pacientes, se
requirió la obtención de un tejido infectado con un aislamiento parasitario portador del virus.
Para ello, se utilizaron dos hámsteres dorados juveniles de especie Mesocricetus auratus,
los cuales fueron inoculados en hocico y almohadillas plantares de las patas traseras con
100 µl de solución salina conteniendo 2 x106 promastigotes de MHOM/BR/75/M4147.
Los animales fueron alojados en una jaula con condiciones estables de temperatura (18-
22 °C) y humedad (50-70 %) y ciclo continuos de luz/oscuridad (12 horas), bajo dieta
supervisada por personal capacitado de agua y alimento concentrado ad libitum. El
seguimiento clínico se realizó a intervalos de 3 días, a partir del día de inoculación de los
parásitos hasta tanto fue evidenciada una lesión en el sitio de la inoculación, momento en
el cual el animal fue sacrificado previamente a la toma de la biopsia siguiendo el protocolo
ético.
El tejido fue fraccionado en varios tubos eppendorf y congelados a -80°C hasta su uso.
Con una fracción del tejido se hizo una aposición en una lámina de vidrio que

51
posteriormente fue coloreada con la colación Giemsa y otro fragmento fue preservado en
formol para posterior coloración Hematoxilina-eosina.
6.7 EXTRACCIÓN DE RNA
Para la extracción de los aislamientos parasitarios se utilizó el kit Direct-zol™ RNA
MiniPrep (Zymo Research). La extracción se hizo a partir de una cantidad de parásitos en
fase estacionaria igual o superior a 1X108. En cuanto a las biopsias, la extracción se hizo
del fragmento disponible utilizando el kit Quick-RNATM MiniPrep Plus (Zymo Research).
Al obtener el RNA se procedió a cuantificar en NanoDrop y se mantuvo guardado a -80°C
hasta su uso.
6.8 RETROTRANSCRIPCIÓN
De los aislamientos parasitarios se hizo la retrotranscripción de 2 µg de RNA, a excepción
de dos de ellos en los que se retrotranscribieron 500 ng debido a la baja concentración de
RNA obtenida. En cuanto a las biopsias se hizo retrotranscripción de 1 µg a excepción del
control negativo en el que se hizo retrotranscripción de 200 ng por la misma causa
mencionada anteriormente.
Las condiciones del ensayo fueron estandarizadas a un volumen final de 30 µl en donde
las concentraciones finales usadas fueron 1X de Buffer de la enzima M-MuLV, 4µM de
random primers, 1mM de dATP, dCTP, dTTP y dGTP y 200 U de transcriptasa reversa M-
MuLV. Se ejecutó el perfil térmico sugerido por la casa comercial de la enzima New
England Biolabs: 70°C por 5 min para todos los reactivos excepto la enzima y su respectivo
buffer; una vez agregados se continuó con 60 min a 42°C y finalmente 10 min a 90°C para
la inactivación de la enzima. El cDNA obtenido fue conservado a -20°C hasta su uso.
6.9 CONFIRMACIÓN DE LA CALIDAD DEL CDNA
Para determinar si el cDNA obtenido era de buena calidad se corrió una RT-PCR para los
aislamientos y una qRT-PCR para las biopsias del gen de la subunidad ribosomal 18S del
parásito.
La RT-PCR se llevó a cabo en un volumen final de 50 µl que contenían 66.6 ng de cDNA,
Buffer KCL 1X, 1.65 mM de MgCl2, 200 μM dATP, dCTP, dTTP y dGTP; 0.4 µM de cada
primer [148] Fw 5’- CCAAAGTGTGGAGATCGAAG -3’ y Rev 5’-

52
GGCCGGTAAAGGCCGAATAG -3’ y 2.5 unidades de Taq polimerasa recombinante. El
perfil térmico de la reacción consistió en un ciclo de 94°C por 5 min y 32 ciclos de 94°C por
30 seg, 65°C por 30 seg y 72°C de 30 seg. Los productos amplificados fueron analizados
mediante electroforesis en gel de agarosa al 2 % en solución amortiguadora TAE 1X a 100
V. La banda de 100 pb aprox. fue revelada con bromuro de etidio a una concentración final
de 0.5µg/ml y visualizada por UV.
La qRT-PCR se realizó a un volumen final de 15 µl que contenían 19.9 ng de cDNA, 1X
SYBR Green Master mix Select y 0.6 µM de los mismos primers reportados anteriormente
para el gen. El perfil térmico de la reacción consistió en un ciclo de 50°C por 2 min, un ciclo
de 95°C por 2 min y 45 ciclos de 95°C por 30 seg y 60°C por 45 seg. Todos los ensayos
incluyeron dos réplicas de cada muestra y control.
6.10 IDENTIFICACIÓN DEL VIRUS EN AISLAMIENTOS PARASITARIOS
La RT-PCR capaz de amplificar 488 pb de la región que corresponde a la cápside viral
(Fig. 10) se estandarizo con un perfil térmico que incluyó un ciclo de 95ºC por 5 min; 33
ciclos de 95°C por 10 seg, 60°C por 10 seg y 72°C por 32 seg. La mezcla de reacción
contenía 133,2 ng de cDNA, 1X Buffer KCL, 2.5mM de MgCl2, 200 μM dATP, dCTP, dTTP
y dGTP; 0.5 µM de cada primer Zangger [10] Fw 5’- TBRTWGCRCACAGTGAYGAAGG -
Fig. 10. Diagrama del genoma viral de LRV1 con la ubicación de los primers utilizados en la
detección para aislamientos parasitarios.

53
3’ y Rev 5’- CWACCCARWACCABGGBGCCA T -3’ y 2.5 unidades de Taq polimerasa
recombinante en un volumen final de 50 µl. Los productos amplificados fueron analizados
mediante electroforesis en las condiciones mencionadas anteriormente.
La qRT-PCR se estandarizó en una reacción con 26.64 ng de cDNA, 1X SYBR Green
Master mix Select y 0.2 µM de cada primer Massayuki [26] Fw 5’-
ATGCCTAAGAGTTTGGATTCG-3’ y Rev 5’- ACAACCAGACGATTGCTGTG-3’. La región
amplificada correspondió a 245 pb del extremo 5´UTR, región ampliamente reportada
como conservada entre las diferentes especies de Leishmaniavirus (Fig. 10). El perfil
térmico de la reacción consistió en un ciclo de 50ºC por 2 min, un ciclo de 95°C por 2 min;
40 ciclos de amplificación de 95°C por 15 seg y 60°C por 1 min.
6.11 IDENTIFICACIÓN DEL VIRUS EN BIOPSIAS
En las biopsias, la identificación del virus fue mediante una PCR anidada que amplifica un
fragmento de la región 5’UTR del virus. Tanto en el primer como en el segundo corrido, la
RT-PCR convencional se realizó con un perfil térmico que incluyó un ciclo de 95 ºC por 3
min; 35 ciclos de 95°C por 30 seg, 55°C por 30 seg y 72°C por 30 seg. La mezcla se realizó
a un volumen final de 50 µl que contenian 1X Buffer KCL, 1.5mM de MgCl2, 200 μM dATP,
dCTP, dTTP y dGTP, 2.5 unidades de Taq polimerasa recombinante, 0.3 µM de cada
primer ANILRV-1 [25] Fw 5’- CTGACTGGACGGGGGGTAAT -3’ y ANILRV-1 Rev 5’-
GCGTAAGGGAGTGTTTTG -3’ en el primer round y ANILRV-2 Fw 5’-
GGTAATCGAGTGGGAGTCC -3’ y ANILRV-2 Rev 5’- GCGGCAGTAACCTGG-3’ en el
segundo round, un volumen de cDNA equivalente a 33.3 ng y 1 µl del primer producto sin
purificar respectivamente.
Los fragmentos amplificados fueron de 125 pb y 90 pb respectivamente y fueron
visualizados en las mismas condiciones mencionadas anteriormente en los otros ensayos
de RT-PCR. El control positivo fue cDNA del aislamiento de L. (V.) guyananesis M4147 y
los controles negativos incluyeron una biopsia de un paciente con diagnóstico de lepra
lepromatosa, DNA de un paciente con diagnóstico de tuberculosis y un control de la mix
(NTC).

54
6.12 IDENTIFICACIÓN DE ESPECIE EN AISLAMIENTOS PARASITARIOS Y BIOPSIAS
La identificación de especie se siguió de acuerdo a lo descrito por Cruz-Barrera y
colaboradores en el 2015 [149].
6.13 ANÁLISIS ESTADÍSTICO
El análisis estadístico se aplicó en los aislamientos de LC en falla terapéutica, para dicho
fin se formaron dos grupos de análisis “LRV positivo” y “LRV negativo”. La variable
presencia o ausencia del virus se categorizó con los valores 1 y 0 respectivamente. Dado
el bajo número de datos y la distribución no paramétrica de los mismos se aplicó una
prueba prueba U de Mann-Whitney entre ambos grupos para las varibles porcentaje de
infectividad in vitro y las concentraciones efectivas 50 para Glucantime® (IC50)
determinadas por Pérez y [147]. El análisis se corrió en Microsoft Excel 2013.
El porcentaje de infección se calculó mediante la visualización microscópica de láminas
teñidas con Giemsa y correspondió al número de macrófagos infectados de 100
macrófagos contados y el IC50 correspondió a la concentración de Glucantime® que redujo
a la mitad la supervivencia de los parásitos de Leishmania analizados [147].
6.14 ELABORACIÓN DEL MAPA DE DISTRIBUCIÓN DE ESPECIES
La base de datos se realizó en Microsoft Excel 2013 e incluyó la presencia o ausencia del
virus y la procedencia geográfica a nivel de municipio. Los municipios fueron
georreferenciados en unidades decimales para su posterior ubicación geográfica. Se uso
como herramienta para la elaboración del mapa el programa Arcgis versión 10.1. Se
incluyó una cobertura deartamental de Colombia y una cobertura de altura sacada de
http://www.worldclim.org/ con una resolución de celda de aproximadamente un kilometro.

