
QUÍMICA FÍSICA - unizar.esunizar.es/jmembid/qf/Guiones%20Pr%E1cticas.pdf · Consta de un tubo...
40
1 QUÍMICA FÍSICA 2º CURSO LICENCIADO EN QUÍMICA Prácticas de Laboratorio
-
Upload
truongthien -
Category
Documents
-
view
239 -
download
0
Transcript of QUÍMICA FÍSICA - unizar.esunizar.es/jmembid/qf/Guiones%20Pr%E1cticas.pdf · Consta de un tubo...

1
QUÍMICA FÍSICA
2º CURSO
LICENCIADO EN QUÍMICA
Prácticas de Laboratorio

2
Índice
Normas a seguir en el laboratorio 3
Prácticas de Electroquímica 5
Práctica EQ1: Determinación de números de transporte por el método de la
interfase móvil 6
Práctica EQ2: Medida de la fuerza electromotriz de algunas pilas sencillas y de
concentración utilizando electrodos reversibles a los iones hidrógeno
y a los iones cloruro 8
Práctica EQ3: Valoración potenciométrica de ácidos 9
Prácticas de Cinética de Reacción 13
Características de los reactivos usados 14
Práctica CR1: Estudio cinético de la iodación de la ciclohexanona en medio ácido
por medidas espectrofotométricas 15
Apéndice 1 20
Práctica CR2: Estudio de la reacción de inversión de la sacarosa catalizada por
Ácidos mediante medidas polarimétricas 22
Apéndice 2 30
Práctica CR3: Estudio cinético de la reacción de saponificación del acetato de
Etilo por medidas conductimétricas 33
Apéndice 3 40

3
NORMAS A SEGUIR EN EL LABORATORIO
Preparación.
Las prácticas se prepararán con antelación, leyendo cuidadosamente el guión antes de acudir al laboratorio.
Para la preparación y realización de las prácticas, en este cuadernillo se incluye una detallada explicación
de todas ellas. Traed hechos los cálculos necesarios para preparar las disoluciones, teniendo en cuenta que
en algunas de las balanzas la pesada se lleva a cabo con cuatro cifras decimales de gramo.
Puntualidad.
La impuntualidad afecta al trabajo de compañeros y profesores por lo que se será muy riguroso con ella.
Limpieza.
Para la obtención de buenos resultados, es fundamental evitar la presencia de sustancias extrañas.
Además, el material e instalaciones deben ser utilizados después por otros compañeros. Así pues, lo
primero y lo último que se hará en el laboratorio es lavar el material. La limpieza incluye no tirar agua por el
suelo. Hay que tener gran cuidado de no contaminar unos reactivos con otros o unas disoluciones con otras,
por lo que tenéis que trabajar de la forma siguiente:
- No meter pipetas en las botellas; Pasad a un vaso limpio y homogeneizado una cantidad adecuada de
reactivo y pipetead del vaso, con una pipeta previamente limpia y homogeneizada. A la vez que se
homogeneiza el vaso, se homogeneiza la pipeta, con el mismo líquido; no homogeneicéis la pipeta con la
pera puesta. Homogeneizad con muy poco líquido, haciéndolo pasar por las paredes con cuidado, 3 ó 4
veces.
- Mucho cuidado de que no entre líquido en las peras de succión: succionar con cuidado y no dejéis las
pipetas con la pera de succión puesta ni inclinadas si la boca de la pipeta queda más baja que la punta,
para evitar que el líquido resbale hacia la pera.
- Los sólidos pesados en exceso no se devuelven al bote; lo que sobra se envuelve en un papel y se tira a
la basura; nunca se tira al suelo ni se deja por la mesa.
- No quitar los tapones de los frascos de agua destilada salvo para rellenarlos y, en cualquier caso, no
dejarlos apoyados en las mesas ni ponerlos en contacto con nada que pudiera contaminarlos; volved a
colocarlos en el frasco en cuanto sea posible.
Utilización de las balanzas.
Las balanzas se deben manejar cuidadosamente. Si la balanza es de pesas, cuando se termine de pesar
las pesas quedarán quitadas (balanza a cero).
- Las puertas de la balanza estarán cerradas durante la pesada y, al finalizar, se dejarán cerradas
igualmente.
- Limpiad inmediatamente lo que se caiga sobre la balanza o la mesa. Tanto la balanza como la mesa y el
suelo quedarán perfectamente limpios al finalizar la pesada.
Seguridad.
- No fumar ni comer en el laboratorio.
- Siempre debe pipetearse utilizando las peras de succión, incluso el agua. No pipetear nunca con la boca.
- Todos los reactivos líquidos (excepto el agua destilada) deben manejarse en una campana extractora
puesta en marcha.
- Ante cualquier problema, haced uso de los elementos de seguridad del laboratorio: lavaojos, ducha,
botiquín. etc., y avisad inmediatamente al profesor.
- El HCl es muy peligroso por lo que su manipulación se realizará tomando las máximas precauciones.

4
Preparación de las disoluciones.
En todas las prácticas, pero especialmente en la número 3 de cinética de reacción, se ajustará lo máximo
posible la pesada o el volumen pipeteado al valor calculado, de modo que la concentración corresponda lo
más exactamente posible con la indicada en el guión. En cualquier caso, se debe calcular la concentración
real de la disolución con la cantidad realmente pesada o pipeteada.
- Disoluciones de un sólido:
l.-Se pesa la cantidad adecuada en un vidrio de reloj, pesasustancias o vaso de precipitados, tomando el
sólido del bote con una espátula limpia y seca. Pesad de menos a más, procurando no pasarse y que no
sobre producto.
2.-Si se ha pesado en un vidrio de reloj, se trasvasa el sólido a un vaso de precipitados utilizando una varilla
y se lava bien con agua destilada el vidrio para arrastrar todos los restos de sólido. Todo este líquido de
lavado debe recogerse, naturalmente, en el vaso de precipitados. Si se ha pesado en un vaso de
precipitados o en un pesasustancias con forma de vaso, este paso es innecesario.
3.-Con el agua que se ha añadido en el apartado anterior, o un poco más si fuera necesario, pero en todo
caso una pequeña cantidad, se disuelve el sólido completamente, utilizando la varilla.
4.-Con la ayuda de un embudo se trasvasa la disolución a un matraz aforado de la capacidad adecuada.
5.- Con pequeñas cantidades de agua, se lavan varias veces el vaso que contenia la disolución y la varilla.
Estas aguas de lavado se añaden al matraz aforado, hasta estar seguros de que todo el reactivo ha pasado
al matraz. Después de añadir cada porción de agua de lavado al matraz aforado se agita éste, para ir
homogeneizando la disolución.
6.-Por la pared interior del embudo, se va añadiendo más agua destilada, en veces sucesivas, agitando a
continuación.
7.-Cuando queda poco volumen para llegar al aforo, enrasad el matraz con un cuentagotas.
8.-Se agita bien el matraz para que la disolución quede homogénea.
- Disoluciones de un líquido:
Para ahorrar reactivo, lavad y homogeneizad un vaso y una pipeta para cada reactivo y usad los mismos
todos los grupos de la mesa.
l.-Se pipetea la cantidad adecuada de líquido. Recordad que la pipeta debe estar en posición vertical y el
enrasado debe realizarse con los ojos al nivel del aforo, que debe coincidir con la parte inferior del menisco.
Para el enrasado de buretas y matraces aforados se aplica el mismo procedimiento.
2.-Se vierte el líquido de la pipeta al matraz aforado (no es necesario usar embudo).
3.-Se realizan los pasos 6, 7 y 8 del apartado anterior.
Otras precauciones.
* El iodo sublima fácilmente, y sus vapores son corrosivos para los mecanismos de las balanzas. Pesadlo
en un pesasustancias cerrado. Además, mancha mucho por lo que todo lo que lleve yodo se manejará con
cuidado y se mantendrá en una bandeja. El ioduro de potasio no mancha, pero sí lo hace cuando se oxida a
iodo por efecto del oxígeno del aire. Manejad con cuidado las disoluciones de ioduro potásico.
* Volved a traer al puesto de la práctica todo el material que os llevéis a las balanzas o campanas.
* No dejar abiertos los frascos ni las botellas.
* No coger nada de las otras mesas. Si necesitáis algo, pedidlo.
* Los electrodos son de vidrio. Sed muy cuidadosos con ellos.
* En estas prácticas nunca se utiliza acetona para lavar.

5
Prácticas de Electroquímica

Química Física Prácticas de Electroquímica
6
Práctica EQ1: DETERMINACIÓN DE NÚMEROS DE TRANSPORTE POR EL MÉTODO DE LA
INTERFASE MÓVIL
Introducción teórica
Conociendo el número de moles de un ion transportados durante la electrolisis en función de
la cantidad total de electricidad que ha pasado, es posible calcular el número de transporte de dicho
ion. En el método de la interfase móvil, el transporte de un ion bajo la influencia de un campo
eléctrico se mide observando el desplazamiento de la interfase formada entre la disolución que
contiene el ion y una disolución indicadora.
Supongamos que disponemos las disoluciones en un tubo de sección A, y que en un tiempo t
la interfase se desplaza una longitud l a lo largo del tubo. En ese tiempo la carga que pasa por el
sistema es:
iii cvFzAt tI Q (1)
I: intensidad de la corriente; t: tiempo; zi: carga del ion i; F: constante de Faraday; vi: velocidad del
ion i; ci: concentración del ion i.
La carga transportada por un determinado ion j será:
j j j j jQ t A z F v c Q t (2)
donde tj es el número del transporte del ion j.
Como la velocidad de los iones alcanza rápidamente un valor constante, podemos escribir:
vi = l/t (para todo ion i), y la fracción de carga que transportan los iones de tipo j, es decir, su
número de transporte, tj, queda:
tI
cFzlAt
jjj
(3)
Preparad la parte teórica de esta práctica con el apartado “Movilidades eléctricas de los iones”,
del libro Fisicoquímica de I.N. Levine.
Parte experimental
La célula se muestra en la figura. Consta de un tubo graduado con una disolución de HCl 0,1
M que establece la comunicación entre el ánodo de Cu y el cátodo de Pt. En determinaciones más
precisas el tubo graduado puede situarse en el interior de otro tubo de mayor diámetro, por el que
circula agua para eliminar el calor generado por la electrolisis y reducir así las variaciones térmicas.
A la disolución del HCl 0,1 M (atención a la concentración exacta que figura en la etiqueta)
se ha añadido una pequeña cantidad de anaranjado de metilo para que su color permita seguir el
movimiento de la interfase. El tubo, previamente lavado con agua destilada, se homogeneiza con
esta disolución y se conecta al resto del dispositivo.

