
Plataformas de Proteómica
52
Instituto de Biotecnología Universidad Nacional Autónoma de México Curso: “Métodos Fisíco-Químicos en Biotecnología” Plataformas de Plataformas de Proteómica Proteómica Presentan: Ing. Daniela Morales Sánchez. Ing. Lilí Esmeralda Gallo Ramírez. 30 de Mayo 2006, Cuernavaca Morelos.
Transcript of Plataformas de Proteómica

Instituto de BiotecnologíaUniversidad Nacional Autónoma de México
Curso:
“Métodos Fisíco-Químicos en Biotecnología”
Plataformas de Plataformas de ProteómicaProteómica
Presentan:
Ing. Daniela Morales Sánchez.
Ing. Lilí Esmeralda Gallo Ramírez.
30 de Mayo 2006, Cuernavaca Morelos.

ÍNDICE
1. Introducción 1.1 Tecnología de la proteómica 1.1.1 Separación de proteínas 1.1.2 Identificación y caracterización de proteínas 2. Fundamentos de la electrofóresis 3. Tipos de electrofóresis 3.1 Electrofóresis libre 3.2 Electrofóresis zonal 3.2.1 Equipamiento de la electrofóresis 3.3 Tipos de soporte 3.3.1 Soportes no restrictivos tipo I 3.3.2 Soportes restrictivos tipo II 3.4 Factores que afectan la electroforesis 3.4.1 Campo eléctrico 3.4.2 Muestra 3.4.3 Tampón 3.4.4 Soporte 3.5 Tipos de electroforesis 3.5.1 Inmunoelectroforesis 3.5.2 Electroforesis en gel de poliacrilamida (PAGE) 3.5.2.1 Formación del gel de poliacrilamida 3.5.2.2 Tipos de monómeros 3.5.2.3 Catalizadores empleados en la formación del gel de
poliacrilamida 3.5.2.4 Sistemas de tampones que se emplean en la electroforesis en
geles de poliacrilamida 3.5.2.5 Formatos de la PAGE 3.5.2.6 PAGE en condiciones no desnaturalizantes 3.5.2.7 PAGE en condiciones desnaturalizantes 3.5.2.8 PAGE-SDS 3.5.3 Electroforesis en geles de gradientes 3.5.4 Electroforesis en geles de azarosa 3.5.5 Electroforesis capilar 3.5.6 Enfoque isoeléctrico 3.5.7 Electroforesis bidimensional 4. Información sobre la estructura de las proteínas 4.1 Fragmentación de proteínas 4.1.1 Proteólisis 4.1.1.1 Digestión en solución 4.1.1.2 Digestión en gel 4.1.2 Rompimiento químico 4.2 Análisis de secuencia amino y carboxilo terminal 4.2.1 Secuenciación del extremo amino terminal 4.2.1.1 Proteínas con modificaciones en el amino terminal 4.2.2 Secuenciación del extremo carboxilo terminal 5. Espectrometría de masas 5.1 Preparación de la muestra 5.2 Ionización de la muestra 5.2.1 MALDI (Matrix Assisted Lasser Desorption Ionization)
1 3 3 5 7 8 8 9 9
10 10 11 11 11 11 12 12 13 14 16 16 18
18
19 19 20 20 21 25 26 26 27 29 30 30 30 31 31 32 33 33 34 35 35 35 35 36

5.2.2 ESI (ElectroSpray Ionization) 5.3 Análisis de masa 5.3.1 Analizador de cuadrupolo 5.3.2 Analizalizador de tiempo de vuelo (Time of Flight, ToF) 5.3.3 Analizador de trampa de iones 5.3.4 Analizadores híbridos. 5.3.5 Analizadores ToF/ToF 5.3.6 FT-ICR (Fourier transform-ion cyclotron resonance) 5.3.7 Fragmentación de iones 6. Identificación de proteínas utilizando bases de datos. 6.1 Huella de masa del péptido (peptide mass fingerprint, PMF) 6.2 Secuenciación utilizando datos generados con la técnica product
ion scan MS/MS 6.3 Secuenciación de novo 7. Identificación de modificaciones post-traduccionales 7.1 Identificación de sitios de fosforilación 7.1.1 Enriquecimiento de fosfoproteínas 7.1.2 Determinación de sitios de fosforilación mediante degradación
de Edman 7.1.3 Determinación de sitios de fosforilación mediante
espectrometría de masas 8. Bibliografía
36 38 38 40 41 41 42 42 43 43 44
45 45 46 47 47
47
47 49