55
7 RESULTADOS
7.1 ESTANDARIZACIÓN DE LA DETECCIÓN DEL VIRUS EN EL LA CEPA DE REFERENCIA M4147.
7.1.1 Deteccion del virus en la cepa de referencia M4147 por PCR convencional.
Usando cDNA del aislamiento de referencia M4147, se realizó un primer en ensayo de
qRT-PCR buscando la amplificación de un fragmento de 448 pb correspondiente a la
cápside. Como controles negativos se usaron las cepas de L. (V.) braziliensis M2903 y L.
(V.) guyanensis LEM85, quienes no han sido reportadas previamente como positivas para
el virus. Los primers utilizados fueron seleccionados debido a que la literatura los señala
como los primers universales para la detección del virus, ya que fueron diseñados a partir
de la región conservada entre los genomas reportados de LRV1 y LRV2 para las cepas L.
(L.) major ASKH, L. (V.) guyanensis CUMC-1A y L. (V.) guyanensis M4147 [10].
En este ensayo se evidenció una curva clara de amplificación pero el sistema no logró
calcular un threshold cycle (Ct) probablemente debido a que el tamaño del amplicon
superaba lo recomendado para la técnica. La reacción mostró una curva melting definida,
una temperatura melting (Tm°) de 84.2°C y al correrse en un gel de agarosa del 2% se
observó una banda del tamaño esperado.
Dados estos hallazgos se sugirió hacer uso de la amplificación de este fragmento mediante
una RT-PCR como primer filtro de detección del virus. Las condiciones estandarizadas se
reportan en la sección de “Metodología”, el perfil térmico utilizado fue el mismo reportado
por Zangger y colaboradores, con la eliminación del ciclo de detección final [142]. Los
resultados obtenidos muestran la amplificación de una única banda del tamaño esperado
(Fig. 11).
En cuanto a la determinación de la cantidad optima de cDNA a utilizar en la RT-PCR, se
realizó una titulación probando 5 cantidades de cDNA: 133.2 ng, 66.6 ng33.3 ng, 19.9 ng
y 6.6 ng. Dado que a la mayor cantidad no se satura la reacción y se observa mayor
definición de la amplificación, se determinó que se utilizaría dicha cantidad para detección
del virus en los aislamientos (Fig. 12).

56
7.1.2 Estandarización de las condiciones de detección del cDNA viral por qRT-
PCR.
Con la idea de ampliar la sensibilidad de detección del virus mediante qRT-PCR, se
seleccionaron otros primers reportados por Massayuki y colaboradores que amplifican una
región del extremo 5´ UTR del genoma viral, correspondiente al ORF1. Para lo anterior se
probaron tres concentraciones de cDNA de la cepa de referencia M4147: 133.2 ng, 66.6
ng y 26.64 ng a una concentración final de primers de 0.2 µM y un con un perfil térmico de
50°C por 2 min, 95°C por 2 min y 40 ciclos de 95°C por 15 seg, 57°C por 15 seg y 72°C
por un 1 min. En este primer ensayo se obtuvo para las tres condiciones una curva melting
definida, Tm° superiores a 82°C que sugieren una amplificación específica y francas curvas
de amplificación, sin embargo los Cts sugieren una probable saturación de cDNA pues la
premisa de la técnica indica que a menor Ct hay mayor disposición del blanco molecular
en la muestra analizada y en el ensayo el menor Ct se obtuvo al utilizar la menor cantidad
de cDNA (Tabla 3).
Fig. 11. Amplificación del fragmento de 488
pb correspondiente a la region de la cápside
mediante qRT-PCR y RT-PCR. MPM:
marcador de peso molecular.
Fig. 12. Titulación de la cantidad de cDNA en
la RT-PCR. A. 133.2 ng, B. 66.6 ng, C. 33.3 ng,
D. 19.9 ng y E. 6.6 ng

57
De acuerdo a lo anterior se realizó una curva de detección en base 10 con siete puntos a
partir de 24.64 ng de cDNA, para determinar el alcance de la prueba. En el ensayo solo se
obtuvo curvas francas de amplificación, adecuados Tm°, curvas melting definidas y Cts
definidos solo para un rango de 26.64 ng a 26 pg de cDNA, equivalentes a un rango de
5x105 a 5x102 parásitos respectivamente (Tabla 4, Fig. 13).
Tabla 3. Ensayo de titulación de cDNA para la estandarización de
la qRT-PCR usando primers de Massayuki y colaboradores.
ID Carga parasitaria
(parásitos) cDNA (ng) Ct promedio Tm°
promedio
1 5 x105 26.64 20.75 83.9
2 5 x104 2.66 24.8 83.9
3 5 x103 0.26 29.9 83.9
4 5 x102 0.026 35.6 83.9
5 5 x101 0.0026 Indeterminado 65.5
6 5 0.00026 Indeterminado 66.3
7 0.5 0.000026 Indeterminado 66.7
8 NTC NTC Indeterminado 66.35
A
B
A. Tabla 4. Cts promerdio y Tm° promedio obtenidos para la curva parasitaria en base 10 para
la detección de LRV con los primers de Masayuki y colaboradores. B. Fig. 13. Plot de
amplificación en RT-qPCR para la curva parasitaria anteriomente mencionada.

58
La eficiencia de la qRT-PCR calculada fue de 59.08% con un R2 de 0.99, hecho que aunque
mostró una adecuada linealidad de la curva estándar, reflejó una baja capacidad de
amplificación. Por tanto, para optimizar la técnica se cambió el perfil térmico al sugerido
por el inserto de la casa comercial Applied Biosystems para primers con Tm° de fabricación
mayores a 60°C y se probaron dos concentraciones finales de primers, 0.2 y 0.4 µM. El
ensayo mostró grandes mejoras al lograr la cobertura de tres puntos mas dentro de la
curva realizada detectando amplificación del blanco viral en un rango de 26.64ng a 0.026
pg de cDNA, equivalentes a un rango de 5x105 a 0.5 parásitos respectivamente (Tabla 5).
Las eficiencias obtenidas fueron de 95.68% y 92.29% con concentraciones finales de
primers 0.2 y 0.4 µM respectivamente, ambas curvas con R2 de 0.99. De lo anterior se
concluyen las condiciones adecuadas para la amplificación del virus y se reportan en la
sección de “Metodología”.
7.2 DETECCIÓN DEL VIRUS EN AISLAMIENTOS CLINICOS DE PACIENTES CON LM Y LC EN FALLA
TERAPÉUTICA.
Para todos los aislamientos clinicos se realizo la RT-PCR convencional según las
condiciones estandarizadas en la sección 7.1.1 del presente capitulo. Del total de 21
aislamientos se obtuvo positividad en 9 de ellos, lo cual corresponde a una frecuencia de
Tabla 5. Cts promerdio y Tm° promedio obtenidos para la curva parasitaria en base 10 para la
detección de al cambiar condiciones del perfil térmico y utilizando dos concentraciones finales
de primers LRV de Masayuki y colaboradoes.

59
presencia viral de 16.6% (1/6) en aislamientos de LM y de 26.6% (4/15) en LC asociada a
falla terapéutica.
Con el animo de corroborar los resultados se procedio a la busquedad de la detección viral
mediante la prueba cuantitativa cuyas condiciones fueron estandarizadas según se
describe en 7.1.2. Como resultado de la qPCR se pudo observar que en 4 aislamientos
negativos para el cDNA por RT-PCR convencional la qRT-PCR fue positiva. Los Cts y las
Tm° de los aislamientos encontrados positivos con la qRT-PCR se señalan a continuación
(Tabla 6). Dos de ellos comprendían a LM y los otros a LC en falla terapéutica. Ademas de
obtener la curva melting definida, los productos se corrieron en un gel del 2% mediante
electroforesis y todos mostraron una banda del tamaño esperado, 245 pb (Fig. 14).
A
B
A. Tabla 6. Cts promerdio y Tm° promedio obtenidos para los aislamientos de LM y LC en falla
terapéutica negativos en la RT-PCR. B. Fig. 14. Ejemplo de resultados obtenidos con los 4
aislamientos negativos, plot de amplificación, curva melting y gel del aislamiento 707 de LM.
Aislamiento Condición Ct promedio Tm° promedio
707 LM 34.4 83.1
384 LM 33.35 82.7
45 LC en falla terapéutica 29.55 83.1
36 LC en falla terapéutica 16.9 82.4
NTC - Indeterminado 67.23
Control de la retrotranscripción - Indeterminado Noisy

60
En esta PCR se usaron como control positivo la cepa MHOM/BR/75/M4147 y como control
negativo la cepa MHOM/PA/71/LS94, se incoporó además un control de la
retrotranscripción sin enzima y un NTC. Todos los ensayos incluyeron dos réplicas de cada
muestra y control.
7.3 ESTANDARIZACIÓN DE LA DETECCIÓN DEL VIRUS EN BIOPSIAS.
Para la estandarización de la detección del virus en las biopsias era necesario trabajar con
una muestra compatible con las que serían analizadas de pacientes con LM, por lo que se
vio la necesidad de generar un control positivo a partir de una modelo animal. Dos
hámsteres sirios fueron inoculados con la cepa de referencia M4147, desarrollaron una
lesión en el hocico a los 17 días de inoculación, en las almohadillas plantares no se observó
la aparición de ningún tipo de lesión. Los animales fueron sacrificados a los 24 días de
inoculación y se obtuvo biopsias de los tejidos comprometidos. Con la coloración Giemsa
de la aposición en lámina de vidrio y la coloración Hematoxilina-eosina de la biopsia en
formol se confirmó la presencia de amastigotes de Leishmania en el tejido (Fig. 15).
Una vez confirmada la infección se utilizo el cDNA obtenido para la estandarización de la
detección en el virus. El primer ensayo realizado fue la verificación de la calidad del cDNA
mediante la PCR convención 18S siguiendo las condiciones mencionadas en el numero
6.9 del capitulo de “Metodología”, sin embargo al probar 4 concentraciones distintas de
cDNA (66.6ng, 133.2ng, 199.8ng y 266.4ng), solo fue ligeramente visible en el gel en las
concentraciones mas altas (Fig. 16). Dado el resultado anterior se decidio utilizar la qRT-
PCR 18S para dicho fin.
Fig. 15. A. Ejemplar murino infectado con lesion en hocico. B. Coloración Giemsa de aposicon
de biopsia en lámina de vidrio. C. Coloración Hematoxilina-eosina de biopsia 64X. ↑ señala
formas amstigotes de Leishmania guyanensis M4147.