Química Física Prácticas de Electroquímica
7
A continuación, succionando por la parte
superior, se llena el tubo con la disolución de HCl. Es
importante cerciorarse de que ni en la superficie
anódica ni en las paredes del capilar queden adheridas
burbujas de aire. Finalmente se coloca el electrodo de
Pt.
El potencial aplicado a la célula ha de ser tal
que la intensidad de la corriente sea de 5 mA. El
ánodo se disolverá y, a consecuencia de la migración
de los iones, la superficie límite entre las disoluciones
de cloruro de cobre y HCl ascenderá a lo largo del
tubo. La posición de la interfase puede observarse
fácilmente gracias al distinto color de las dos
disoluciones. A medida que la experiencia avanza,
variará la resistencia del electrolito y, para mantener
constante la intensidad de la corriente en los 5 mA
iniciales, será preciso ir variando la resistencia interna
de la fuente.
Anótese el tiempo que tarda la interfase en
pasar por cada una de las divisiones de 0,1 ml del tubo
(un mínimo de 8 medidas buenas) y utilizando el valor medio y la ecuación (3), calcular los
números de transporte de los iones hidrógeno y cloruro en el HCl 0,1 M a la temperatura de la
experiencia.

Química Física Prácticas de Electroquímica
8
Práctica EQ2: MEDIDA DE LA FUERZA ELECTROMOTRIZ DE ALGUNAS PILAS SENCILLAS Y DE
CONCENTRACIÓN UTILIZANDO ELECTRODOS REVERSIBLES A LOS IONES HIDRÓGENO Y A LOS
IONES CLORURO.
Se procederá a medir la f.e.m. de las siguientes pilas y cumplimentar, con los resultados
obtenidos, la hoja de resultados. Antes de ir al laboratorio se determinará el cátodo y el ánodo y se
escribirán las expresiones de la f.e.m. para todas las pilas.
1) Pt | Q, QH2, HCl (0,01 m) HCl (0,01 m) | AgCl(s) | Ag 2) Pt | Q, QH2, HCl (0,1 m) HCl (0,1 m) | AgCl(s) | Ag 3) Pt | Q, QH2, HCl (0,01 m) HCl (0,01 m) | AgCl(s) | Ag _______________________________| | Ag | AgCl(s) | HCl (0,1 m) HCl (0,1 m), Q, QH2 | Pt 4) Pt | Q, QH2, HCl (0,01 m) HCl (0,10 m), QH2, Q | Pt 5) Ag | AgCl(s) | HCl (0,1 m) HCl (0,010 m) | AgCl(s) | Ag 6) Ag | AgCl(s) | HCl (0,1 m) HCl (0,01 m) | AgCl(s) | Ag Todas las disoluciones de quinhidrona deben estar saturadas (es poco soluble).
Las pilas 1 y 2 son pilas galvánicas típicas para las que se pueden determinar los cambios de
energía libre (debido a que el tabique poroso separa dos disoluciones de la misma concentración, el
potencial de unión liquida es prácticamente cero).
La pila 3 es una pila de concentración sin transporte.
Las pilas 4 y 5 son pilas de concentración con transporte, con electrodos reversibles a los
cationes y los aniones respectivamente.
En la pila 6 las disoluciones están separadas por un puente salino, el cual elimina casi por
completo, el potencial de difusión, siendo la f.e.m. igual a la diferencia de los potenciales de los dos
electrodos.

Química Física Prácticas de Electroquímica
9
Práctica EQ3: VALORACIÓN POTENCIOMÉTRICA DE ÁCIDOS.
Para llevar a cabo esta práctica es necesario llevar al laboratorio 4 hojas de papel milimetrado
por grupo de trabajo (2 personas), un lápiz, una goma de borrar y una calculadora (científica).
Además, es necesario traer los siguientes datos, con indicación de las fuentes bibliográficas de las
que se han obtenido:
Valor del potencial normal del electrodo de quinhidrona a 25oC.
Valor del potencial normal del electrodo de AgCl/Ag a 25oC.
Solubilidad (mol/l) del KCl a 25oC.
Actividad de los iones Cl- a 25oC en la disolución saturada de KCl, calculada a partir de los
valores de la molalidad y del coeficiente de actividad de este ion, usando para ello la
expresión de Debye-Hückel para disoluciones concentradas:
log 0,51 | |⁄
1 ⁄ 0,30
Este trabajo es obligatorio elaborarlo previamente a la realización de la práctica y que lo aportéis
al laboratorio para adjuntarlo al informe-resumen que presentaréis al finalizar misma.
Introducción
El punto de equivalencia de una valoración es el punto en el cual las cantidades de sustancia
valorada y de agente valorante añadido están en proporción estequiométrica.
Para determinar el punto de equivalencia en una valoración ácido-base se puede usar un
indicador que cambie de color a un valor del pH correspondiente al del punto de equivalencia. Este
tipo de valoraciones son muy subjetivas, y escasamente precisas si la disolución es turbia. Una
alternativa a este problema consiste en determinar, a lo largo de la valoración, una propiedad que
esté directamente relacionada con el pH.
Se llama curva de valoración a la gráfica que representa la variación de la propiedad
estudiada durante una valoración en función de la cantidad de agente valorante añadido. El punto de
equivalencia viene dado por el punto de inflexión de la curva de valoración.
En el caso de una valoración ácido-base, una posibilidad es utilizar la disolución que se
valora como parte de una pila cuya f.e.m. dependa de la concentración de iones hidrógeno, y
determinar a lo largo de la valoración. Las valoraciones en las que se utiliza para determinar el
punto equivalencia se denominan valoraciones potenciométricas. Las valoraciones potenciométricas
no se limitan a valoraciones ácido-base, sino que son posibles otros tipos; basta con que en la
reacción se forme o desaparezca alguna sustancia para la cual se disponga de un electrodo
reversible.

Química Física Prácticas de Electroquímica
10
Objetivo de la práctica
El objeto de la práctica es valorar potenciométricamente con NaOH 0,1 M una disolución de
ácido clorhídrico. Para ello necesitaremos un electrodo reversible a los iones H+, y para completar
la pila, puesto que no se pueden medir potenciales de electrodo, deberá usarse un electrodo de
referencia cuyo potencial no cambie a lo largo de la valoración. Como electrodo de referencia,
usaremos un electrodo de plata-cloruro de plata saturado (con disolución saturada de cloruro de
potasio) y como electrodo reversible a los iones H+, usaremos un electrodo de quinhidrona
(complejo equimolecular que en disolución se disocia dando quinona e hidroquinona).
Utilizaremos en la práctica lo que se llama un electrodo combinado, que integra en un solo
dispositivo un electrodo de plata-cloruro de plata, con su disolución saturada de cloruro de potasio
incluida, y un hilo de platino, aislados el uno del otro. Consta de dos tubos concéntricos que no se
comunican entre sí. El tubo interior simplemente aísla y lleva el hilo de Pt hasta hacerlo sobresalir
por el extremo del electrodo. Este es el platino con el que se construirá el electrodo de quinhidrona.
En el espacio entre el tubo interior y el exterior se encuentra el electrodo de plata-cloruro de plata
completo, electrolito incluido; cerca del lugar de salida del hilo de platino hay un tabique poroso
que comunica la disolución de cloruros del electrodo de plata-cloruro de plata con el exterior. La
disolución de quinhidrona (que debe estar saturada) para el electrodo de quinhidrona se prepara
disolviendo quinhidrona hasta saturación en el ácido a valorar. Para construir la pila, basta con
introducir el electrodo combinado en la disolución de ácido + quinhidrona, cuidando de que queden
cubiertos por el líquido tanto el trozo de hilo de platino que sobresale como el tabique poroso por el
que se han de comunicar la disolución de ácido + quinhidrona y la de cloruro de potasio del interior
del electrodo combinado. Puesto que el potencial del electrodo de plata-cloruro de plata permanece
constante a lo largo de la valoración, de la pila dependerá exclusivamente de la concentración de
iones H+ en la disolución del ácido + quinhidrona.
La pila es, por tanto:
Pt | Q(ac.), QH2(ac.), H+(ac.) KCl(ac., sat.) | AgCl(s) | Ag
cuya se medirá con un multímetro digital.
Procedimiento experimental
Como agente valorante se usará NaOH de concentración conocida (previamente valorada
con ftalato ácido de potasio); la concentración se indica en la botella.
Se pipetean 30 cm3 de la disolución ácida correspondiente y se vierten en un vaso de 100
cm3 en el que se llevará a cabo la valoración. Colocando el vaso en un agitador magnético se añade
quinhidrona hasta saturación (es poco soluble).

Química Física Prácticas de Electroquímica
11
Luego se introduce el electrodo; esta operación debe realizarse con mucho cuidado de no
golpear el fondo con el electrodo y con el agitador apagado, para evitar que golpee al electrodo. Se
toma el valor de cuando todavía no se ha añadido base y se saca el electrodo.
Entonces se procede a la valoración, que llevaremos a cabo en dos fases. En una primera
fase, cuyo objetivo es localizar la zona en la que está el punto de equivalencia, realizaremos 5
adiciones sucesivas de 5 ml de la disolución de NaOH, agitando al mismo tiempo. Después de cada
adición se apaga el agitador, se introduce el electrodo, se mide de la pila y se vuelve a sacar el
electrodo. Continuaremos las medidas con nuevas adiciones de valorante, ahora de 2 ml hasta haber
rebasado claramente el punto de equivalencia. A continuación se procederá a representar frente al
volumen de valorante añadido, lo que permitirá determinar la zona en la que se encuentra el punto
de equivalencia. Seguidamente realizaremos una segunda valoración que comenzaremos añadiendo,
en una sola vez, valorante hasta quedarnos aproximadamente 2,5 ml antes del punto de
equivalencia, llevando a cabo la medida de con el procedimiento ya descrito en la primera fase.
Seguidamente realizaremos 4 medidas añadiendo el valorante de 0,4 en 0,4 ml, con lo que nos
quedaremos, aproximadamente, a 1 ml del punto de equivalencia. Durante los siguientes 2 ml (uno
antes y uno después del punto de equivalencia) las adiciones de valorante serán de 0,2 en 0,2 ml y
terminaremos con 4 nuevas medidas realizadas con adiciones de 0,4 en 0,4 ml.
Con los datos obtenidos en la segunda valoración se realizará una nueva representación de
frente al volumen de valorante añadido (V) y de /V frente a V para determinar el punto de
equivalencia de forma exacta.
Advertencia: Los electrodos deben dejarse tal como se encontraron.
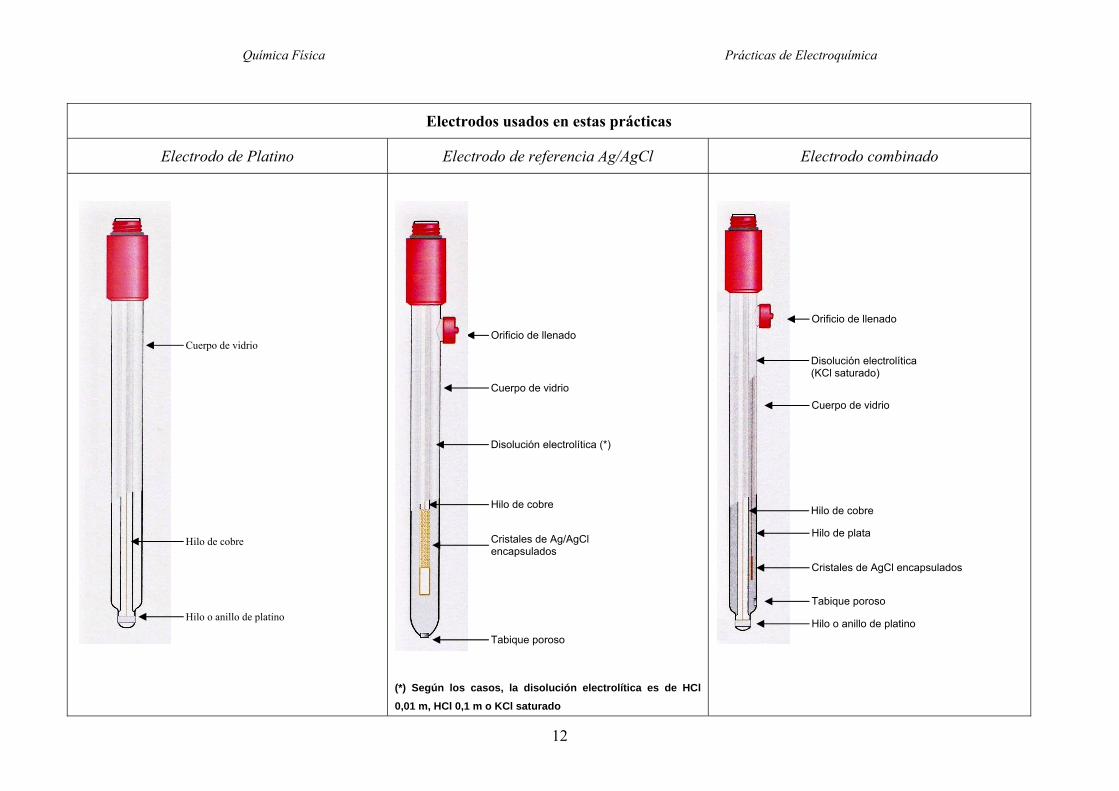
Química Física Prácticas de Electroquímica
12
Electrodos usados en estas prácticas
Electrodo de Platino Electrodo de referencia Ag/AgCl Electrodo combinado
(*) Según los casos, la disolución electrolítica es de HCl
0,01 m, HCl 0,1 m o KCl saturado
Cuerpo de vidrio
Hilo de cobre
Orificio de llenado
Disolución electrolítica (KCl saturado)
Cristales de AgCl encapsulados
Tabique poroso
Hilo de plata
Hilo o anillo de platino
Cuerpo de vidrio
Hilo de cobre
Orificio de llenado
Disolución electrolítica (*)
Cristales de Ag/AgCl encapsulados
Tabique poroso
Hilo de cobre
Hilo o anillo de platino
Cuerpo de vidrio