1
1. INTRODUCCIÓN Los rápidos avances conseguidos en la tecnología del DNA están permitiendo la secuenciación sistemática de los genomas de diversos organismos. Los proyectos de secuenciación a gran escala están proporcionando una ingente cantidad de secuencias de DNA. Sin embargo, aún se desconoce la función biológica de la mayoría de las proteínas codificadas por los genes detectados. Así pues, el siguiente paso en la era post-genómica debe ser el estudio funcional de todos estos genes. Se puede decir que hubo tres factores decisivos para el desarrollo de la proteómica:
• La secuenciación de genomas a gran escala y el desarrollo de bases de datos de proteínas.
• El desarrollo de técnicas de espectrometría de masas para analizar proteínas y péptidos.
• Los avances realizados en la separación de proteínas mediante 2D-PAGE con la introducción de los gradientes de pH inmovilizados (IPGs).
La proteómica es uno de los campos que puede ayudar a establecer una conexión
entre las secuencias genómicas y su comportamiento biológico, constituyendo una herramienta importante en el análisis funcional de genes de función desconocida.
El término “Proteoma” fue usado por vez primera en 1995 para describir el
conjunto de PROTEínas de un genOMA, en una célula o un tejido. De forma imperceptible, la palabra proteoma dio lugar a una nueva disciplina, la “Proteómica”. Pero, ¿qué es la proteómica? Esencialmente la proteómica es el estudio a gran escala de los productos génicos de un genoma mediante métodos bioquímicos, con el fin de obtener una visión global integrada de los procesos celulares. El término proteómica se ha asociado tradicionalmente con la separación de un gran número de proteínas de una célula u organismo mediante 2D-PAGE. Según esto, la proteómica comienzo en los años setenta cuando se empezaron a construir bases de datos de proteínas utilizando la electroforesis bidimensional. Sin embargo, la identificación de las proteínas era difícil debido a la falta de métodos analíticos rápidos y sensibles para la caracterización de las proteínas. En los años noventa la espectrometría de masas surge como un método analítico muy poderoso, ya que limita la mayoría de las limitaciones del análisis de proteínas. Este desarrollo, junto con la disponibilidad de los genomas secuenciados marca el comienzo de una nueva era. Actualmente muchas áreas de estudio han sido agrupadas dentro de la proteómica. Se pueden incluir, entre otros, los estudios de interacciones de proteínas, de modificaciones pos-traduccionales, el análisis funcional de las proteínas y estudios de localización.
Se puede hablar de dos tipos de proteómica: proteómica de expresión y
proteómica del mapa celular. La proteómica de expresión es el estudio cuantitativo de la expresión de proteínas
entre muestras que difieren en alguna variable. En esta estrategia se compara la expresión del proteoma total o de subproteómas entre diferentes muestras. La información obtenida puede permitir la identificación de nuevas proteínas implicadas

2
en transducción de señales, la identificación de proteínas específicas de una enfermedad y proteínas de interés en microbiología médica.
La proteómica del mapa celular o estructural es el estudio de la localización
subcelular de las proteínas y de las interacciones proteína-proteína, mediante la purificación de orgánulos o complejos y la posterior identificación de sus componentes mediante espectrometría de masas.
También se utiliza el término de proteómica funcional para referirse a diversas
aproximaciones proteómicas que permiten el estudio y caracterización de un grupo de proteínas determinado proporcionando información importante sobre señalización, mecanismos de la enfermedad o interacciones proteína-fármaco.
Fig. 1. Estrategias utilizadas en proteómica para la identificación y el análisis de proteínas. Siglas: (SEC) Size-exclusion chromatography; (IEX) ion-exchange chromatography; (HIC) hidrophobic interaction chromatography; (FFE) free flow electrophoresis; (CZE) capillary zone electrophoresis; (FRET) flourescence resonance energy transfer.
Para poder desarrollar los ambiciosos objetivos de la proteómica se requiere la
implicación de diversas disciplinas como biología molecular, bioquímica, microbiología y bioinformática. Siendo esta ultima de enorme importancia, ya que será necesario desarrollar ordenadores con una gran capacidad para organizar la gran cantidad de información que se generara en estas investigaciones.
Para caracterizar un proteoma de una célula es importante tener en cuenta que el
proteoma es dinámico y que es reflejo del medio ambiente en el que es estudiado.
Separación de Proteínas
Mapeo de Péptidos
Identificación
Caracterización
Cromatografía: RP-HPLC,
afinidad, IEX, SEC, HIC
Fragmentación de Proteínas: en gel vs. en solución • Enzimática: tripsina, V8, quimiotripsina, pepsina, etc. • Química: CNBr, hidrólisis ácida, etc.
Análisis de secuencias Amino y Carboxilo terminal
Modificaciones posttraduccionales:
fosforilación, glicosilación, metilación, acetilación
Técnicas Electroforéticas: E-2D, E-1D, FFE,
CZE
Aproximaciones de afinidad • Inmunoblot • Captura por afinidad (E-1D
y E-2D)
Mapeo de péptidos
Espectrometría de masas • Peptide mass
fingerprinting MS/MS
Modificaciones covalentes Uniones por enlaces disulfuro
Proteólisis
Conformación de proteínas
Presencia de ligandos Localización celular in vivo, inmunolocalización por FRET
Variantes de empalme
alternativo Pr
oteó
mic
a Es
truc
tura
l Pr
oteó
mic
a Fu
ncio
nal
Reducción y Alquilación