61
La qRT-PCR 18S se realizó usando las condiciones propuestas por Van de Bogaart y
colaboradores [148] y una concentración final de primers de 0.6 µM. En el ensayo se
probaron 6 concentraciones de cDNA 6.6ng, 19.9ng, 33.3ng, 66.6ng, 133.2ng y 266.4ng.
Los Cts obtenidos mostraron que usando las cantidades 66.6ng, 133.2ng y 266.4ng la
reacción se saturaba y usando 6.6 ng el Ct era prolongado respecto a las dos
concentraciones anteriores, por tanto las concentraciones 19.9ng y 33.3ng mostraron
mejores resultados. Dada la cantidad en volumen requerida se decidió utilizar para las
biopsias 19.9ng (Tabla 7).
El siguiente paso una vez confirmada la calidad del cDNA y la presencia detectable del
parásito, se continuo con la detección viral, para lo cual se realizó un ensayo preliminar
Fig. 16. Titulacion de la cantidad de cDNA de biopsia infectada con L. (V.)
guyanensis M4147 para la verifficacion de la calidad del mismo en la RT-PCR 18S.
cDNA biopsia hámster (ng)
Ct promedio Tm° promedio
266.4 38.57 84.3
133.2 38.8 84.3
66.6 34.5 84.3
33.3 28.02 84.3
19.9 27.94 84.5
6.6 38.26 84.7
NTC Indeterminado 65.5
Tabla 7. Cts promerdio y Tm° promedio obtenidos para
la curva del gen 18S usando cDNA proveniente de el
modelo infectado con la cepa L. (V.) guyanensis M4147.

62
usando 3 concentraciones de cDNA de la biopsia control 66.6ng, 133.2 ng y 199.8 ng en
la qRT-PCR del extremo 5´UTR estandarizada previamente para los aislamientos, sin
embargo no se obtuvo amplificación en ninguno de los casos. Con este resultado fue
necesario el uso de otra herramienta de detección para lo que se decidio utilizar una RT-
PCR anidada propuesta por Ramos y colaboradores [25].
En el proceso de estandarizacion de las condiciones de la RT-PCR, el primer ensayo
realizó con el aislamiento de M4147 con el fin de garantizar la amplificación y no consumir
en vano el control generado con el modelo animal. El perfil térmico sugerido por la literatura
funciono adecuadamente, en cuanto a las concentraciones de los reactivos se probaron
las condiciones de la RT-PCR utilizada en la detección del virus en aislamientos, pero se
obtuvo además de la banda de interés varias inespecifidades por lo que fue necesario
hacer una titulación del MgCl2, la cual resulto efectiva. Las condiciones finales del ensayo
se informan en la sección de “Metodología”.
Para la titulación del cDNA se probaron 6 cantidades de cDNA del fragmento de hocico del
hámster sirio infectado con la cepa de referencia M4147, 6.6ng, 19.9ng, 33.3ng, 66.6ng,
99.8ng y 333ng. Ninguna de dichas cantidades arrojó una banda contundente en el gel
(Fig. 17) y se procedio a hacer el anidamientos de las tres concentraciones mas bajas, de
las cuales se utilizo para el segundo round de la RT-PCR 2µl de una dilución 1/10. Como
parte del seguimiento al primer round de la RT-PCR se anidaron tanto el control positivo
como el negativo; el volumen utilizado del control positivo del primer round que
corresponde al cDNA del aislamiento de L. (V.) guyanensis M4147 y del control negativo
de reacción se utilizó 1µl puro (Fig. 18).
Los resultados obtenidos mostraron productos con banda única de 90 pb en las 3
cantidades de cDNA de las biopsias del animal y los controles tanto negativos, como
positivos dieron el resultado esperado. De manera interesante se evidencia que no hay
inhibición al usar 1µl puro de cDNA del control del aislamiento al momento de anidarlo,
razon por la cual se decidió utilizar 1 µl puro de los productos del primer round de las
biopsias objeto de estudio con el fin de aumentar la probabilidad de detección del virus en
el segundo round.

63
7.4 ALGORTIMO DE DETECCIÓN DEL VIRUS EN MUESTRAS CLINICAS DE PACIENTES CON LM Ó
LC EN FALLA TERAPÉUTICA.
Dadas las condiciones estandarizadas para la detección del virus tanto en aislamientos
como en biopsias se sugiere el siguiente algoritmo de detección:
Fig. 19. Algortimo de detección de LRV en aislamientos y biopsias.
Fig. 17. Primer round de la RT-PCR anidada
usando diferentes concentraciones de cDNA
procedente de biopsia del modelo murino
infectado con L. (V.) guyanensis M4147.
Fig. 18. Segundo round de la RT-PCR anidada de los
productos del primer round de las concentraciones
6.6ng, 19.98ng y 33.3ng.

64
7.5 FRECUENCIA DEL VIRUS
7.5.1 Frecuencia en aislamientos parasitarios y biopsias.
Una vez aplicado el algoritmo completo tanto a aislamientos como a biopsias se obtuvo
una frecuencia total del virus de 56.7% (17/30) en las muestras clínicas. En las biopsias,
todas procedentes de pacientes con diagnóstico confirmado de LM, aunque fueron
seleccionadas porque en el examen histopatológico se reportaron amastigotes, se les hizo
la verficación de la presencia del parásito mediante la qRT-PCR 18S, las biopsias M351A
y M359 fueron negativas, por lo que se hizo un segundo ensayo doblando la cantidad de
cDNA utilizado, solo la biopsia M351A resulto positiva tras la modificación y por tanto, la
biopsia M359 fue excluida del protocolo de detección viral. La prevalencia en biopsias del
virus fue del 80% (8/10) (Tabla 8).
En cuando a los aislamientos la prevalencia general fue del 42.85% (9/21), en los
aislamientos de LM fue del 50% (3/6) y en los aislamientos de LC en falla terapéutica del
40% (6/15). La verificación parasitaria fue innecesaria dado que todos provenían de cultivo.
El uso secuencial de la RT-PCR y la qRT-PCR logró rescatar 4 aislamientos que habrían
quedado como negativos si solo se hubiera considerado la RT-PCR de la cápside viral. En
la tabla 8 se presentan los resultados obtenidos aplicando el algoritimo para la detección
de LRV en aislamientos y biopsias de LM ó LC en falla terapéutica:
Muestra ID
Tipo de Muestra
Condición RT-PCR qRT-PCR
qRT-PCR 18S
RT-PCR nested
Round 1
RT-PCR nested
Round 2
LRV Final
576 Aislamiento LM Positivo NA NA NA NA Positivo
707 Aislamiento LM Negativo Positivo NA NA NA Positivo
127 Aislamiento LM Negativo Negativo NA NA NA Negativo
463 Aislamiento LM Negativo Negativo NA NA NA Negativo
384 Aislamiento LM Negativo Positivo NA NA NA Positivo
235 Aislamiento LM Negativo Negativo NA NA NA Negativo
2 Aislamiento Falla
terapéutica Positivo NA
NA NA NA Positivo
11 Aislamiento Falla
terapéutica Negativo Negativo
NA NA NA Negativo
18 Aislamiento Falla
terapéutica Positivo NA
NA NA NA Positivo
41 Aislamiento Falla
terapéutica Negativo Negativo
NA NA NA Negativo
8 Aislamiento Falla
terapéutica Negativo Negativo
NA NA NA Negativo

65
9 Aislamiento Falla
terapéutica Negativo Negativo
NA NA NA Negativo
23 Aislamiento Recaida Negativo Negativo NA NA NA Negativo
26 Aislamiento Falla
terapéutica Negativo Negativo
NA NA NA Negativo
44 Aislamiento Falla
terapéutica Negativo Negativo
NA NA NA Negativo
36 Aislamiento Falla
terapéutica Negativo Positivo
NA NA NA Positivo
45 Aislamiento Falla
terapéutica Negativo Positivo
NA NA NA Positivo
60 Aislamiento Falla
terapéutica Negativo Negativo
NA NA NA Negativo
53 Aislamiento Falla
terapéutica Positivo NA
NA NA NA Positivo
61 Aislamiento Recaida Positivo NA NA NA NA Positivo
63 Aislamiento Recaida Negativo Negativo NA NA NA Negativo
M312 Biopsia LM NA NA Positivo Negativo Positivo Positivo
M323 Biopsia LM NA NA Positivo Negativo Positivo Positivo
M326 Biopsia LM NA NA Positivo Negativo Positivo Positivo
M333 Biopsia LM NA NA Positivo Negativo Positivo Positivo
M340 Biopsia LM NA NA Positivo Negativo Positivo Positivo
M343 Biopsia LM NA NA Positivo Negativo Positivo Positivo
M354 Biopsia LM NA NA Positivo Negativo Positivo Positivo
M351A Biopsia LM NA NA Positivo* Negativo Negativo Negativo
M359 Biopsia LM NA NA Negativo NA NA NA
M348 Biopsia LM NA NA Positivo Negativo Positivo Positivo
7.6 TIPIFICACIÓN DE ESPECIES
El 95.5% de los aislamientos eran L. (V.) braziliensis el 4.5% restante era L. (V.)
panamensis. Todos los aislamientos positivos para el virus correspondieron a L. (V.)
braziliensis.
En cuanto a las biopsias, el 55.5% correspondio a L. (V.) braziliensis, el 22.2% a L. (V.)
panamensis y 22.2% a Leishmania spp. La única muestra negativa para el virus
correspondia a la especia L. (V.) braziliensis.
Tabla 8. Datos obtenidos aplicando el algoritimo para la detección de LRV en aislamientos y
biopsias de LM ó LC en falla terapéutica.

66
7.7 ANÁLISIS DE OTRAS VARIABLES.
7.7.1 Análisis estadístico.
En la prueba U de Mann-Whitney, al comparar la distribución de los datos en los grupos
de aislamientos de LC en falla terapéutica “LRV positivo” y “LRV negativo, no se encontró
diferencias significativas para el porcentaje de infección in vitro ni para el IC50 para
Glucantime®. Las probabilidades obtenidas fueron de 0.5237 y 0.7280 respectivamente
(Tabla 9 y 10) (Fig. 20).
05
01
00
150
200
0 1
Ic50(ug/ml) % Infectivi
ID IC 50(ug/ml) % INFECTIVIDAD LRV
2 103 52 1
8 133 38 0
9 174 36 0
18 174 33 1
23 98 42 0
26 71 64 0
44 122 57 0
36 160 51 1
41 132 40 0
45 124 54 1
53 145 44 1
61 119 40 1
63 127 46 0
60 103 36 0
1 26 60 1
% INFECTIVIDAD
LRV Media SD Mediana IQR
0 44,875 10,35702 41 14,5
1 47,71429 9,214378 51 14
Total 46,2 9,600595 44 16
IC50
LRV Media SD Mediana IQR
0 120 30,36916 124,5 32
1 121,5714 48,76084 124 57
Total 120,7333 38,48092 124 42
A B
C D
A. Tabla 9. Datos de % de infectividad in vitro, IC50 para Glucantime y presencia o ausencia del LRV en
aislamientos de LC en falla terapéutica. B. Fig. 20. Gráfica de biogotes obtenida del análisis de la prueba U
de Mann-Whitney para los grupos “LRV positivo” (1) y “LRV negativo” (0) respecto a las variables % de
infectividad e IC50. C. Tabla 10. Datos de media, mediana, SD e IQR para la varible IC50. D. Tabla 11. Datos
de media, mediana, SD e IQR para la varible %de infectividad. SD: Desviación estándar, IQR: Rango
intercuartilico.