Química Física Prácticas de Cinética de Reacción
13
Prácticas de Cinética de Reacción
Química Física Prácticas de Cinética de Reacción
14
Características de los reactivos utilizados en las prácticas de Cinética de Reacción
A continuación se indican las características de los reactivos utilizados en las prácticas. Para
ahorrar tiempo en el laboratorio, es necesario traer hechos los cálculos de las cantidades de
reactivos que hay que pesar (gramos) o pipetear (ml) para preparar las disoluciones que se
necesitan en cada práctica.
Las pesadas se llevarán a cabo con balanzas cuya precisión es, en ocasiones, la milésima de
gramo; por tanto hay que calcular las masas en gramos y con tres cifras decimales.
La precisión de las pipetas graduadas es la décima de ml.
*Antes de empezar la práctica, enseñad al profesor los resultados de los cálculos.
Masa molar (g/mol)
Pureza (% en masa)
Densidad de los líquidos
(g/ml)
Práctica 1: Iodación de la ciclohexanona.
Ciclohexanona 98,15 99,5% 0,946
Ácido clorhídrico 36,47 33% 1,17
Iodo 253,81 99%
Práctica 2: Inversión de la sacarosa.
Sacarosa 342,30 100%
Ácido clorhídrico 36,47 33% 1,17
Práctica 3: Saponificación del acetato de etilo.
Acetato de etilo 88,10 99,5% 0,903
Hidróxido de sodio 39,997 99%
Acetato de sodio 82,04 99%

Química Física Prácticas de Cinética de Reacción
15
Práctica CR1: ESTUDIO CINÉTICO DE LA IODACIÓN DE LA CICLOHEXANONA EN MEDIO ÁCIDO
POR MEDIDAS ESPECTROFOTOMÉTRICAS.
Introducción
El objetivo de la práctica es el estudio experimental de la cinética de la reacción entre iodo
(I2 e -3I ) y ciclohexanona en disolución acuosa y medio ácido, determinando experimentalmente la
ecuación de velocidad del proceso (órdenes parciales y constante de velocidad) y proponiendo un
mecanismo que justifique la ecuación cinética obtenida.
La reacción bajo estudio se puede representar como:
(I2 + I3-) + + H+ + (I- + 2I-)
En una disolución acuosa de I2 y I- se establece muy rápidamente el siguiente equilibrio:
I2 + I- -3I (K = 710 dm3 mol-1 a 250C) (1)
donde K es la constante de equilibrio. Así pues, el iodo estará presente en dos formas, I2 e -3I ,
siendo ambas especies capaces de reaccionar con la ciclohexanona, de manera que la concentración
total de iodo, bajo el punto de vista cinético, será la suma de las correspondientes a las dos especies.
El estudio de la reacción se hará a través de medidas espectrofotométricas en la zona
espectral correspondiente al visible. La absorción de radiación por una sustancia viene regida por la
ley de Beer-Lambert:
A = c l (2)
donde A es la absorbancia, el coeficiente de absorción molar (constante característica de cada
sustancia a una longitud de onda dada), c la concentración de la muestra y l la longitud de muestra
atravesada por la radiación. La sustancia cuya absorbancia seguiremos a lo largo de la reacción es el
iodo y, para facilitar el estudio, aprovecharemos el hecho de que a dos longitudes de onda (565 y
468 nm) los coeficientes de absorción molar de las especies I2 e -3I son iguales (puntos
isosbésticos). Además, ninguna otra de las especies presentes absorbe radiación de forma apreciable
a esas longitudes de onda, de forma que, puesto que la absorbancia es una propiedad aditiva:
A = A(I2) +A(-3I ) =(I2) [I2] l + ( -
3I ) [-3I ] l = ([I2] + [
-3I ]) l (3)
Así pues, a cualquiera de estas dos longitudes de onda la concentración total de iodo
presente está directamente relacionada con la absorbancia, eligiéndose la de 565 nm, ya que en la
región de 468 nm cambia muy rápidamente con la longitud de onda, lo que puede ocasionar
errores.

Química Física Prácticas de Cinética de Reacción
16
Para la reacción química que estamos estudiando se sabe que la ecuación cinética es del tipo:
-2 3 - +
2 3
I + II + I ciclohexanona H
a cbd
v kdt
(4)
siendo v la velocidad de reacción por unidad de volumen, k la constante de velocidad, ([I2] + [-3I ])
la concentración total de iodo y a, b y c los órdenes parciales de reacción respecto del iodo, la
ciclohexanona y los iones H+ respectivamente. Teniendo en cuenta la expresión (3) podemos
escribir:
-2 3I + I 1d dA
vdt l dt
(5)
y, por tanto:
- +2 3I + I ciclohexanona H
a cbdAlk
dt (6)
Será necesario planificar y elegir convenientemente los experimentos a realizar y las
condiciones de los mismos para que a partir de los datos experimentales obtenidos puedan
determinarse los órdenes parciales respecto al iodo, a la ciclohexanona y a los iones H+, así como la
constante de velocidad del proceso.
De los posibles métodos para la determinar de la ecuación cinética (método del aislamiento,
método de las velocidades iniciales, de Powell, etc.) se ha elegido el método de las velocidades
iniciales. Para determinar el orden parcial respecto de una sustancia A es necesario obtener datos
experimentales, en este caso de absorbancias, para distintas mezclas de reacción preparadas con
diferente concentración inicial de A, pero concentraciones iniciales del resto de los reactivos iguales
en todas ellas. Esto marca la pauta de los experimentos que deberán realizarse en el caso objeto de
estudio, en el que se debe determinar el orden de reacción respecto a tres especies químicas.
Finalmente, una vez que se conozca la ecuación de velocidad experimental se buscará y
propondrá un mecanismo que justifique la ecuación cinética obtenida.
Procedimiento experimental
En primer lugar se deben preparar, en sendos matraces aforados:
- 100 cm3 de una disolución de ciclohexanona en agua, de molaridad igual a 0,46 mol dm-3.
- 100 cm3 de una disolución de HCl en agua, de molaridad 0,5 mol dm-3.
- 50 cm3 de una disolución 0,05 M de iodo en una disolución al 10% en masa de KI. (La disolución
de KI al 10% en masa es el disolvente, y se proporciona preparada).
No introducir pipetas en las botellas. Pasar a un vaso limpio y homogeneizado una cantidad
adecuada del producto y pipetear del vaso, con pipetas limpias y homogeneizadas. El HCl y la
cic1ohexanona se deben manipular en el interior de una campana extractora.