67
7.7.2 Análisis geográfico.
En la figura 21 se georreferencia el posible origen geográfico de la infeccion por
Leishmania de todos los pacientes incluidos en el estudio, solo uno de los datos fue
excluido ya que no era claro el sitio de adquisición de la enfermedad. De las 17 muestras
clinicas positivas, se evidencia que el 35.2% (6/17) provenian de la la región Andina, el
35.2% (6/17) de la Orinoquía, el 23.5% (4/17) de la Amazonía y el 5.8% (1/17) de la región
Caribe (Fig. 24).
Fig. 21. Mapa de distribución geográfica de los aislamientos y
biopsias negativos y positivos paral LRV. Rojo: positivos para LRV,
azul: negativos para LRV.

68
8 DISCUSIÓN
Alrededor de 15 estudios se han realizado para la detección de LRV, las frecuencias del
virus en muestras clínicas oscilan entre 0 - 87%, lo que puede corresponder a múltiples
causas.
El componente técnico es una variable de peso a tener en cuenta debido a que se observan
diferencias de las frecuencias respecto al tipo de muestra y la técnica empleada. En el
2013, en el estudio de Zangger y colaboradores se comparan diferentes técnicas para la
detección del virus. La frecuencia osciló entre el 50% y 71.4%. El mayor porcentaje de
detección fue observado al utilizar extracción del dsRNA correspondiente al acido nucleico
viral y su posterior separación electroforética. Sin embargo, requirió para el análisis de por
lo menos 108 parásitos y una alta carga viral, lo cual en términos clínicos no tiene mayor
aplicabilidad. El siguiente porcentaje más alto lo obtuvo la aplicación de la técnica de dot
blot usando el anticuerpo J2, que detectó el virus tanto en biopsia como en aislamientos
en una alta frecuencia, sin embargo el anticuerpo no es especifico para el virus y al tener
la capacidad de reconocer cualquier RNA de doble banda, las coinfecciones con otros virus
podrían llegar a generar falsos positivos en el caso de usar como muestra las biopsias
[142].
En cuanto a la qRT-PCR, la frecuencia obtenida en dicha disertación fue de 55.6% usando
primers dirigidos a la region 5´UTR y a la polimerasa, sin embargo se discute que aunque
puede ser altamente sensible y permitiría la cuantificación de la carga viral, podría
presentar el inconveniente de perder capacidad de detección en cepas del virus que
presenten polimorfismos en la región amplificada. La posibilidad de no detectar el virus por
sus polimorfismos también se vio reflejada en el ensayo de western blot cuyo blanco de
detección era la cápside [142], hecho que años antes se había visto en ensayos de
hibridización [6].
En nuestro estudio al aplicar como primer filtro una RT-PCR de la cápside con los primers
universales, no fueron identificados 4 aislamientos que fueron captados en el segundo
filtro, la qRT-PCR del 5´UTR. Lo anterior señala de manera importante que los primers
considerados universales no reconocen variantes del virus que podrían presentar
polimorfimos en la secuencia de la cápside, y por ende que los resultados obtenidos en

69
estudios como el de Zangger y colaboradores del 2014 [10], Adaui y colaboradores [28] y
Macedo y colaboradores [29] se deben reconsiderar a la luz de lo que el presente estudio
informa, ya que al usar estos primers pudieron obtener falsos negativos que explicarian las
bajas frecuencias informadas para LRV (0-33%).
De acuerdo a esto se propone un algoritmo de detección del virus en aislamientos y
biopsias (Fig. 19) que incluye en este último caso la comprobación previa de la presencia
del parásito por medio de una qPCR, ya que al analizar muestras clínicas con cargas
parasitarias indetectable es improbable detectar el cDNA de LRV.
La detección del virus en las biopsias presenta una mayor dificultad, pues la muestra
incluye el material genético presente del huésped mamífero y en el parasito, disminuyendo
así la probabilidad de encontrar el del virus. En nuestro estudio no se logró detectar el virus
en las biopsias de pacientes con LM a través de la qRT-PCR del 5´UTR usando los primers
sugeridos por Massayuki y colaboradores, lo que difiere de la frecuencia más alta obtenida
en biopsias (58.7%) lograda por Bourreau y colaboradores haciendo uso de una qRT-PCR
con primers dirigidos también a la region 5´UTR [30], sin embargo cabe resaltar que la
metodología utilizada incluyo el uso de sondas, a diferencia de nuestro ensayo que utilizó
la metodología SYBR Green.
En consecuencia, se eligió la metodología de Ramos y colaboradores que incluia el uso
de un PCR anidada, aunque la frecuencia reportada para el virus solo fue del 4.2% [25].
Optimizando dicha metodología se logró una frecuencia del virus del 80%. Algunos
estudios señalan que para la detección de microorganismos, la PCR anidada respecto a
la qPCR podría llegar a ser más [150, 151] ó igual de sensible [152], sin embargo otros
atribuyen mayor sensibilidad a la qPCR y consideran otros beneficios como la posibilidad
de la cuantificación, mayor especificidad, la reducción de tiempo empleado en los
procedimientos y la disminución de problemas de contaminación [153-156].
Aunque la presencia del virus ha sido asociada en modelos murinos a un desarrollo
metastásico de la enfermedad cutánea [116], el estudio en muestras clínicas con
condiciones complicadas de la LC tales como la LM y la falla terapéutica no se han
encontrado positivas en su totalidad. En cuanto a la LM, la frecuencia de detección en el
es estudio de Massayuki y colaboradores que incluyó únicamente pacientes con LM

70
confirmada y compromiso nasal fue del 70.3%, los cuales además correspondían a los
estadios mas avanzados del compromiso nasal según Lessa y colaboradores III, IV y V.
Sin embargo, no hubo diferencias significativas en la clasificación de las lesiones entre los
grupos LRV-positivos y LRV-negativos. El estudio mostró una asociación significativa entre
la presencia del virus y LM con riesgo relativo (RR) de 2.67 [26].
Por otra parte, Cantanhêde y colaboradores encontraron una frecuencia de LRV del 71.1%
en pacientes con LM y 36.7% en pacientes con LC, encontrando una asociación
significativa entre la presencia de lesiones mucosas y del virus con un RR de 2.93 [27]. En
contraste, en los estudios de Ogg y Ramos, ninguna de las muestras correspondientes a
pacientes con LM portaba el virus [25, 144]. La frecuencia del virus en LM encontrada en
nuestro estudio entre biopsias y aislamientos fue del 68.7%, siendo mayor la frecuencia
obtenida en las biopsias (80%) que en aislamientos (50%). Lo anterior podría deberse
probablemente a que durante el cultivo, dada la ubicación citoplasmática del virus, el
número de pases influya aleatoriamente en la pérdida de parasitos portadores del virus.
Respecto a la presencia de LRV en muestras clínicas clasificadas dentro del grupo de falla
terapéutica, los estudios de Adaui y colaboradores reportan una frecuencia del virus del
33%, en donde el 53% correspondia a pacientes en falla terapéutica, ya sea por no
presentar una respuesta primaria efectiva o por tener recaida. El análisis estadístico arrojó
una asociación significativa entre la presencia del virus y la falla terapéutica, con un OR de
3.5 [28]. En el mismo año, Bourreau y colaboradores reportan una frecuencia del virus en
pacientes con diagnostico de LCL del 58%, de los cuales el 27% presentó una infección
persistente y recaida sintomática que exigió la extencion de la terapia antileismania y el
uso de una segunda línea de tratamiento. Hubo asociación significativa entre la presencia
del virus y la recaida sintomática, además de que la presencia del virus fue ligada a un
incremento de los niveles de marcadores inflamatorios como CCL5, CXCL10 e IL6 [30].
En nuestro estudio reportamos una frecuencia del virus del 40% en aislamientos de LC en
falla terapéutica que incluyeron pacientes que presentaron falla al tratamiento con
antimonio al no mostrar cura de la lesion a las seis semanas posteriores al inicio del
tratamiento ó recaida al presentar una nueva lesion dentro de los bordes de una cicatriz
previa dentro de los seis meses después de la cura [147]. El análisis estadístico no mostró
diferencias significativas entre el grupo de LRV-positivo y LRV-negativo ni para el

71
porcentaje de infección ni para el IC50 para Glucantime®. Esto es coherente con lo que
reporta Adaui y colaboradores, quienes no encontraron asociación significativa entre la
presencia del virus y la resistencia al antimonio in vitro, insinuando que el virus no
proporciona por si mismo resistencia del parásito al medicamento de manera intrínseca en
macrófagos infectados, sino que debe haber factores del huésped mamífero implicados
[28].
Si bien se ha mencionado, el desenlace de las Leishmaniasis es producto de una
combinación de diferentes factores tanto del parásito como del huésped [157] y la
presencia del virus es solo uno de ellos, que dados los hallazgos no puede considerarse
causa necesaria de las complicaciones de la LC; incluso en algunos estudios el virus ha
sido encontrado en pacientes con diagnostico de LCL [24, 25, 27, 28, 30, 130, 143, 144].
De otro modo, el seguimiento de estos pacientes que han sido identificados como
portadores del virus en un epidosido de Leishmaniasis cutánea no complicada podría
revelar si tiempo después llegarian a desarrolar complicaciones quizá mediante
mecanismos ya descritos en modelos murinos como el fomento de la persistencia de los
parásitos mediante el incremento de la supervivencia de los macrófagos infectados [23] ó
la producción de IL-17 [145].
Algunos estudios han revelado que mutaciones el TLR predisponen al desarrollo de ciertas
enfermedades, en el caso del TLR3, receptor endosomal implicado en la detección del
genoma viral, se han detectado polimorfimos que se asocian con la infección con Hepatitis
B [158] y con la encefalitis japonesa [159]. Sin embargo, en ambos casos la presencia del
polimorfismo se relacionó con una defectuosa respuesta inmune, hecho que se contradice
con el modelo propuesto por Ives y colaboradores, quienes sugieren que el efecto del
reconocimiento viral por parte del receptor endosomal exacerba la respuesta inmune
inflamatoria en el huésped mamífero generando una destrucción del tejido reflejada en
lesiones necróticas como las conocidas en LM [116].
El modelo mencionado fue construido a partir de experimentos usando L. (V.) guyanensis
M4147. LRV1 ha sido detectado en las especies del subgénero Viannia excepto de L. (V.)
panamensis y LRV2 en L. (L.) major y L. (L.) aethiopica. Un solo reporte ha encontrado el
virus en dos pacientes infectados por L. (L.) amazonensis y L. (V.) lainsoni. En este estudio
todas las muestras clínicas positivas para el virus correspondieron a parásitos de la

72
especie L. (V.) braziliensis. El hecho de que unos parasitos contengan el virus y otros no,
incluso dentro de la misma especie, puede atribuirse por ejemplo a la forma de
reproducción del parásito, pues la división celular podría conllevar a una aleatorización de
la presencia de LRV; incluso, si la reproducción del parásito se diera por via sexual, como
se ha visto en algunos estudios en vectores, la fusión citoplasmática contribuiría a dicho
fenómeno.
En tanto, otro fenómeno que podría estar relacionado con la variación en la frecuencia de
detección del virus es la presencia de un mecanismo eficiente de RNA de interferencia
(RNAi) en el parásito que le permita la eliminación de dichos dsRNA virales exógenos. Por
ello, Nicolás y colaboradores sugieren que la proliferación del virus dentro del parásito esta
asociada a la ausencia de una via de RNAi [160]. En los parásitos del Leishmania,
únicamente en L. (V.) braziliensis M2903 se ha demostrado una fuerte actividad RNAi
[161], a la fecha negativa para el virus [29, 121].
Finalmente, respecto al componente geográfico, la presencia del virus se ha visto
primordialmente en regiones de la cuenca amazónica lo que podría sugerir un patrón
geográfico de distribución [24, 29, 143]. Salinas y colaboradores aportaron el primer mapa
de distribución del virus en Colombia, mostrando que en el set de muestras analizado, el
virus se encontró en aquellas provenientes del sureste del país, específicamente de los
departamentos de Caqueta, Putumayo y Guanía, sugiriendo en su discusión que podría
haber factores ecológicos y epidemilógicos que podrían determinar la distribución de las
especies de Leishmania portadoras del virus y que por tanto el encontrar pacientes
infectados con Leishmania-LRV en regiones distintas a la amazonia podría revelar una
introducción de la enfermedad por diversos factores [24].
El estudio de Ramos y colaboradores concuerda con lo mencionado anteriormente pues
al evaluar muestras clínicas del sureste, norte y noreste de Brazil, los dos casos positivos
provenían del estado de Amazonas de la region norte y no encontraron casos LRV-
positivos en Rio de Janeiro, lugar endémico para Leishmaniasis del sureste del país [25].
Dado que los estudios de detección del virus en muestras clínicas se han hecho
principalmente en Brazil seguido de Perú, Guayana Francesa y Bolivia, podría haber un
sesgo en el muestreo que sugiera que su distribución puede estar limitada
geográficamente; además el hecho de que en la misma área geográfica coexistan casos

73
positivos y negativos para el virus [28, 29, 143, 144], indica que deben haber otras causas
asociadas a la presencia del mismo.
En este estudio, el virus fue detectado en porcentajes iguales en la región Andina y
Orinoquía y en un porcentaje mas bajo en la Amazonia; al georreferenciar los casos
positivos y negativos para el virus no se encontró un patrón definido de distribución ni se
visualiza que las cordilleras pudieran haber funcionado como barrera geográfica. En
departamentos como Cundinamarca, Guanía, Meta, Caquetá y Vichada se encontraron
casos tanto positivos como negativos.

74
9 CONCLUSIONES
La frecuencia del virus encontrada en los aislamientos de LM y LC en falla
terapéutica fue de 42.86% y en biopsias de LM de 88.8%.
Se estableció un algoritmo de detección dependiendo de la muestra disponible para
dicho fin, que en el caso de los aislamientos sugiere la reevaluación de tres estudios
previos que hacen identificación del virus.
Todas las muestras clasificadas en este estudio como portadoras del virus
corresponden a parasitos de las especies L. (V.) braziliensis, L. (V.) panamensis y
Leismania sp.
La distribución del virus no presenta ningún patrón especifico y muestra una
distribución semejante entre la region Andina y Orinoquía.
Al comparar aislamientos de L. (V.) braziliensis asociados a cuadros de LC en falla
terapéutica, coinfectados con LRV y no co-infectados, no se evidenciaron
diferencias en el porcentaje de infección en macrofagos humanos ni en la
susceptibilidad "in vitro" de amastigotes intracelulares. Esto sugiere que la presencia
del virus en el parasito no afecta su capacidad infectiva ni cambia su perfil de
susceptibilidad a antimoniales.

75
10 BIBLIOGRAFÍA
1. Lessa, H.A., et al., A proposed new clinical staging system for patients with mucosal leishmaniasis. Trans R Soc Trop Med Hyg, 2012. 106(6): p. 376-81.
2. Cuervo, P., G.B. Domont, and J.B. De Jesus, Proteomics of trypanosomatids of human medical importance. Journal of Proteomics, 2010. 73(5): p. 845-867.
3. Rodriguez, G., et al., Las Leishmaniasis atlas y texto. Hospita Universitario Centro Dermatológico Federico Lleras Acosta, 2016.
4. Hartley, M.A., et al., Leishmania RNA virus: when the host pays the toll. Front Cell Infect Microbiol, 2012. 2: p. 99.
5. Schlein, Y., Leishmania and sandflies: interactions in the life cycle and transmission. Parasitology Today, 1993. 9(7): p. 255-258.
6. Guilbride, L., P.J. Myler, and K. Stuart, Distribution and sequence divergence of LRV1 viruses among different Leishmania species. Molecular and biochemical parasitology, 1992. 54(1): p. 101-104.
7. Iniesta, F.M. and F.M. Luengo, Manual para el diagnóstico de Leishmaniasis. 1982: Secretariado de Publicaciones, Universidad de Murcia.
8. Oliveira, F., et al., Sand flies, Leishmania, and transcriptome-borne solutions. Parasitology international, 2009. 58(1): p. 1-5.
9. MacBeth, K.J. and J.L. Patterson, Single-site cleavage in the 5'-untranslated region of Leishmaniavirus RNA is mediated by the viral capsid protein. Proceedings of the National Academy of Sciences, 1995. 92(19): p. 8994-8998.
10. Zangger, H., et al., Leishmania aethiopica field isolates bearing an endosymbiontic dsRNA virus induce pro-inflammatory cytokine response. PLoS Negl Trop Dis, 2014. 8(4): p. e2836.
11. Ronet, C., S.M. Beverley, and N. Fasel, Muco-cutaneous leishmaniasis in the New World: the ultimate subversion. Virulence, 2011. 2(6): p. 547-52.
12. Gallego, M., Zoonosis emergente por patógenos parásitos: Las Leishmaniosis. Laboratorio Veterinario Avedila, 2005(33): p. 13-26.
13. SIVIGILA, SIVIGILA 2015 PERIODO IX. 2015. 14. Disch, J., et al., Leishmania (Viannia) subgenus kDNA amplification for the
diagnosis of mucosal leishmaniasis. Diagn Microbiol Infect Dis, 2005. 51(3): p. 185-90.
15. David, C.V. and N. Craft, Cutaneous and mucocutaneous leishmaniasis. Dermatologic therapy, 2009. 22(6): p. 491-502.
16. Ovalle, C.E., et al., Distribución geográfica de especies de Leishmania aisladas de pacientes consultantes al Instituto Nacional de Dermatología Federico Lleras Acosta, ESE, 1995-2005. Biomédica, 2006. 26(1): p. 145-51.
17. Echeverry, M.C., et al., Guía de atención de la leishmaniasis. 2007: p. 175-213. 18. Santrich, C., et al., Mucosal disease caused by Leishmania braziliensis guyanensis.
The American journal of tropical medicine and hygiene, 1990. 42(1): p. 51-55. 19. Saravia, N.G., et al., Mucocutaneous leishmaniasis in Colombia: Leishmania
braziliensis subspecies diversity. The American journal of tropical medicine and hygiene, 1985. 34(4): p. 714-720.
20. OMS, Control de la Leishmaniasis. 2010. 21. Hartley, M.A., et al., The immunological, environmental, and phylogenetic
perpetrators of metastatic leishmaniasis. Trends Parasitol, 2014. 30(8): p. 412-22.

76
22. Machado-Coelho, G.L.L., et al., Risk factors for mucosal manifestation of American cutaneous leishmaniasis. Transactions of the Royal Society of Tropical Medicine and Hygiene, 2005. 99(1): p. 55-61.
23. Eren, R.O., et al., Mammalian Innate Immune Response to a Leishmania-Resident RNA Virus Increases Macrophage Survival to Promote Parasite Persistence. Cell Host & Microbe, 2016. 20(3): p. 318-328.
24. Salinas, G., et al., Leishmania RNA viruses in Leishmania of the Viannia subgenus. The American journal of tropical medicine and hygiene, 1996. 54(4): p. 425-429.
25. Ramos Pereira, L.d.O., et al., Severity of tegumentary leishmaniasis is not exclusively associated with Leishmania RNA virus 1 infection in Brazil. Memórias do Instituto Oswaldo Cruz, 2013. 108(5): p. 665-667.
26. Ito, M.M., et al., Correlation between presence of Leishmania RNA virus 1 and clinical characteristics of nasal mucosal leishmaniosis. Brazilian Journal of Otorhinolaryngology, 2015.
27. Cantanhêde, L.M., et al., Further Evidence of an Association between the Presence of Leishmania RNA Virus 1 and the Mucosal Manifestations in Tegumentary Leishmaniasis Patients. PLOS Negl Trop Dis, 2015. 9(9): p. e0004079.
28. Adaui, V., et al., Association of the Endobiont Double-Stranded RNA Virus LRV1 With Treatment Failure for Human Leishmaniasis Caused by Leishmania braziliensis in Peru and Bolivia. Journal of Infectious Diseases, 2015: p. jiv354.
29. Macedo, D.H., et al., Low frequency of LRV1 in Leishmania braziliensis strains isolated from typical and atypical lesions in the State of Minas Gerais, Brazil. Molecular and Biochemical Parasitology, 2016.
30. Bourreau, E., M. Marine Ginouves, and G. Prévot, Leishmania-RNA virus presence in L. guyanensis parasites increases the risk of first-line treatment failure and symptomatic relapse. J Infect Dis, 2015.
31. Zambrano, P., Protocolo de Vigilancia en Salud Pública en Leishmaniasis. 2014. p. 30.
32. Muskus, C.E. and M.M. Villa, Metaciclogénesis: un proceso fundamental en la biología de Leishmania. Biomédica, 2002. 22(2): p. 167-77.
33. Tropicales, P.d.E.y.C.d.E., Manual de diagnóstico y control de la leishmaniasis en Centroamérica. 2010, Universidad de Antioquia Medellín.
34. Killick, K.R. and J. Rioux, Intravectorial cycle of Leishmania in sandflies. Annales de parasitologie humaine et comparee, 1990. 66: p. 71-74.
35. Kreutzer, R.D., et al., Evidence of sexual reproduction in the protozoan parasite Leishmania (Kinetoplastida: Trypanosomatidae). The American journal of tropical medicine and hygiene, 1994. 51(3): p. 301-307.
36. Rougeron, V., et al., Reproductive strategies and population structure in Leishmania: substantial amount of sex in Leishmania Viannia guyanensis. Molecular ecology, 2011. 20(15): p. 3116-3127.
37. Ash, L.R. and T.C. Orihel, Atlas de parasitologia humana / Atlas of Human Parasitology. 2010: Editorial Medica Panamericana Sa de.
38. Stuart, K. and J.E. Feagin, Mitochondrial DNA of kinetoplastids. International review of cytology, 1992. 141: p. 65-88.
39. de Bruijn, M.H. and D.C. Barker, Diagnosis of New World leishmaniasis: specific detection of species of the Leishmania braziliensis complex by amplification of kinetoplast DNA. Acta tropica, 1992. 52(1): p. 45-58.
40. Lopez, M., et al., Diagnosis of Leishmania using the polymerase chain reaction: A simplified procedure for field work. The American journal of tropical medicine and hygiene, 1993. 49(3): p. 348-356.