Química Física Prácticas de Cinética de Reacción
17
Ajuste de cero de los aparatos
Para eliminar la posible contribución a la absorbancia por parte de aquellas sustancias
distintas de I2 e -3I que van a estar presentes en la disolución reaccionante, se prepara en un
erlenmeyer previamente limpio y seco (si no está seco, las gotas de agua destilada que queden harán
variar la concentración) una disolución de las mismas y con ella se ajusta el cero de absorbancia del
espectrofotómetro. Esta disolución "cero" se prepara tomando disolución de ciclohexanona, agua y
disolución de ácido en proporción 1:1:3. Hay que comprobar el ajuste de cero varias veces a lo
largo de la práctica, por lo que hay que guardar la disolución "cero".
Ajuste del cero
Lan Optics
Heios
Seleccionar =565 nm pulsando Seleccionar =565 nm con el pulsador y las teclas numéricas
Seleccionar Absorbancia pulsando repetidamente el botón MODE
Seleccionar Absorbancia con el pulsador MODE y las flechas
Pasar la disolución "cero" a una cubeta limpia y homogeneizada con la disolución, secar la cubeta por fuera, colocar la cubeta en el portacubetas* del espectrofotómetro y cerrar la tapa
Poner a cero pulsando 100%T Poner a cero pulsando ZERO BASE
*Las cubetas se manipulan por las caras estriadas para evitar su deterioro y se colocan en el espectrofotómetro con las caras lisas orientadas en la dirección de paso de la radiación. Estudio cinético
Una vez ajustado el cero, se prepara en un matraz erlenmeyer limpio y seco la primera mezcla de
reacción (véase tabla 1) añadiendo las disoluciones de los reactivos en el siguiente orden: agua,
HCl, ciclohexanona e iodo. El tiempo se empezará a contar cuando se ha añadido la mitad de la
disolución de iodo. Cuando se ha añadido toda la disolución de iodo, y actuando con rapidez, se
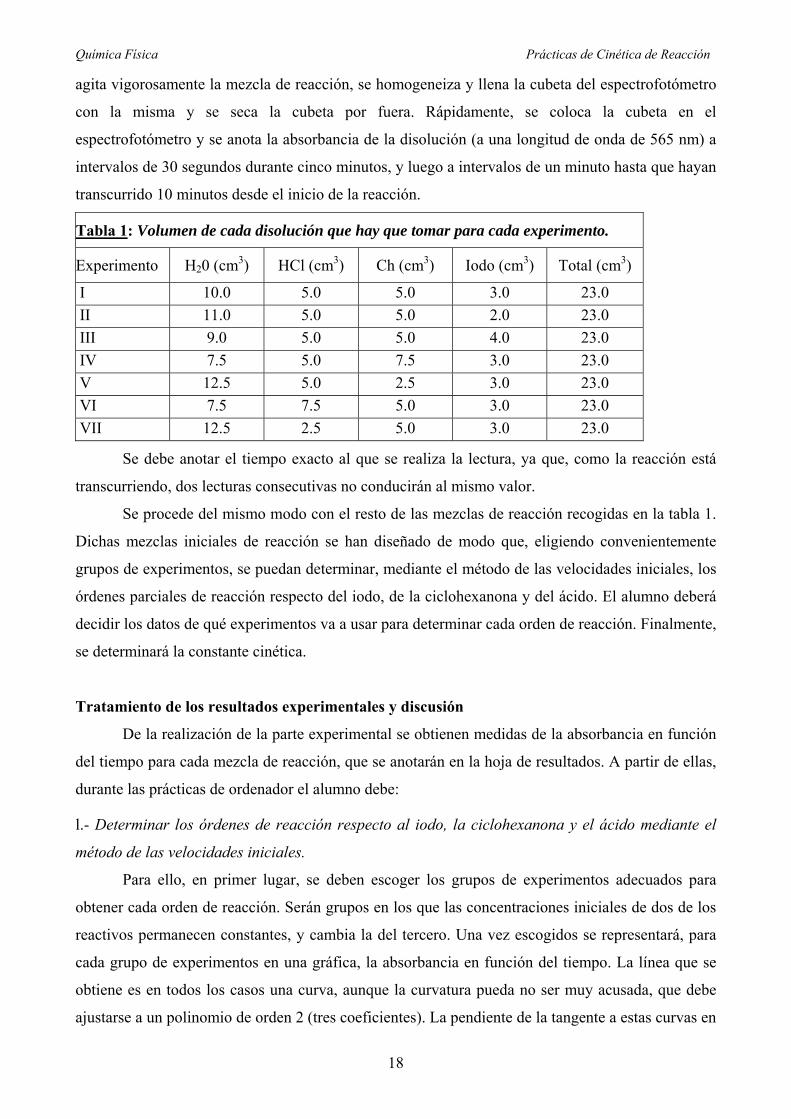
Química Física Prácticas de Cinética de Reacción
18
agita vigorosamente la mezcla de reacción, se homogeneiza y llena la cubeta del espectrofotómetro
con la misma y se seca la cubeta por fuera. Rápidamente, se coloca la cubeta en el
espectrofotómetro y se anota la absorbancia de la disolución (a una longitud de onda de 565 nm) a
intervalos de 30 segundos durante cinco minutos, y luego a intervalos de un minuto hasta que hayan
transcurrido 10 minutos desde el inicio de la reacción.
Tabla 1: Volumen de cada disolución que hay que tomar para cada experimento.
Experimento H20 (cm3) HCl (cm3) Ch (cm3) Iodo (cm3) Total (cm3)
I 10.0 5.0 5.0 3.0 23.0
II 11.0 5.0 5.0 2.0 23.0
III 9.0 5.0 5.0 4.0 23.0
IV 7.5 5.0 7.5 3.0 23.0
V 12.5 5.0 2.5 3.0 23.0
VI 7.5 7.5 5.0 3.0 23.0
VII 12.5 2.5 5.0 3.0 23.0
Se debe anotar el tiempo exacto al que se realiza la lectura, ya que, como la reacción está
transcurriendo, dos lecturas consecutivas no conducirán al mismo valor.
Se procede del mismo modo con el resto de las mezclas de reacción recogidas en la tabla 1.
Dichas mezclas iniciales de reacción se han diseñado de modo que, eligiendo convenientemente
grupos de experimentos, se puedan determinar, mediante el método de las velocidades iniciales, los
órdenes parciales de reacción respecto del iodo, de la ciclohexanona y del ácido. El alumno deberá
decidir los datos de qué experimentos va a usar para determinar cada orden de reacción. Finalmente,
se determinará la constante cinética.
Tratamiento de los resultados experimentales y discusión
De la realización de la parte experimental se obtienen medidas de la absorbancia en función
del tiempo para cada mezcla de reacción, que se anotarán en la hoja de resultados. A partir de ellas,
durante las prácticas de ordenador el alumno debe:
l.- Determinar los órdenes de reacción respecto al iodo, la ciclohexanona y el ácido mediante el
método de las velocidades iniciales.
Para ello, en primer lugar, se deben escoger los grupos de experimentos adecuados para
obtener cada orden de reacción. Serán grupos en los que las concentraciones iniciales de dos de los
reactivos permanecen constantes, y cambia la del tercero. Una vez escogidos se representará, para
cada grupo de experimentos en una gráfica, la absorbancia en función del tiempo. La línea que se
obtiene es en todos los casos una curva, aunque la curvatura pueda no ser muy acusada, que debe
ajustarse a un polinomio de orden 2 (tres coeficientes). La pendiente de la tangente a estas curvas en

Química Física Prácticas de Cinética de Reacción
19
tiempo cero es:
- +2 3 00 000
I + I ciclohexanona Ha cbdA
lk lvdt
(7)
donde el subíndice cero indica tiempo cero, es decir, velocidad de reacción inicial, concentraciones
iniciales de los reactivos y pendiente para t = 0. En cada grupo de experimentos las concentraciones
de dos de los tres reactivos serán las mismas para todos los experimentos. Para el caso del H+, por
ejemplo, obtendremos varias ecuaciones del tipo:
+
00
' HcdA
kdt
(8)
donde
-2 3 00
' I + I ciclohexanonaa b
k lk (9)
será la misma para todas ellas.
Si tomamos logaritmos en las ecuaciones (8):
+
00
ln ln ' ln HdA
k cdt
(10)
Representando ln(-dA/dt)0 frente a ln[H+]0 para el grupo de experimentos obtendremos una
recta de pendiente e (orden de reacción respecto a H+) y de ordenada en el origen k'.
Procediendo de forma similar se determinan los otros órdenes parciales de reacción.
2.- Determinar el coeficiente de absorción molar de I2 e -3I a 565 nm, especificando sus unidades.
A = c l. El punto de corte de las curvas de absorbancia frente a tiempo con el eje de absorbancias
será la absorbancia correspondiente a la concentración inicial de iodo (tiempo cero), que conocemos
para todos los experimentos: A0 = l ([I2]+[-3I ])0. Representando A0 frente a ([I2]+[
-3I ])0 obtenemos
una recta que debe pasar por el origen (si la concentración de iodo es cero la absorbancia también lo
es) y cuya pendiente es l, de donde obtenemos . El valor de l es 1 cm para las cubetas prismáticas
de los espectrofotómetro Pye Unicam y Perkin Elmer y 1,15 cm para los tubos del Spectronic.
3.- Determinar el valor de la constante de velocidad k, especificando sus unidades.
Se calcula a partir de las k' obtenidas en el apartado 1 (ecuación 9). Conocidas las
concentraciones iniciales para cada grupo de experimentos y los valores de y l, obtendremos
varios valores para k, que deben ser iguales entre sí (o muy similares, teniendo en cuenta el error
experimental). El valor definitivo de k será el valor medio de todos ellos.
Con todos estos resultados se completará la hoja de resultados que, junto con los ajustes y
las gráficas realizadas se entregarán al finalizar las sesiones de prácticas de ordenador.

Química Física Prácticas de Cinética de Reacción
20
APÉNDICE 1
Absorción de la radiación por parte de la materia
Como consecuencia de la interacción de la radiación electromagnética con la materia, la
primera puede resultar transmitida, absorbida, reflejada o dispersada.
Cuando la radiación electromagnética atraviesa una muestra, de sólido, líquido o gas, se
observa en el análisis posterior que desaparecen selectivamente ciertas frecuencias. Esta energía
absorbida se ha empleado en pasar las partículas de la muestra desde su estado de más baja energía
(estado fundamental) a estados de mayor energía (estados excitados).
Tanto los átomos como las moléculas o los iones tienen un número limitado de niveles de
energía posibles, esto es, la energía está cuantizada. Para que se produzca absorción de radiación, la
energía de ésta (la energía del fotón excitante) debe ser igual a la diferencia de energía entre el
estado fundamental de la sustancia absorbente y uno de sus estados excitados.
Como la secuencia de niveles de energía es única para cada sustancia, también lo serán las
separaciones entre dichos niveles y, por tanto, el conjunto de frecuencias a las que absorbe
radiación también será característico de la sustancia.
La ley de Beer-Lambert indica la cantidad de radiación absorbida por una muestra. Se define
una magnitud, A, llamada absorbancia como:
0logI
AI
siendo I0 la intensidad del rayo antes de entrar en la muestra e I la intensidad después de haberla
atravesado. Nótese que siempre I ≤ I0 con lo que la absorbancia es siempre positiva.
La ley de Beer-Lambert se formula de la siguiente manera:
A=cl
c es la concentración de la especie absorbente en la muestra, l la longitud de la muestra atravesada
por la radiación (anchura de la cubeta a través de la cual pasa la luz) y es el coeficiente de
absorción molar, magnitud característica de la especie absorbente y dependiente de la frecuencia.
La absorbancia es una magnitud aditiva, por tanto, si existen varias especies absorbentes:
A= Al + A2 + A3 +...= 1c1l + 2c2l + 3c3l +...
E0
E1
E3
E2 E2 - E0 = h2
E1 - E0 = h1
E3 - E0 = h3
1, 2, 3 son frecuencias características de la especie absorbente (átomo, molécula, ión, etc.)