77
41. Brenière, S.F., et al., Polymerase chain reaction-based identification of New World Leishmania species complexes by specific kDNA probes. Acta tropica, 1999. 73(3): p. 283-293.
42. Ovalle Bracho, C., et al., Polymerase chain reaction with two molecular targets in mucosal leishmaniasis' diagnosis: a validation study. Memórias do Instituto Oswaldo Cruz, 2007. 102(5): p. 549-554.
43. Ritter, U., F. Frischknecht, and G. van Zandbergen, Are neutrophils important host cells for Leishmania parasites? Trends in parasitology, 2009. 25(11): p. 505-510.
44. Rios Yuil, J.M. and O. Sousa, Inmunología en la infección por Leishmania: Conceptos actuales. Revista Medico Científica, 2010: p. 19-31.
45. Sacks, D.L., Leishmania–sand fly interactions controlling species‐specific vector competence. Cellular microbiology, 2001. 3(4): p. 189-196.
46. Hernández, M.Á.M. and J.Á. Ezquerra, EPIDEMIOLOGÍA MOLECULAR DE LA COINFECCIÓN POR “LEISHMANIA”-VIH. 2002.
47. Bates, P.A., Leishmania sand fly interaction: progress and challenges. Curr Opin Microbiol, 2008. 11(4): p. 340-4.
48. Dostálová, A. and P. Volf, Leishmania development in sand flies: parasite-vector interactions overview. Parasites & vectors, 2012. 5(1): p. 1.
49. Myler, P.J. and N. Fasel, Leishmania: after the genome. 2008: Horizon Scientific Press.
50. Kamhawi, S., Phlebotomine sand flies and Leishmania parasites: friends or foes? Trends Parasitol, 2006. 22(9): p. 439-45.
51. Bañuls, A.-L., M. Hide, and F. Prugnolle, Leishmania and the Leishmaniases: A Parasite Genetic Update and Advances in Taxonomy, Epidemiology and Pathogenicity in Humans. 2007. 64: p. 1-458.
52. Lainson, R., et al., Evolution, classification and geographical distribution. 1987: Academic Press.
53. Montalvo, A.M., et al., PCR-RFLP/HsP70 para identificar y tipificar Leishmania de la región neotropical. Revista Cubana de Medicina Tropical, 2006. 58(3): p. 0-0.
54. Berzunza-Cruz, M., et al., PCR for identification of species causing American cutaneous leishmaniasis. Parasitology research, 2009. 104(3): p. 691-699.
55. Marco, J.D., et al., Polymorphism-specific PCR enhances the diagnostic performance of American tegumentary leishmaniasis and allows the rapid identification of Leishmania species from Argentina. BMC infectious diseases, 2012. 12(1): p. 191.
56. Ovalle‐Bracho, C., Y.R. Díaz‐Toro, and S. Muvdi‐Arenas, Polymerase chain reaction–miniexon: a promising diagnostic method for mucocutaneous leishmaniasis. International journal of dermatology, 2015.
57. WHO, Status of endemicity of cutaneous leishmaniasis worldwine, 2012. 2012. 58. Corredor, A., et al., Distribution and etiology of leishmaniasis in Colombia. The
American journal of tropical medicine and hygiene, 1990. 42(3): p. 206-214. 59. Ramírez, J.D., et al., Taxonomy, diversity, temporal and geographical distribution of
Cutaneous Leishmaniasis in Colombia: A retrospective study. Scientific Reports, 2016. 6.
60. Osorio, L.E., C.M. Castillo, and M.T. Ochoa, Mucosal leishmaniasis due to Leishmania (Viannia) panamensis in Colombia: clinical characteristics. The American journal of tropical medicine and hygiene, 1998. 59(1): p. 49-52.
61. Scott, P. and F.O. Novais, Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nature Reviews Immunology, 2016. 16(9): p. 581-592.

78
62. Bogdan, C. and M. Röllinghoff, The immune response to Leishmania: mechanisms of parasite control and evasion. International journal for parasitology, 1998. 28(1): p. 121-134.
63. Lenis, A.M., La respuesta celular inmune en la leishmaniasis cutánea americana. Biomédica, 1998. 18(4): p. 274-84.
64. Qureshi, A.A., et al., Immunomodulatory properties of maxadilan, the vasodilator peptide from sand fly salivary gland extracts. The American journal of tropical medicine and hygiene, 1996. 54(6): p. 665-671.
65. Mougneau, E., F. Bihl, and N. Glaichenhaus, Cell biology and immunology of Leishmania. Immunological reviews, 2011. 240(1): p. 286-296.
66. Ehrchen, J., et al., Keratinocytes Determine Th1 Immunity during Early Experimental. 2010.
67. Awasthi, A., R.K. Mathur, and B. Saha, Immune response to Leishmania infection. Indian Journal of Medical Research, 2004. 119(6): p. 238.
68. Rojas, W., et al., Inmunología de rojas. Medellín: Corporación para Investigaciones Biológicas, 2012.
69. Vélez Tobón, G.J., et al., Función del sistema NADPH oxidasa en la formación de trampas extracelulares de los neutrófilos (NETs). Revista Cubana de Hematología, Inmunología y Hemoterapia, 2016. 32(1): p. 43-56.
70. Gabelloni, M.L., et al., Trampas extracelulares de neutrófilos: una novedosa estrategia antiinfecciosa empleando moléculas antimicrobianas largamente conocidas. Química Viva, 2013. 12(1): p. 3-13.
71. Guimarães-Costa, A.B., et al., Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proceedings of the National Academy of Sciences, 2009. 106(16): p. 6748-6753.
72. Tuon, F.F., et al., Toll-like receptors and leishmaniasis. Infect Immun, 2008. 76(3): p. 866-72.
73. Faria, M.S., F.C. Reis, and A.P.C. Lima, Toll-like receptors in leishmania infections: guardians or promoters? Journal of parasitology research, 2012. 2012.
74. Becker, I., et al., Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Molecular and biochemical parasitology, 2003. 130(2): p. 65-74.
75. Tuon, F.F., et al., Expression of TLR2 and TLR4 in lesions of patients with tegumentary American leishmaniasis. Revista do Instituto de Medicina Tropical de São Paulo, 2012. 54(3): p. 159-164.
76. Kropf, P., et al., Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infection and immunity, 2004. 72(4): p. 1920-1928.
77. Liese, J., U. Schleicher, and C. Bogdan, TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. European journal of immunology, 2007. 37(12): p. 3424-3434.
78. Sacramento, L., et al., Toll-Like Receptor 9 Signaling in Dendritic Cells Regulates Neutrophil Recruitment to Inflammatory Foci following Leishmania infantum Infection. Infection and immunity, 2015. 83(12): p. 4604-4616.
79. de Morais, C.G.V., et al., The Dialogue of the Host-Parasite Relationship: Leishmania spp. and Trypanosoma cruzi Infection. BioMed Research International, 2014.
80. Bogdan, C., et al., Invasion, control and persistence of Leishmania parasites. Current opinion in immunology, 1996. 8(4): p. 517-525.
81. Ponte-Sucre, A., E. Diaz, and M. Padrón-Nieves, Drug Resistance in Leishmania Parasites: Consequences, Molecular Mechanisms and Possible Treatments. 2012: Springer Science & Business Media.

79
82. EL MINISTRO, D.S., RESOLUCION NUMERO 412 DE 2000. 83. Tuon, F.F., et al., Treatment of New World cutaneous leishmaniasis–a systematic
review with a meta‐analysis. International journal of dermatology, 2008. 47(2): p. 109-124.
84. Llanos-Cuentas, A., et al., Clinical and parasite species risk factors for pentavalent antimonial treatment failure in cutaneous leishmaniasis in Peru. Clinical infectious diseases, 2008. 46(2): p. 223-231.
85. Chulay, J., L. Fleckenstein, and D. Smith, Pharmacokinetics of antimony during treatment of visceral leishmaniasis with sodium stibogluconate or meglumine antimoniate. Transactions of the Royal Society of Tropical Medicine and Hygiene, 1988. 82(1): p. 69-72.
86. Frézard, F., et al., Chemistry of antimony-based drugs in biological systems and studies of their mechanism of action. Reviews in Inorganic Chemistry, 2013. 33(1): p. 1-12.
87. Haldar, A.K., P. Sen, and S. Roy, Use of antimony in the treatment of leishmaniasis: current status and future directions. Molecular biology international, 2011. 2011.
88. Croft, S.L., S. Sundar, and A.H. Fairlamb, Drug resistance in leishmaniasis. Clinical microbiology reviews, 2006. 19(1): p. 111-126.
89. Sereno, D., et al., Antimonial-Mediated DNA Fragmentation inLeishmania infantum Amastigotes. Antimicrobial agents and chemotherapy, 2001. 45(7): p. 2064-2069.
90. Henao, H.H., et al., Eficacia y toxicidad de los antimoniales pentavalentes (Glucantime® y Pentostam®) en un modelo animal de leishmaniasis cutánea americana: aplicación de la luminometría. Biomédica, 2004. 24(4): p. 393-402.
91. Arevalo, J., et al., Influence of Leishmania (Viannia) species on the response to antimonial treatment in patients with American tegumentary leishmaniasis. Journal of Infectious Diseases, 2007. 195(12): p. 1846-1851.
92. PAHO, Guia-Leishmaniasis-Americas. 2013. 93. de Saldanha, R.R., et al., Meglumine antimonate treatment enhances phagocytosis
and TNF-α production by monocytes in human cutaneous leishmaniasis. Transactions of the Royal Society of Tropical Medicine and Hygiene, 2012. 106(10): p. 596-603.
94. da Costa, D.C.S., et al., Oral Manifestations in the American Tegumentary Leishmaniasis. PloS one, 2014. 9(11): p. e109790.
95. González, U., et al., Interventions for American cutaneous and mucocutaneous leishmaniasis (Review). Cochrane Database Syst Rev, 2009. 2: p. CD004834.
96. Amato, V.S., H.F. de Andrade Jr, and M.I.S. Duarte, Mucosal leishmaniasis: in situ characterization of the host inflammatory response, before and after treatment. Acta tropica, 2003. 85(1): p. 39-49.
97. Tuon, F.F., et al., Local immunological factors associated with recurrence of mucosal leishmaniasis. Clin Immunol, 2008. 128(3): p. 442-6.
98. Strazzulla, A., et al., Mucosal leishmaniasis: an underestimated presentation of a neglected disease. Biomed Res Int, 2013. 2013: p. 805108.
99. Ives, A., et al., MyD88 and TLR9 dependent immune responses mediate resistance to Leishmania guyanensis infections, irrespective of Leishmania RNA virus burden. PLoS One, 2014. 9(5): p. e96766.
100. Carvalho, A.M., et al., Age Modifies the Immunologic Response and Clinical Presentation of American Tegumentary Leishmaniasis. The American journal of tropical medicine and hygiene, 2015. 92(6): p. 1173-1177.
101. Jirmanus, L., et al., Epidemiological and clinical changes in American tegumentary leishmaniasis in an area of Leishmania (Viannia) braziliensis transmission over a