Química Física Prácticas de Cinética de Reacción
21
Componentes de los instrumentos para medir absorción de radiación
Los componentes básicos de un espectrofotómetro son:
1) Una fuente estable de energía radiante que puede variar de intensidad.
2) Un artificio que permite el empleo de una región restringida de longitudes de onda.
3) Recipientes transparentes para la muestra.
4) Un detector de radiación que convierte la energía radiante en una señal mensurable,
generalmente eléctrica.
5) Un indicador de señal (registrador gráfico, pantalla, etc.) donde se lee la absorbancia de la
muestra.
lector muestra
detector
monocromador
rejilla
rejilla
fuente de radiación

Química Física Prácticas de Cinética de Reacción
22
Práctica CR2: ESTUDIO DE LA REACCIÓN DE INVERSIÓN DE LA SACAROSA CATALIZADA POR
ÁCIDOS MEDIANTE MEDIDAS POLARIMÉTRICAS.
Introducción
El objetivo de esta práctica es la determinación de la constante de velocidad del proceso:
sacarosa + H2O + H+ → glucosa + fructosa + H+ (1)
La ecuación cinética es del tipo:
ankdt
dv HOHsacarosa
sacarosa2 (2)
donde v es la velocidad por unidad de volumen de la reacción, k la constante de velocidad, n el
orden parcial de la reacción respecto al agua y a el orden parcial de la reacción respecto de los
iones H+.
Durante la reacción la concentración de agua permanece prácticamente constante, ya que es
mucho mayor que la concentración de sacarosa empleada normalmente. Por tanto, la velocidad de
reacción por unidad de volumen se puede considerar independiente de la concentración de agua.
Por su parte, los iones hidrógeno actúan como catalizador, lo que significa que su
concentración no cambia con el tiempo. Por consiguiente, a una temperatura y una concentración
de iones H+ dadas, la velocidad de reacción por unidad de volumen sólo depende de la
concentración de sacarosa:
sacarosasacarosa
'kdt
dv (3)
siendo:
ank'k HOH2 (4)
La reacción es, por tanto, de pseudoprimer orden desde el punto de vista experimental. Si
representamos por c la concentración de sacarosa, la ecuación (3) puede escribirse:
c'kdt
dc (5)
que integrada entre el instante inicial, t = 0, y un tiempo t cualquiera conduce a:
t'kxc
cln
c
cln
0
00 (6)
donde representamos por c0 la concentración inicial de sacarosa y por c0-x la concentración
presente cuando ha transcurrido un tiempo t.

Química Física Prácticas de Cinética de Reacción
23
Estudio de la reacción
Aprovechando que los hidratos de carbono que intervienen en la reacción son ópticamente
activos, el seguimiento de la reacción lo realizaremos a través del ángulo de rotación óptica, ,
definido como el ángulo que se desvía el plano de polarización de la luz polarizada linealmente
cuando ésta atraviesa la muestra. Trabajando en disolución, para una sustancia depende de:
- La naturaleza de la sustancia.
- La concentración de la disolución.
- La longitud de la trayectoria de la radiación a través de la disolución.
- La longitud de onda de la radiación empleada.
- La temperatura.
Para poder comparar la actividad óptica de diversas sustancias hay que fijar las demás
variables anteriores, para lo cual se define la rotación específica de una sustancia, que es la
desviación sufrida por el plano de polarización de la luz polarizada linealmente cuando atraviesa 1
dm de longitud de una disolución que contiene 1 g/cm3 de esa sustancia a una temperatura dada y
para un tipo de radiación determinado. Así, por ejemplo 20D
representa la rotación específica de
una sustancia a 20°C y utilizando como fuente luminosa la línea D del sodio. Así pues:
lc M20D (7)
donde c es la concentración de la sustancia en mol/dm3, M la masa molar en g/mol y l la longitud
de muestra atravesada por la luz en dm.
Dado que las rotaciones específicas de las sustancias que intervienen en la reacción son:
sacarosa glucosa fructosa
20D
66,50 52,50 -92,00
y teniendo en cuenta que la propiedad es aditiva, al llevar a cabo la reacción será positivo al
principio debido a la presencia mayoritaria de sacarosa, disolución dextrógira, pero según
transcurre la reacción irá disminuyendo y acabará siendo negativo, disolución levógira, debido al
progresivo aumento de la concentración de fructosa, fuertemente levógira.
Utilizando la ecuación (7), podemos reescribir la ecuación (6) en función de los ángulos de
rotación óptica (ver Apéndice) obteniendo:
t'klnt
0 (8)
ecuación de una recta de pendiente k', donde 0 es el ángulo de rotación óptica en el instante inicial
(t = 0), t es el ángulo de rotación óptica en un instante t y ∞ es el ángulo de rotación óptica a
tiempo infinito, es decir, cuando la reacción ha terminado. Así pues, el cálculo de k' requiere
conocer 0, ∞ y diversos valores de t para poder representar la recta dada por la ecuación (8).

Química Física Prácticas de Cinética de Reacción
24
Descripción de los polarímetros
Los ángulos de rotación óptica se miden con un polarímetro, cuyo esquema consta de:
Figura 1. Esquema de un polarímetro
-Una fuente luminosa, habitualmente una lámpara de vapor de sodio que proporciona luz de dos
longitudes de onda muy próximas entre sí, 589,0 y 589,6 nm, de un característico color amarillo. Es
la llamada línea D del sodio. Esta luz no está polarizada.
-Un prisma de Nicol fijo llamado polarizador, que transforma la luz normal en luz polarizada.
-Una cámara para colocar un tubo con la muestra.
-Un prisma de Nicol móvil, el analizador, cuyo giro permite de la muestra.
-Un ocular a través del cual se ve un círculo dividido en dos o tres partes, según el aparato usado y,
en algunos casos, la escala de medida del ángulo.
-Una escala graduada o una pantalla digital sobre la que se lee el ángulo.
Dirección de propagación
Fuente de radiación
Radiación normal
Polarizador Analizador Radiación plana
polarizada
Radiación plana
polarizada
Muestra ópticamente
activa

Química Física Prácticas de Cinética de Reacción
25
Lectura en la escala graduada
El polarímetro Zeiss posee una escala que se ve a través del ocular, por debajo del círculo de
ajuste, mientras que el Zuzi posee dos escalas idénticas, una a cada lado del exterior del ocular.
Este diseño permite leer indistintamente con uno u otro ojo sin levantarlo del ocular tras el ajuste.
La escala de lectura consta realmente de dos escalas, una fija que va de 0 a 10 y una móvil que se
desplaza con el giro del analizador.
Lectura de ángulos positivos tras el ajuste: -Parte entera de la medida: la más pequeña de las dos divisiones de la escala móvil entre las que se ha quedado el 0 de la escala fija. -Parte decimal de la medida: la división de la escala fija que ha quedado mejor alineada con cualquiera de las de la escala móvil.
Figura 2a. Lecturas positivas Ejemplo: la lectura es 7,600, por estar el 0 de la escala fija entre el 7 y el 8 de la móvil y ser la división de la escala fija que lleva el número 6 la que mejor coincide con una de las divisiones de la escala móvil.
Lectura de ángulos negativos tras el ajuste: -Parte entera de la medida: la de menor valor absoluto de las dos divisiones de la escala móvil entre las que se ha quedado el 0 de la escala fija. -Parte decimal de la medida: 100 menos el valor leído siguiendo el procedimiento de las lecturas positivas.
Figura 2b. Lecturas negativas Ejemplo: la lectura es -2,200, por estar el 0 de la escala fija entre el -2 y el -3 de la móvil y ser la división número 8 de la escala fija la que mejor coincide con una de las divisiones de la escala móvil (100-80=20).
A continuación se describen los distintos polarímetros usados en la práctica junto con las
instrucciones para su manejo.
(a) Polarímetro Zeiss
1- Interruptor de encendido del aparato 2- Lámpara de sodio 3- Tapa de la cámara de muestras 4- Mando de giro del analizador 5- Ocular
Encendido del aparato - Pulsar el interruptor de encendido del aparato.
Puesta a cero *No es necesario.* Al estar unidos el analizador y la escala mecánicamente, un ángulo cero indica que el analizador no se ha movido y, por tanto, está alineado con el polarizador, por lo que el campo visual estará en la posición de la figura 3aB. Medida - Lavar bien el tubo del polarímetro y llenarlo
con la muestra cuyo ángulo se quiere medir. - Colocar el tubo en la cámara de muestra. - Con el mando de giro del analizador ajustar el campo visual a través del ocular hasta llevarlo a la posición de la figura 3aB.
- Leer el ángulo de rotación con su signo en la escala.
Apagado del aparato - Pulsar el interruptor de encendido del aparato.
4
1
3
2
5
0 10 1 2 3 4 5 6 7 8 9
10 0
0 10 1 2 3 4 5 6 7 8 9
20 10

Química Física Prácticas de Cinética de Reacción
26
(b) Polarímetro Zuzi Manual
1- Interruptor de encendido del aparato 2- Lámpara de sodio 3- Tapa de la cámara de muestras 4- Escala para leer el ángulo 5- Ocular 6- Mando de giro del analizador 7- Mando de giro del analizador (fino)
Encendido del aparato - Pulsar el interruptor de encendido del aparato.
Puesta a cero *No es necesario.* Al estar unidos el analizador y la escala mecánicamente, un ángulo cero indica que el analizador no se ha movido y, por tanto, está alineado con el polarizador, por lo que el campo visual estará en la posición de la figura 3aB.
Medida - Lavar bien el tubo del polarímetro y llenarlo
con la muestra cuyo ángulo se quiere medir. - Colocar el tubo en la cámara de muestra. - Con los mandos de giro del analizador ajustar el campo visual a través del ocular hasta llevarlo a la posición de la figura 3aB.
- Leer el ángulo de rotación con su signo en la escala.
Apagado del aparato - Pulsar el interruptor de encendido del aparato.
(d) Polarímetro Zuzi automático
1- Interruptor de encendido del aparato 2- Interruptor de encendido de la lámpara 3- Interruptor de medida 4- Botón de selección 5- Botón de puesta a cero 6- Tapa de la cámara de muestras 7- Pantalla digital
Encendido del aparato - Activar el interruptor de encendido del aparato. - Esperar 5 minutos. - Activar el interruptor de la lámpara.
Puesta a cero - Lavar bien el tubo del polarímetro y llenarlo con agua destilada.
- Colocar el tubo en la cámara de muestra. - Activar el interruptor de medida. - Esperar a que la lectura se estabilice. - Pulsar el botón de puesta a cero; la lectura en la pantalla digital pasa a ser 00.000
- Desactivar el interruptor de medida.
Medida - Lavar bien el tubo del polarímetro y llenarlo
con la muestra cuyo ángulo se quiere medir. - Colocar el tubo en la cámara de muestra. - Activar el interruptor de medida. - Esperar a que la lectura se estabilice. - Leer el ángulo de rotación con su signo en la. - Desactivar el interruptor de medida.
Apagado del aparato - Desactivar el interruptor de la lámpara. - Desactivar el interruptor de encendido del
aparato.
7
1
6
2 3 4 5
4
1
3
2
5
6
7

Química Física Prácticas de Cinética de Reacción
27
Manejo de los polarímetros
Si el polarizador y el analizador están alineados, cuando una muestra no es ópticamente
activa, el rayo de luz polarizada procedente del polarizador la atraviesa sin modificar su plano de
vibración, y sale por el analizador. El círculo que aparece en el campo visual presenta una
luminosidad no muy brillante y uniforme en todas sus partes (Fig.3aB y 3bB), y el ángulo de
rotación óptica que aparece en la escala o en la pantalla debe ser 0°.
Figura 3. Campo visual: (a) polarímetro de tres zonas; (b) polarímetro de dos zonas.
Sin embargo, cuando la muestra es ópticamente activa, el plano de polarización de la luz se
modifica al atravesarla, y al llegar al analizador se encuentra vibrando en un plano distinto al de la
alineación inicial. El círculo que aparece en el campo visual no es uniforme, sino que presenta
una(s) zona(s) de brillo y otra(s) de sombra (Fig. 3aA, 3aC, 3bA, 3bC). Cuál sea la zona de brillo y
cuál la de sombra depende de que el plano se haya desviado hacia la derecha (ángulo positivo,
sustancia dextrógira, Fig. 3aA y 3bA) o hacia la izquierda (ángulo negativo, sustancia levógira, Fig.
3aC, 3bC). Para tomar la medida del ángulo de rotación óptica hay que hacer girar el analizador
hasta alinearlo con el nuevo plano de vibración de la luz que le llega. Al ir girando el analizador en
el sentido correcto, las partes del círculo van igualando su luminosidad, hasta quedar iguales y con
una luminosidad intermedia, momento en que el analizador está alineado con el plano de vibración
de la luz que recibe (Fig. 3aB y 3bB). Como inicialmente el analizador estaba alineado con el
polarizador, el ángulo que hemos tenido que girar el analizador es el mismo ángulo que ha girado el
plano de vibración de la luz polarizada al atravesar la muestra, es decir, el ángulo de rotación óptica
de la muestra (Fig. 1). El citado ángulo se lee sobre la escala graduada o la pantalla digital.
Si se gira el analizador un ángulo mayor del necesario, se observa un intercambio de las
zonas de luz y sombra: por ejemplo, de la situación de las figuras 3aA o 3bA se pasa a las de las
figuras 3aC o 3bC. En tal caso, hay que retroceder hasta conseguir el ajuste. En realidad, esta es la
forma más fiable de realizar las medidas: girar primero el analizador y localizar la zona donde se
produce el intercambio de campos de luz y sombra y continuar girando el analizador
alternativamente hacia la derecha y hacia la izquierda del punto de intercambio, reduciendo cada
vez más la amplitud del giro, hasta localizar el punto exacto en el que se produce el intercambio, en
el cual las partes del círculo son iguales.
(b)
C B A A B C
(a)

Química Física Prácticas de Cinética de Reacción
28
Hay que tener mucho cuidado con no salirse demasiado de la zona de la medida, ya que en
ese caso las partes del círculo se ven iguales, pero muy brillantes. Además, girando el analizador
hacia la derecha o hacia la izquierda de este punto no hay cambio apreciable en la luminosidad.
Este no es el punto de medida del ángulo. En el punto correcto la luminosidad de los campos es
débil, y además, girando un poco el analizador se aprecia claramente cambio en la luminosidad.
Tubos de observación para la muestra y su manipulación
Las muestras líquidas se introducen en tubos especiales de vidrio que se colocan sobre la
bandeja que hay en el interior de la cámara del polarímetro, entre el polarizador y el analizador. Los
tubos tienen longitudes estándar; las más frecuentes son 50, 100 y 200 mm.
Los tubos se desmontan, se lavan las piezas por separado con agua y agua destilada y se
vuelven a montar. Es necesario llenar el tubo sin que queden burbujas en su interior, ya que éstas
interfieren el haz de luz polarizada y alteran el proceso de medida.
Para proceder al llenado se desenrosca el anillo de seguridad de uno de los extremos y se
retira la ventana de vidrio, construida con un material que no afecta a la polarización de la luz. Se
coloca el tubo en posición vertical y se llena totalmente, hasta que un menisco de líquido
sobresalga por encima de la boca del tubo. Entonces, se apoya el borde de la ventana sobre el borde
de la boca del tubo y se desliza lateralmente sobre la boca del tubo. De esta forma el tubo queda
cerrado y sin burbujas en su interior. A continuación, se vuelve a colocar la junta de goma y el
anillo de seguridad, y se seca cuidadosamente el exterior del tubo.
Si, a pesar de todo, ha quedado una pequeña burbuja, se hace oscilar el tubo en horizontal
para conseguir que la burbuja vaya a parar al abultamiento que tiene el tubo.
Procedimiento experimental
La realización de la práctica requiere la preparación de dos disoluciones:
-100 ml de disolución que contenga 20 g de sacarosa.
-100 ml de disolución de HCl 4 M.
Realización de las medidas
Medida del ángulo de rotación óptica a tiempo cero, 0
No se puede medir 0 una vez realizada la mezcla de reacción, ya que el tiempo transcurrido
hasta que se introduce el tubo en el polarímetro y se toma la medida hace que ya no estemos a
tiempo cero. Puesto que el HCl no es ópticamente activo y sin él no comienza la reacción,
prepararemos una disolución en la que la sacarosa se encuentre en igual concentración que en la
mezcla de reacción, pero sustituyendo el HCl por agua destilada, con lo que las características
ópticas son idénticas a las de la mezcla de reacción en el instante inicial pero la reacción no
comienza y se puede medir el valor de 0 con toda tranquilidad.

Química Física Prácticas de Cinética de Reacción
29
Se preparará en un erlenmeyer limpio y seco una disolución con 25 cm3 de la disolución de
sacarosa y 25 cm3 de agua destilada. Se agita para conseguir una buena mezcla, se homogeneiza el
tubo del polarímetro, previamente limpio, con la disolución, se llena, se introduce el tubo en el
polarímetro y se realiza la medida tal como se ha indicado.
* Antes de continuar, debe enseñarse al profesor el valor obtenido para 0*
Medida del ángulo de rotación óptica en función del tiempo, t
Prepararemos la mezcla de reacción con 25 cm3 de la disolución de sacarosa y 25 cm3 de la
disolución de HCl, al tiempo que ponemos el reloj en marcha cuando se ha añadido la mitad de la
segunda disolución. Se agita fuertemente para obtener una mezcla homogénea.
El tubo del polarímetro, previamente lavado, se homogeneiza y se llena con esta mezcla,
haciéndose una primera lectura del ángulo de rotación lo antes posible. A partir de este momento se
siguen realizando lecturas con la siguiente pauta:
- Lecturas cada 2 minutos hasta el minuto 12.
- Lecturas cada 3 minutos hasta el minuto 30.
- Lecturas cada 5 minutos hasta el 60.
- Lecturas cada 10 minutos hasta que la lectura permanezca constante durante 30 minutos.
Hay que anotar el tiempo exacto en el que se realiza la medida, ya que al estar la reacción en
marcha dos medidas consecutivas no conducirán al mismo valor.
El polarímetro debe permanecer encendido durante todo el tiempo que dure la práctica, pero
el tubo se mantendrá fuera cuando no se estén realizando medidas, para evitar el calentamiento de
la disolución, ya que se trata de una reacción muy sensible a la temperatura.
Medida del ángulo de rotación óptica a tiempo infinito ∞
Corresponde al momento en que la reacción ha terminado y toda la sacarosa se ha
desdoblado en glucosa y fructosa. Su medida se realiza cuando el ángulo de rotación óptica
permanece constante a lo largo del tiempo.
Cálculos y resultados
En las prácticas de ordenador se hará el tratamiento de los resultados experimentales,
calculando para cada medida
tln 0
de modo que representando estos valores frente al tiempo debemos obtener una recta (8), lo que
confirmará el pseudoorden 1 que se ha supuesto, y su pendiente nos proporcionará k’.

Química Física Prácticas de Cinética de Reacción
30
APÉNDICE 2
Polarimetría
Luz polarizada
La luz natural es una radiación electromagnética formada por un campo eléctrico y un
campo magnético oscilantes, perpendiculares entre sí y perpendiculares a la dirección de
propagación de la onda.
Figura 1A. Diagrama de una onda electromagnética.
Una onda electromagnética cuyo campo eléctrico (omitiremos el magnético por
simplicidad) oscila en un solo plano, por ejemplo el plano vertical en la figura 1A, se dice que está
polarizada en un plano, polarizada linealmente o, simplemente, polarizada. El origen de las ondas
luminosas son las moléculas o átomos de las fuentes de luz, que adquieren energía y luego la
emiten en forma de radiación electromagnética. Las ondas procedentes de una molécula o átomo
cualquiera están polarizadas pero dado que una fuente luminosa natural contiene un número
enorme de átomos o moléculas orientados al azar, la luz emitida es una mezcla de ondas
polarizadas en todas las direcciones del espacio.
Figura 2A. Diagrama esquemático de la luz ordinaria y la luz polarizada.
Cuando todas las ondas de un rayo de luz vibran en el mismo plano, se dice que la luz está
polarizada en un plano, polarizada linealmente o, simplemente, polarizada.
Existen varios métodos que permiten obtener un rayo de luz polarizada a partir de un rayo
de luz natural. El utilizado habitualmente en los polarímetros es el prisma de Nicol.
La radiación oscila en todas las direcciones
La radiación oscila en un solo plano
Dirección de propagación
La radiación oscila en todas las direcciones
Campo magnético
Campo eléctrico
Dirección de propagación

Química Física Prácticas de Cinética de Reacción
31
Prisma de Nicol
Existen sustancias cristalinas transparentes que, siendo homogéneas, son anisótropas; es
decir, la velocidad de una onda luminosa que se propaga en ellas no es la misma en todas las
direcciones. Los cristales que poseen esta propiedad se llaman birrefringentes. Cuando un rayo de
luz entra en uno de estos cristales se divide en dos rayos, cada uno de los cuales está polarizado
perpendicularmente respecto al otro. En consecuencia, si podemos separar ambos rayos, puede
utilizarse un cristal birrefringente para obtener luz polarizada a partir de luz natural. Este es el
principio del funcionamiento del prisma de Nicol.
El prisma de Nicol se prepara a partir de un cristal de calcita CaCO3. Se tallan las caras
externas con un ángulo obtuso de 680 y, seguidamente, se corta el cristal a lo largo de la diagonal y
se pega de nuevo con bálsamo del Canadá. El índice de refracción del bálsamo del Canadá tiene un
valor tal que uno de los dos rayos se refleja totalmente, mientras que el otro se transmite.
(a) (b) Figura 3A. (a) Cristal natural de calcita. (b) Prisma de Nicol.
Actividad óptica
Cuando un rayo de luz polarizada linealmente atraviesa cierto tipo de cristales, líquidos o
disoluciones, se encuentra que la luz emergente también está polarizada linealmente, pero vibra en
un plano diferente al inicial, es decir, el plano de polarización de la luz ha girado un determinado
ángulo, llamado ángulo de rotación óptica, . A este fenómeno se le llama rotación del plano de
polarización, y las sustancias que presentan tal efecto se denominan ópticamente activas.
Cuando el giro del plano de polarización es hacia la derecha mirando a lo largo del haz de
luz en el sentido del avance, las sustancias se llaman dextrógiras, y los correspondientes valores de
se consideran positivos. Si el giro es hacia la izquierda se denominan levógiras, y los valores de
son negativos.
La actividad óptica puede deberse a la estructura molecular de la sustancia, como ocurre con
muchos compuestos orgánicos naturales, en cuyo caso se mantiene cuando ésta se funde o se
disuelve. También puede constituir una propiedad del cristal en su conjunto, con lo que desaparece
con la fusión o disolución.
Polarización por birrefringencia o doble polarización
Calcita (CaCO3)
Polarización por birrefringencia o doble polarización
Calcita (CaCO3)