80
20-year period. The American journal of tropical medicine and hygiene, 2012. 86(3): p. 426-433.
102. Pérez, H., I. Malavé, and B. Arredondo, The effects of protein malnutrition on the course of Leishmania mexicana infection in C57Bl/6 mice: nutrition and susceptibility to leishmaniasis. Clinical and experimental immunology, 1979. 38(3): p. 453.
103. Oliveira, A.G., et al., Influence of the nutritional status in the clinical and therapeutical evolution in adults and elderly with American Tegumentary Leishmaniasis. Acta Trop, 2013. 128(1): p. 36-40.
104. Fakher, F.H.A., et al., TLR9-dependent activation of dendritic cells by DNA from Leishmania major favors Th1 cell development and the resolution of lesions. The Journal of Immunology, 2009. 182(3): p. 1386-1396.
105. Weinkopff, T., et al., Role of Toll-like receptor 9 signaling in experimental Leishmania braziliensis infection. Infection and immunity, 2013. 81(5): p. 1575-1584.
106. Roiko, M.S., et al., An unusual presentation of leishmaniasis in a human immunodeficiency virus-positive individual. JMM Case Reports, 2016. 3(1).
107. Lauletta, J.A., Leishmaniasis–HIV coinfection: current challenges. 2016. 108. Gois, L., et al., Immune response to Leishmania antigens in an AIDS patient with
mucocutaneous leishmaniasis as a manifestation of immune reconstitution inflammatory syndrome (IRIS): a case report. BMC infectious diseases, 2015. 15(1): p. 1.
109. Goto, H. and J.A. Lauletta Lindoso, Cutaneous and mucocutaneous leishmaniasis. Infect Dis Clin North Am, 2012. 26(2): p. 293-307.
110. de Oliveira Guerra, J.A., et al., Mucosal leishmaniasis caused by Leishmania (Viannia) braziliensis and Leishmania (Viannia) guyanensis in the Brazilian Amazon. PLoS Negl Trop Dis, 2011. 5(3): p. e980.
111. McCall, L.I. and J.H. McKerrow, Determinants of disease phenotype in trypanosomatid parasites. Trends Parasitol, 2014. 30(7): p. 342-9.
112. Oliveira, F.S., et al., American tegumentary leishmaniasis caused by Leishmania (Viannia) braziliensis: assessment of parasite genetic variability at intra-and inter-patient levels. Parasites & vectors, 2013. 6(1): p. 1.
113. Rogers, M.B., et al., Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome research, 2011. 21(12): p. 2129-2142.
114. Peacock, C.S., et al., Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nature genetics, 2007. 39(7): p. 839-847.
115. Gómez-Arreaza, A., et al., Viruses of parasites as actors in the parasite-host relationship: A “ménage à trois”. Acta Tropica, 2017. 166: p. 126-132.
116. Ives, A., et al., Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science, 2011. 331(6018): p. 775-8.
117. Amato, V.S., et al., Mucosal leishmaniasis: Current scenario and prospects for treatment. Acta Tropica, 2008. 105(1): p. 1-9.
118. Molyneux, D., Virus-like particles in Leishmania parasites. 1974. 119. Diamond, L.S. and C.F.T. Mattern, Protozoal Viruses, in Advances in Virus
Research, F.B.B.K.M. Max A. Lauffer and M.S. Kenneth, Editors. 1976, Academic Press. p. 87-112.
120. Tarr, P.I., et al., LR1: a candidate RNA virus of Leishmania. Proceedings of the National Academy of Sciences, 1988. 85(24): p. 9572-9575.
121. Widmer, G., et al., Characterization of a RNA virus from the parasite Leishmania. Proceedings of the National Academy of Sciences, 1989. 86(15): p. 5979-5982.

81
122. Widmer, G., M.C. Keenan, and J.L. Patterson, RNA polymerase activity is associated with viral particles isolated from Leishmania braziliensis subsp. guyanensis. Journal of virology, 1990. 64(8): p. 3712-3715.
123. Weeks, R., et al., LRV1 viral particles in Leishmania guyanensis contain double-stranded or single-stranded RNA. Journal of virology, 1992. 66(3): p. 1389-1393.
124. Stuart, K.D., et al., Molecular organization of Leishmania RNA virus 1. Proceedings of the National Academy of Sciences, 1992. 89(18): p. 8596-8600.
125. Scheffter, S., G. Widmer, and J.L. Patterson, Complete sequence of Leishmania RNA virus 1-4 and identification of conserved sequences. Virology, 1994. 199(2): p. 479-483.
126. Lee, S.E., et al., Identification of a ribosomal frameshift in Leishmania RNA virus 1–4. Journal of biochemistry, 1996. 120(1): p. 22-25.
127. Scheffter, S.M., et al., The complete sequence of Leishmania RNA virus LRV2-1, a virus of an Old World parasite strain. Virology, 1995. 212(1): p. 84-90.
128. Widmer, G. and S. Dooley, Phylogenetic analysis of Leishmania RNA virus and Leishmania suggests ancient virus-parasite association. Nucleic acids research, 1995. 23(12): p. 2300-2304.
129. Gupta, V., J.L. Patterson, and Y.-T. Ro, Leishmaniavirus, in The Springer Index of Viruses. 2011, Springer. p. 1931-1936.
130. Saiz, M., et al., Short report: detection of Leishmaniavirus in human biopsy samples of leishmaniasis from Peru. The American journal of tropical medicine and hygiene, 1998. 58(2): p. 192-194.
131. Gupta, V. and A. Deep, An insight into the Leishmania RNA virus. Indian journal of medical microbiology, 2007. 25(1): p. 7.
132. MacBeth, K. and J. Patterson, The short transcript of Leishmania RNA virus is generated by RNA cleavage. Journal of virology, 1995. 69(6): p. 3458-3464.
133. Chung, I., et al., Generation of the short RNA transcript in Leishmaniavirus correlates with the growth of its parasite host, Leishmania. Molecules and cells, 1998. 8(1): p. 54-61.
134. Macbeth, K.J. and J.L. Patterson, Overview of the Leishmaniavirusendoribonuclease and functions of other endoribonucleases affecting viral gene expression. Journal of Experimental Zoology, 1998. 282(1‐2): p. 254-260.
135. Weeks, R.S., et al., Transcribing and replicating particles in a double-stranded RNA virus from Leishmania. Molecular and Biochemical Parasitology, 1992. 52(2): p. 207-213.
136. Maga, J.A., G. Widmer, and J.H. LeBowitz, Leishmania RNA virus 1-mediated cap-independent translation. Molecular and cellular biology, 1995. 15(9): p. 4884-4889.
137. Ro, Y.-T., S.M. Scheffter, and J.L. Patterson, Specific in vitro cleavage of a Leishmania virus capsid-RNA-dependent RNA polymerase polyprotein by a host cysteine-like protease. Journal of virology, 1997. 71(12): p. 8983-8990.
138. Sen, G.C. and S.N. Sarkar, Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev, 2005. 16(1): p. 1-14.
139. Matsumoto, M. and T. Seya, TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv Drug Deliv Rev, 2008. 60(7): p. 805-12.
140. Hartley, M.-A., C. Ronet, and N. Fasel, Backseat drivers: the hidden influence of microbial viruses on disease. Current opinion in microbiology, 2012. 15(4): p. 538-545.
141. Alves-Ferreira, E.V.C., et al., Differential Gene Expression and Infection Profiles of Cutaneous and Mucosal <italic>Leishmania braziliensis</italic> Isolates from the Same Patient. PLoS Negl Trop Dis, 2015. 9(9): p. e0004018.