Química Física Prácticas de Cinética de Reacción
32
Obtención de la ecuación cinética en función de los ángulos de rotación óptica.
El ángulo de rotación óptica que presenta la mezcla de reacción en cada momento es la
suma de las contribuciones de la sacarosa, glucosa y fructosa, ya que las tres especies son
ópticamente activas y la actividad óptica es una propiedad aditiva:
fgs (A1)
Cada contribución se puede poner en función de la concentración de la correspondiente
especie utilizando la ecuación (7) del guión:
lB;cBlc ssDssssssDs M M (A2) lB;cBlc ggDggggggDg M M (A3) lB;cBlc ffDffffffDs M M (A4)
Según esto, procederemos a escribir las concentraciones de cada especie para tiempo cero, para
cualquier tiempo intermedio t y para tiempo ∞, junto con el ángulo de rotación óptica de la muestra
obtenido como suma de las contribuciones de las especies presentes (ecuación es Al y A2, A3, A4):
Tiempo Concentraciones Ángulo de rotación óptica
sacarosa → glucosa + fructosa
t = 0 c0 0 0 000s0 cB (A5) t = t c0 - x x x xBxBxcBt fg0s (A6) t = ∞ 0 c0 c0 0f0g0 cBcB (A7)
Utilizando las ecuaciones A5, A6 y A7 obtenemos:
fgs00 BBBc (A8)
fgs0 BBBxct (A9)
de donde:
txc
c 0
0 (A10)
y por tanto, la ecuación cinética:
t'kxc
cln
0 (A11)
puede escribirse como:
tk'lnt
0 (A12)

Química Física Prácticas de Cinética de Reacción
33
Práctica CR3: ESTUDIO CINÉTICO DE LA REACCIÓN DE SAPONIFICACIÓN DEL ACETATO DE
ETILO POR MEDIDAS CONDUCTIMÉTRICAS.
Introducción
La saponificación del acetato de etilo en medio alcalino transcurre según la reacción:
AcEt + NaOH → AcNa + EtOH
a una velocidad apreciable a temperatura ambiente.
La finalidad de esta práctica es comprobar que se trata de un proceso de primer orden con
respecto a cada uno de los reaccionantes y calcular la constante de velocidad.
La velocidad por unidad de volumen de esta reacción se puede expresar como el producto de
una constante por las concentraciones de los reaccionantes elevadas a sus correspondientes órdenes
de reacción:
v = k[AcEt]m [NaOH]n
donde k es la constante de velocidad, m y n los órdenes de reacción respecto de AcEt y NaOH
respectivamente.
Esta velocidad se puede expresar como concentración de producto que aparece en la unidad
de tiempo o como concentración de reactivo que desaparece en la unidad de tiempo. Tomando esta
última opción, en el caso de que la reacción sea de primer orden respecto al AcEt y al NaOH,
podemos escribir:
NaOHAcEtkdt
AcEtdv (1)
Si partimos de concentraciones iniciales iguales, c0, de NaOH y AcEt, entonces las
concentraciones de estos reactivos serán iguales en cada momento, [NaOH] = [AcEt] = c , ya que la
reacción se realiza mol a mol. Según esto, podemos escribir:
2
dt
dck
c (2)
que integrada conduce a:
t11
t1
dt d
dt d
00022
00
kcc
kc
kc
ck
c
c tc
c
tc
c
(3)
Si partimos de una concentración inicial igual, c0, de ambos reactivos, en un tiempo
cualquiera t se habrán transformado parte de los reaccionantes y se habrán formado productos cuya
concentración denominaremos x. La concentración de reactivos será c = c0 - x . Para t = ∞, cuando
la reacción ha terminado, todos los reaccionantes se han transformado en productos, y las
concentraciones de éstos serán c0.
AcEt + NaOH → AcNa + EtOH t = 0 c0 c0 0 0 t = t c0-x c0-x x x t =∞ 0 0 c0 c0

Química Física Prácticas de Cinética de Reacción
34
Sustituyendo en (3):
xckc
cxck
00
00
xt
11t (4)
En esta práctica a realizar la determinación de k y la comprobación de que la reacción se
ajusta a estas ecuaciones. No mediremos directamente cómo varía la concentración de las especies
químicas en función del tiempo, sino cómo varía una propiedad física de éstas relacionada
directamente con la concentración, la conductividad específica de la disolución.
La conductividad eléctrica
La conducción de la corriente eléctrica en el seno de las disoluciones electrolíticas se debe al
transporte de los iones. La conductividad específica de una disolución se mide introduciendo en la
disolución una célula de conductividades y haciendo pasar corriente alterna (la corriente continua
produciría electrolisis). Una célula de conductividades consiste en dos láminas iguales y paralelas
de platino o grafito, de área A y colocadas una frente a otra a una distancia l, entre las que se hace
circular la corriente a través de la disolución.
La resistencia que opone la disolución al paso de la corriente será:
A
lR ( = resistividad específica de la disolución) (5)
o bien
A
lR
1 ( = conductividad específica de la disolución) (6)
A
l
R
1 = -1m-1 (Sistema Internacional) o bien -1cm-1, ya que (7)
l/A es un parámetro característico de cada célula, que depende de su forma geométrica y se
denomina constante de la célula. Sus unidades son, habitualmente cm-1.
La conductividad específica es una propiedad aditiva, de forma que la para una disolución de
varios electrolitos, se puede calcular como la suma de las conductividades específicas de todos los
componentes de la disolución, incluido el disolvente si, como es el caso del agua, produce iones. En
esta práctica no tendremos en cuenta la contribución del disolvente por ser mucho más pequeña que
la de las demás sustancias.
Se define la conductividad molar de un electrolito, , como:
c
donde c es la concentración estequiométrica del electrolito.
Así pues, es la conductividad específica debida al electrolito en una disolución de
concentración estequiométrica c de dicho electrolito; en nuestro caso, se identifica con la de la
disolución, ya que despreciamos la contribución del disolvente.

Química Física Prácticas de Cinética de Reacción
35
Si viene expresada en -1cm-1 habrá que usar c en mol/cm3, y entonces las unidades de
serán -1cm2mol-1. Si, como es habitual, expresamos la concentración en mol/dm3, entonces:
211333
11cmmolΩ
1000
)/1000cm(dm)dm(mol
)cm(Ω
cc
se puede descomponer en suma de las conductividades molares correspondientes a cada
especie iónica presente. Para electrolitos fuertes, totalmente disociados:
i
ii1
cc
donde i es la conductividad iónica molar del ión i y ci la concentración molar del ión i.
Utilizando las ecuaciones (9) y (10) se puede escribir la ecuación cinética (4) en función de
las conductividades de la mezcla reactiva en el instante inicial (tiempo 0), 0, en tiempos t
intermedios, t, y cuando la reacción ha terminado (tiempo ∞), ∞ De este modo se obtiene (ver
Apéndice):
t
ttkc 00 (11)
Por tanto, para realizar el estudio cinético de la reacción necesitaremos determinar la
conductividad de la mezcla reactiva a tiempo cero, a tiempo infinito y a varios tiempos t mientras la
reacción transcurre.
Representando el segundo miembro de la ecuación 11 frente al tiempo se debe obtener una
recta, lo que indica que la reacción es de orden 1 respecto de cada uno de los dos reactivos, tal como
se ha supuesto. La pendiente de la recta es (c0 k), de donde obtendremos k. Obsérvese que la línea
debe pasar por el origen de coordenadas.
Medida de la conductividad
Los conductímetros permiten realizar medidas de conductividad específica de disoluciones
si el aparato dispone de la constante de la célula. Asimismo, se pueden realizar medidas de
constantes de las células de conductividades si se dispone de una disolución patrón de
conductividad conocida. Para ello, tienen un selector de tipo de medida. En esta práctica se
determinará, en primer lugar, la constante de la célula, utilizando una disolución patrón de
conductividad conocida. Una vez conocida la constante de la célula y calibrado el aparato en
relación a ella, se realizarán las medidas de conductividad.
El conductímetro utilizado en esta práctica es el Crisol modelo GLP 31 que, además de la
célula de conductividades incorpora un termómetro para medir la temperatura de la disolución
(Figura 1). La selección del rango de medida es automática.

Química Física Prácticas de Cinética de Reacción
36
(a) (b) Figura 1. Célula de conductividad y termómetro. (a) modelo independiente; (b) modelo integrado.
Todas las medidas de conductividad y de determinación de constantes de las células se
llevarán a cabo con las disoluciones correspondientes colocadas en vasos termostatizados a 25°C,
por lo que habrá que esperar unos minutos después de colocar las disoluciones en el vaso hasta que
éstas alcancen la temperatura de termostatización.
Determinación de la constante de la célula
En el caso de que no se conozca la constante de la célula, que es el nuestro, ésta se
determina con una disolución de KCl 0,01 M, que es el patrón habitual, cuya conductividad
específica está perfectamente determinada. Durante el calibrado sumergiremos la célula del
aparato en esta disolución a 25oC de modo que el aparato recalculara la constante para que la
medida proporcione el valor de conductividad que tiene almacenada para esta disolución. En un
vaso limpio, homogeneizado y termostatizado se pone la disolución de KCl 0,01 M y se introduce
en él la célula de conductividades previamente limpia, seca y homogeneizada, agitando un poco
para evitar que queden atrapadas burbujas de aire que interfieran el paso de la corriente. A
continuación se procede del modo siguiente:
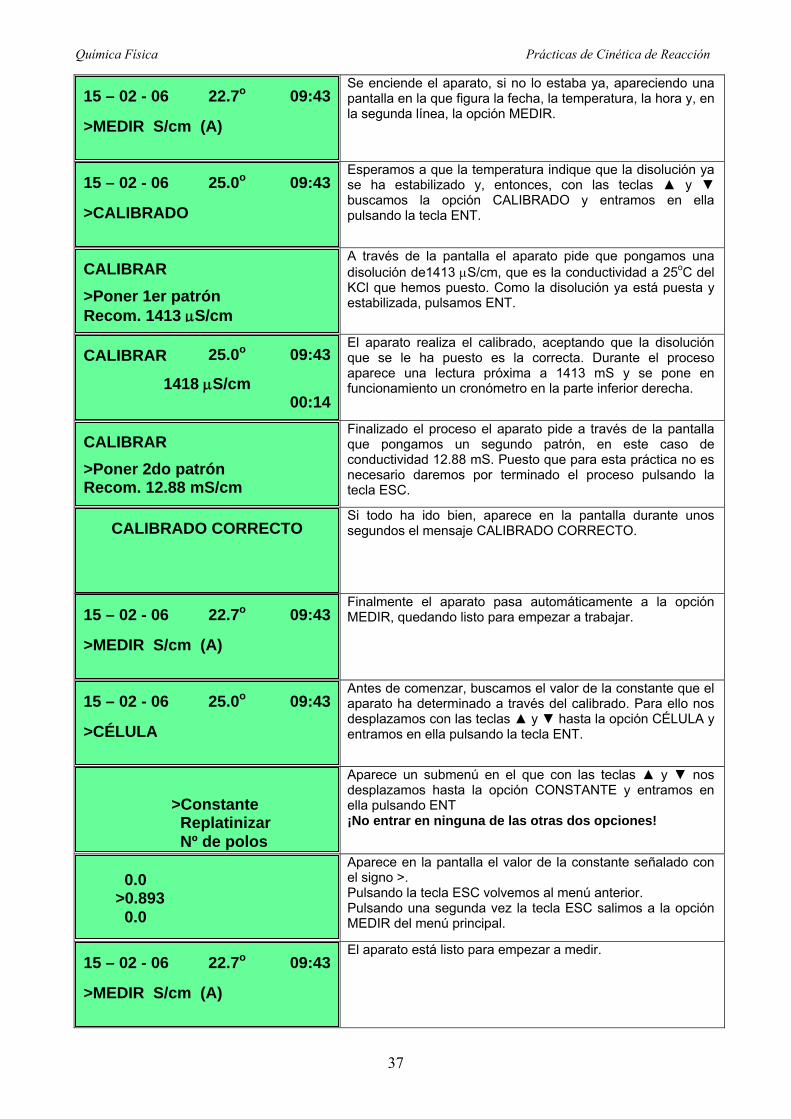
Química Física Prácticas de Cinética de Reacción
37
Se enciende el aparato, si no lo estaba ya, apareciendo una pantalla en la que figura la fecha, la temperatura, la hora y, en la segunda línea, la opción MEDIR.
Esperamos a que la temperatura indique que la disolución ya se ha estabilizado y, entonces, con las teclas y buscamos la opción CALIBRADO y entramos en ella pulsando la tecla ENT.
A través de la pantalla el aparato pide que pongamos una disolución de1413 S/cm, que es la conductividad a 25oC del KCl que hemos puesto. Como la disolución ya está puesta y estabilizada, pulsamos ENT.
El aparato realiza el calibrado, aceptando que la disolución que se le ha puesto es la correcta. Durante el proceso aparece una lectura próxima a 1413 mS y se pone en funcionamiento un cronómetro en la parte inferior derecha.
Finalizado el proceso el aparato pide a través de la pantalla que pongamos un segundo patrón, en este caso de conductividad 12.88 mS. Puesto que para esta práctica no es necesario daremos por terminado el proceso pulsando la tecla ESC.
Si todo ha ido bien, aparece en la pantalla durante unos segundos el mensaje CALIBRADO CORRECTO.
Finalmente el aparato pasa automáticamente a la opción MEDIR, quedando listo para empezar a trabajar.
Antes de comenzar, buscamos el valor de la constante que el aparato ha determinado a través del calibrado. Para ello nos desplazamos con las teclas y hasta la opción CÉLULA y entramos en ella pulsando la tecla ENT.
Aparece un submenú en el que con las teclas y nos desplazamos hasta la opción CONSTANTE y entramos en ella pulsando ENT ¡No entrar en ninguna de las otras dos opciones!
Aparece en la pantalla el valor de la constante señalado con el signo >. Pulsando la tecla ESC volvemos al menú anterior. Pulsando una segunda vez la tecla ESC salimos a la opción MEDIR del menú principal.
El aparato está listo para empezar a medir. 15 – 02 - 06 22.7o 09:43
>MEDIR S/cm (A)
0.0 >0.893 0.0
>Constante Replatinizar Nº de polos
15 – 02 - 06 25.0o 09:43
>CÉLULA
15 – 02 - 06 22.7o 09:43
>MEDIR S/cm (A)
CALIBRADO CORRECTO
CALIBRAR
>Poner 2do patrón Recom. 12.88 mS/cm
CALIBRAR
1418 S/cm 00:14
25.0o 09:43
CALIBRAR
>Poner 1er patrón Recom. 1413 S/cm
15 – 02 - 06 25.0o 09:43
>CALIBRADO
15 – 02 - 06 22.7o 09:43
>MEDIR S/cm (A)

Química Física Prácticas de Cinética de Reacción
38
Medida de la conductividad específica de una disolución
Una vez calibrado el aparato, basta introducir la célula, previamente limpia con agua
destilada, seca y homogeneizada, en la disolución cuya conductividad específica queremos medir.
Situados en la opción MEDIR se pulsa ENT y el aparato empieza a medir de forma continua. En la pantalla aparece la palabra MIDIENDO y el valor de la conductividad. El cronómetro se pone en marcha en la esquina inferior derecha.
Si se pulsa ESC, el aparato deja de medir y queda fija en la pantalla la última medida que había antes de pulsar; la palabra MIDIENDO desaparece y el cronómetro se detiene. Si se pulsa ENT el aparato vuelve a medir nuevamente y el cronómetro vuelve a funcionar.
Pulsando ESC por segunda vez, se vuelve a la opción MEDIR del menú principal. Si se vuelve a pulsar ENT el aparato vuelve a medir nuevamente y el cronómetro comienza a funcionar.
Procedimiento experimental
Preparar, en matraces aforados, las siguientes disoluciones:
(a) 50 ml de AcEt 0,2 M (b) 250 ml de NaOH 0,05 M (c) 250 m de AcNa 0,01 M
Es importante que las concentraciones de las disoluciones sean lo más exactas posible, ya
que, al preparar la mezcla de reacción, debemos obtener concentraciones iniciales iguales de los
dos reactivos para poder aplicar las ecuaciones cinéticas desarrolladas anteriormente.
Las conductividades correspondientes a tiempo cero y tiempo infinito no se pueden medir
directamente en la mezcla de reacción. En el primer caso, porque la primera medida que se pueda
hacer, por muy rápida que sea, no será exactamente tiempo cero. En el segundo, porque habría que
esperar mucho tiempo hasta estar seguros de que la reacción se ha completado. Por esta razón, se
prepararán disoluciones especiales para medir dichas conductividades. Estas disoluciones tendrán
exactamente la misma conductividad que la mezcla de reacción en tiempo cero e infinito, pero no
serán mezclas reaccionantes, con lo que sus conductividades podrán determinarse sin problemas.
Medida de 0
Tomar 20 ml de la disolución b (NaOH 0,05 M) y diluir hasta 100 ml en un matraz aforado
de 100 ml. (Recordad que no se pipetea directamente del matraz, sino que se pasa una cantidad
adecuada de disolución a un vaso limpio y homogeneizado y se coge del vaso). Sumergir en esta
nueva disolución, previamente pasada a un vaso limpio, homogeneizado y termostatizado, la
célula de conductividades y el termómetro, previamente limpios, secos y homogeneizados. Agitar
un poco la célula para eliminar posibles burbujas de aire, esperar a que la disolución alcance los
25°C y tomar la medida de la conductividad. En la mezcla de reacción, para t = 0 el NaOH es el
15 – 02 - 06 22.7o 09:43
>MEDIR S/cm (A)
22.7o 09:43
1012 S/ cm (C) 03.47
MIDIENDO 22.7o 09:43
1012 S/ cm (C) 03.47

Química Física Prácticas de Cinética de Reacción
39
único compuesto que conduce la corriente eléctrica y su concentración será [NaOH]0 = 0'01 M,
idénticas características que la disolución que hemos preparado, en la que la concentración de
NaOH es la misma y tampoco hay más sustancias conductoras. Así pues, la conductividad medida
para esta disolución debe ser la misma que la de la mezcla de reacción a t = 0.
Medida de ∞
En un vaso termostatizado, limpio y homogeneizado, se pone una cantidad suficiente de la
disolución c (AcNa 0'01 M) ya que para t = ∞ sería esta sustancia y en esa concentración la única
conductora. Se introduce la célula de conductividades y el termómetro, limpios, secos y
homogeneizados, se agita un poco, se espera a que la disolución esté a 25oC y se toma la medida.
* Antes de continuar, enseñar al profesor los valores obtenidos para 0 y ∞*
Medida de t
Preparamos la mezcla de reacción:
a) Se toman 10 ml de la disolución b (NaOH 0,05 M) y se ponen en un vaso limpio, seco (para
evitar que las gotas de agua destilada puedan modificar las concentraciones) y termostatizado. No
homogeneizarlo, ya que en la mezcla de reacción las concentraciones iniciales deben ser exactas.
b) Se añaden 37,5 ml de agua destilada con una bureta.
En este momento se introduce en la disolución la célula de conductividades y el termómetro,
limpios y secos (no homogeneizados), se agita y se espera unos minutos hasta que alcance los 25oC.
c) Se añaden 2,5 ml de la disolución a (AcEt 0,2 M).
En el momento de añadirlos, cuando haya caído aproximadamente la mitad, se pone en
marcha el cronómetro. De esta forma, tenemos un volumen total exacto de 50 ml, y las
concentraciones iniciales de los reactivos son [AcEt]0 = [NaOH]0 = 0'01 M. Rápidamente, se agita
bien, tanto para eliminar burbujas como para mezclar bien las tres disoluciones añadidas, y se mide
la conductividad de la mezcla reaccionante cada 2 minutos. Se realizan medidas durante una hora.
La célula se lava exclusivamente con agua destilada, abundante, y se seca cuidadosamente con
papel y se homogeneiza cuando se la cambie de disolución; cuando se vaya a introducir en la
mezcla de reacción no se homogeneiza, pero sí debe estar limpia y seca.
Cálculos y resultados
Durante las prácticas de ordenador, utilizando los resultados experimentales que habrán sido
convenientemente anotados en la hoja de resultados se calculará para cada medida:
t
t0
y se ajustará frente al tiempo. Si el resultado es una recta (ecuación 11) quedará confirmada la
suposición realizada sobre los órdenes de reacción, y la pendiente permitirá calcular k.

Química Física Prácticas de Cinética de Reacción
40
APÉNDICE 3 Obtención de la ecuación cinética en función de las conductividades
Vamos a obtener expresiones para la conductividad de la disolución a tiempo 0, tiempo t y
tiempo ∞.en estos tres momentos, a partir de las ecuaciones (9) y (10), teniendo en cuenta que la
conductividad especifica de una disolución es la suma de las contribuciones de los electrolitos
presentes (despreciamos la contribución del agua). NaOH y AcNa son electrolitos fuertes, que se
encuentran totalmente disociados en iones en la disolución. En cambio, AcEt y EtOH son
electrolitos muy débiles, que apenas se disocian, y por tanto no contribuyen apreciablemente a la
conductividad de la disolución.
concentraciones conductividad
t=0 AcEt + NaOH → AcNa + EtOH c0 c0 0 0
(A1) 1000
11000
OH0Na00
OH0Na00
NaOH
NaOH00
cc
ccc
c
t=t AcEt + NaOH → AcNa + EtOH c0-x c0-x x x
(A2) x)(x1000
xxx
1
x)(x)(x)(
11000
x
1000
x)(
OH0AcNa0
NaAcAcNa
OH0Na00
NaOH
AcNaNaOH0
cc
ccc
c
t
t
t=∞ AcEt + NaOH → AcNa + EtOH 0 0 c0 c0
(A3) 1000
11000
Na0Ac0
Na0Ac00
AcNa
AcNa0
cc
ccc
c
Restando (A1)-(A2) y (A2)-(A3), obtenemos:
)x()(1000AcOH0 t
)x)(-()(1000AcOH0 ct
y dividiendo (A4)/(A5):
x
x
0
0
ct
t (A6)
Con lo que la ecuación 4 se convierte en:
t
ttkc 00 (A7)


