82
142. Zangger, H., et al., Detection of Leishmania RNA virus in Leishmania parasites. PLoS Negl Trop Dis, 2013. 7(1): p. e2006.
143. Ginouvès, M., et al., Prevalence and Distribution of Leishmania RNA Virus 1 in Leishmania Parasites from French Guiana. The American Journal of Tropical Medicine and Hygiene, 2015: p. 15-0419.
144. Ogg, M.M., et al., Short report: quantification of leishmaniavirus RNA in clinical samples and its possible role in pathogenesis. The American journal of tropical medicine and hygiene, 2003. 69(3): p. 309-313.
145. Hartley, M.-A., et al., Leishmaniavirus-dependent metastatic leishmaniasis is prevented by blocking IL-17A. PLoS Pathog, 2016. 12(9): p. e1005852.
146. Hajjaran, H., et al., Detection and molecular identification of leishmania RNA virus (LRV) in Iranian Leishmania species. Archives of virology, 2016. 161(12): p. 3385-3390.
147. Perez-Franco, J.E., et al., Clinical and Parasitological Features of Patients with American Cutaneous Leishmaniasis that Did Not Respond to Treatment with Meglumine Antimoniate. PLoS Negl Trop Dis, 2016. 10(5): p. e0004739.
148. van den Bogaart, E., et al., Duplex quantitative reverse-transcriptase PCR for simultaneous assessment of drug activity against Leishmania intracellular amastigotes and their host cells. International Journal for Parasitology: Drugs and Drug Resistance, 2014. 4(1): p. 14-19.
149. Cruz Barrera, M., et al., Improving Leishmania Species Identification in Different Types of Samples from Cutaneous Lesions. Journal of Clinical Microbiology, 2015. 53.
150. Hafez, H., et al., Comparison of the specificity and sensitivity of PCR, nested PCR, and real-time PCR for the diagnosis of histomoniasis. Avian diseases, 2005. 49(3): p. 366-370.
151. Perrott, P., et al., A nested real‐time PCR assay has an increased sensitivity suitable for detection of viruses in aerosol studies. Journal of applied microbiology, 2009. 106(5): p. 1438-1447.
152. Drago, L., et al., Comparison of nested PCR and real time PCR of Herpesvirus infections of central nervous system in HIV patients. BMC infectious diseases, 2004. 4(1): p. 55.
153. Alvarez-Martínez, M.J., et al., Sensitivity and specificity of nested and real-time PCR for the detection of Pneumocystis jiroveci in clinical specimens. Diagnostic microbiology and infectious disease, 2006. 56(2): p. 153-160.
154. Calderaro, A., et al., Comparison between two real-time PCR assays and a nested-PCR for the detection of Toxoplasma gondii. Acta Biomedica-Ateneo Parmense, 2006. 77(2): p. 75.
155. Kawada, J.i., et al., Comparison of real‐time and nested PCR assays for detection of Herpes simplex virus DNA. Microbiology and immunology, 2004. 48(5): p. 411-415.
156. Khlif, M., et al., Evaluation of nested and real‐time PCR assays in the diagnosis of candidaemia. Clinical Microbiology and Infection, 2009. 15(7): p. 656-661.
157. Chang, K.-P. and B.S. McGwire, Molecular determinants and regulation of Leishmania virulence. Kinetoplastid biology and disease, 2002. 1(1): p. 1.
158. Al‐Qahtani, A., et al., Toll‐like receptor 3 polymorphism and its association with hepatitis B virus infection in Saudi Arabian patients. Journal of medical virology, 2012. 84(9): p. 1353-1359.
159. Biyani, S., et al., Toll-like receptor-3 gene polymorphism in patients with Japanese encephalitis. Journal of neuroimmunology, 2015. 286: p. 71-76.

83
160. Nicolas, F.E., S. Torres-Martínez, and R.M. Ruiz-Vazquez, Loss and retention of RNA interference in fungi and parasites. PLoS Pathog, 2013. 9(1): p. e1003089.
161. Lye, L.-F., et al., Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog, 2010. 6(10): p. e1001161.
162. Scientific, T. Real-Time PCR Handbook. 2014; Available from: https://www.thermofisher.com/co/en/home/life-science/pcr/real-time-pcr/qpcr-education/real-time-pcr-handbook.html.

84
11 ANEXOS
11.1 GLOSARIO
11.1.1 Eficiencia
Es la medida que indica en una qPCR cuanto se obtiene en cada ciclo térmico a partir de
una hebra molde del DNA o cDNA, lo ideal es que en cada ciclo ésta se duplique. La
eficiencia puede obtenerse a partir de una curva estándar de diluciones en base 10 del
templete en estudio. Para calcularse se grafican los Ct obtenidos y se analizan en una
función lineal que genera una ecuación Y=mX+B, en donde m (pendiente) se relaciona
directamente con la eficiencia, si la m de dicha curva estándar es -3.32 la eficiencia es de
1 (100%). Para calcular la eficiencia entonces se utiliza la siguiente ecuación: 10(-1/m) -1.
Son aceptables eficiencias entre 0.9-1.1 [162].
11.1.2 Coeficiente de determinación (R2)
Estadísticamente es el valor que indica la capacidad del modelo de predecir los valores de
X a partir del valor de Y. En cuanto más se acerque a 1 mayor capacidad tendrá y en
cuanto más se aleje menor capacidad tendrá. Valores menores a 0.95 evidencian
problemas de pipeteo o de la dilución de los estándares. En qPCR el valor de Y
corresponde al Ct [162].
11.1.3 Baseline
Es el promedio del background calculado de acuerdo al nivel de ruido en los ciclos
tempranos de la qPCR cuando no hay un incremento de la fluorescencia a expensas de
los productos de la amplificación [162].
11.1.4 Thershold cycle (Ct)
Durante la qPCR es el ciclo en el que la fluorescencia logra cruzar el threshold. Es
considerado una medida relativa de la concentración del blanco en la muestra sujeta a la
reacción de la PCR en una relación inversamente proporcional; a menor Ct mayor cantidad
del blanco en la muestra [162].

85
11.1.5 Thershold
Es el nivel de fluorescencia que está por encima de la línea de base. Dicho límite diferencia
una señal de amplificación del ruido de fondo de la reacción [162].
11.2 PREPARACIÓN MEDIO DE CULTIVO SENEKJIE
Para preparar 100 mL:
Bacto Beef extract …………………………………………. .. 0.3 g
Agar-agar ……………………………………………... 2 g
Bacto peptona ……………………………………………... 2 g
Cloruro de sodio (NaCl) …………………………………………… 0.5 g
Agua destilada desionizada ………………………………………….100 mL
Gentamicina 10mg/mL ………………………………………….....1 mL
Sangre desfibrinada de conejo …………………………………………...15 mL
MATERIALES
Placa agitadora e imanes
Probeta
Vaso precipitado grande capacidad 500 mL
Vaso precipitado grande capacidad 200 mL
Frascos estériles de 500 mL
Tubos de vidrio 13X150 mm tapa rosca
PROCEDIMIENTO
1. Disolver el Bacto Beef extract, Agar –agar, Bacto peptona y NaCl en 100 mL de
agua destilada desionizada. Calentar sin permitir la ebullición.
2. Ajustar el pH a 7.2 con NaOH 1N y/o HCl 1N. Nuevamente poner a calentar sin
permitir la ebullición.
3. Esterilizar en autoclave a 121°C y 15 libras de presión por 40 min. Luego dejar
enfriar el medio a 55-60°C.

86
4. En cabina de flujo laminar, adicionar el medio al vaso de precipitado de menor
capacidad y agregar la Gentamicina y la sangre de conejo desfibrinada. Mezclar
bien moviendo el envase en forma circular.
5. Servir aproximadamente 3 mL del medio por la boquilla del vaso de precipitado a
los tubos de vidrio y mantener inclinados para formar pico de flauta.
11.3 PREPARACIÓN DE MEDIO SCHNEIDER
Para preparar 1 L:
Schneider ………………………………………... 24.89 g
Bicarbonato de sodio (NaHCO3) …………………………………………… 0.4 g
Cloruro de calcio (CaCl2) ……………………………………………... 2 g
Agua destilada desionizada ………………………………………..1000 mL
MATERIALES
Placa agitadora e imanes
Probeta
Vaso precipitado grande capacidad 2 L
Frascos estériles de 500 mL
PROCEDIMIENTO
1. Medir el 80% del volumen final requerido de agua. La temperatura del agua debe
ser de 15-20 ° C.
2. Mientras se agita suavemente el agua, añadir el medio en polvo [24,89 g/L]. Se
agita hasta que se disuelva, sin embargo el material no irá en solución por
completo. No calentar el agua.
3. Añadir 0,4 g de NaHCO3 o 5,3 ml de solución de NaHCO3 [7,5% w/v] para cada litro
de volumen final de medio que está preparando. Agite hasta que se disuelva.
4. Mientras se agita, se ajusta el pH a por lo menos 9,2 ± 0,2 con NaOH 1N. Se agita
durante un mínimo de 10 minutos. La solución puede llegar a ser turbia.
5. Mientras se agita, ajustar el pH a 6,7 ± 0,2 con HCl 1N, la solución se volverá clara.

87
6. Preparar una solución de CaCl2 mediante la disolución de 0,6 g de cloruro de calcio
anhidro en 50 mL de agua por cada litro de volumen final de medio que está siendo
preparado.
7. Añadir lentamente la solución gota a gota de CaCl2 al medio con mezcla rápida para
evitar la formación de precipitado.
8. Mientras se agita, se ajusta el pH del medio a 0,1-0,3 unidades de pH por debajo
del pH deseado, ya que puede aumentar durante la filtración [pH 7,2]. Se
recomienda el uso de HCl 1N o NaOH 1N.
9. Añadir agua adicional para llevar la solución hasta el volumen final. Esterilizar
inmediatamente por filtración utilizando una membrana con una porosidad de 0,22
micras o menos y asépticamente dispensar medio en el recipiente estéril.
11.4 PREPARACIÓN DEL DE AGAROSA
Para preparar un gel al 2%:
Agarosa ………………………………………....... 0.8 g
TAE 1X ………………………………………….. 40 mL
Bromuro de etidio (BrEt) ……………………………………………...2 µl
MATERIALES
Probeta
Erlenmeyer pequeño
Molde para gel y peines
PROCEDIMIENTO
1. Preparar el molde para el gel y usar el peine que tenga los pozos requeridos.
2. Pesar 0.8 g de agarosa y diluir en los 40 mL de TAE 1X en el erlenmeyer.
3. Llevar al microondas y fundir la agarosa. Estará listo cuando a contraluz no se
observen partículas del polímero. No dejar ebullir.

88
4. Dejar enfriar y agregar 2 µl de BrEt con el fin de que este queda a una concentración
final de 0.5 µg/mL. Agitar suavemente y servir en el molde hasta su solidificación.
11.5 PREPARACIÓN TAE 50X
Para preparar 1 L:
Tris base …………………………………………. 242 g
Ácido acético glacial ……………………………………… 57.1 mL
Ácido etilendiaminotetraacético (EDTA) …………………………………………...37. g
Agua destilada desionizada ………………………………………1000 mL
MATERIALES
Placa agitadora e imanes
Probeta
Vaso precipitado grande capacidad 2 L
Frascos estériles de 1 L
PROCEDIMIENTO
1. Medir el 80% del volumen total requerido de agua destilada desionizada (800 ml)
en un vaso de precipitado con capacidad de 2 L.
2. Agregar lentamente los reactivos mientras se agita suavemente hasta disolver.
Mientras ajustar el pH a 8.5±0.2 con NaOH 1N y/o HCl 1N.
3. Agregar e volumen restante de agua para completar el volumen final.
4. Almacenar a temperatura ambiente y al momento de utilizar diluya a 1X para
trabajar en corridos electroforéticos.