3
Como respuesta a estímulos externos e internos, las proteínas pueden ser modificadas post-traduccionalmente, translocadas, sintetizadas o degradadas (Fig. 2).
Fig. 2 Mecanismos por los que un gen puede dar lugar a múltiples productos génicos.
1.1 Tecnología de la proteómica El desarrollo de la proteómica se debe en parte a los avances importantes realizados en la tecnología de proteínas. Sin embargo, la metodología disponible tiene sus limitaciones y actualmente todavía no es posible realizar muchos tipos de proteómica. Es necesario mejorar algunas de estas limitaciones y desarrollar nuevas tecnologías para conseguir el máximo partido. Las cuatro plataformas de la tecnología proteómica son:
• Preparación y manejo de la muestra
• Determinación de la información de la secuencia parcial de aminoácidos
• Identificación y cuantificación de proteínas
• Mapeo celular 1.1.1 Separación de proteínas La tecnología mas usada para la separación de proteínas es la electroforesis en geles de poliacrilamida. Esta técnica es la más eficaz para resolver mezclas complejas de proteínas. Para muchas aplicaciones proteómicas, la electroforesis en una dimensión es el método de elección. Las proteínas se separan de acuerdo a su masa y como las proteínas son solubilizadas en dodecil sulfato sódico (SDS), no suele haber problemas de solubilización. Es una técnica sencilla, reproducible y permite la separación de proteínas de 10-300 KDa. Una de las aplicaciones mas comunes de la 1-DE es la caracterización de proteínas después de realizar algún tipo de purificación previa. La electroforesis bidimensional 2D-PAGE permite separar hasta miles de proteínas en un solo experimento, y constituye actualmente el método más eficiente para la separación de mezclas muy complejas de proteínas. Esta basada en una separación de proteínas en función de la carga, seguida de una separación de las proteínas en función de su masa molecular. La separación de la primera dimensión se

4
realiza mediante isoelectroenfoque, durante el cual las proteínas son separadas en un gradiente de pH hasta alcanzar una posición en la que su carga neta es cero, es decir, su punto isoeléctrico. En una segunda dimensión, las proteínas son separadas mediante electroforesis en presencia de SDS. La alta resolución de la técnica se basa en que las dos separaciones se basan en parámetros independientes. La innovación clave para la 2D-PAGE fue el desarrollo de geles con un gradiente de pH inmovilizado (IPG). El gradiente de pH inmovilizado elimina los problemas de inestabilidad del gradiente y baja capacidad de carga que iban asociados a los gradientes de pH preparados con anfolitos acarreadores. En los geles IPG, el gradiente es generado por las llamadas “inmobilinas” y está copolimerizado con la matriz de acrilamida del gel. Este sistema ha permitido mejorar la resolución y reproducibilidad de los geles así como aumentar la cantidad de proteína que puede ser cargada. La reproducibilidad conseguida con los IPGs ha hecho posible la comparación de mapas entre distintos laboratorios, facilitando así el intercambio de información. En cuanto a la detección de proteínas, tradicionalmente se ha venido empleando el marcaje radioactivo o la tinción con azul de de Coomassie, o con plata, para conseguir mayor sensibilidad. También se ha desarrollado un método de tinción de plata superficial compatible con la digestión proteica y la espectrometría de masas, aunque el umbral de detección no es tan sensible como el seguido con los protocolos de tinción de plata mas utilizados. Así mismo, se han comenzado a utilizar tinciones y marcajes fluorescentes (Sypro-Ruby, Cy3, Cy5…) que presentan un sensibilidad comparable a la plata y también permiten el análisis posterior de las proteínas mediante espectrometría de masas. También se han desarrollado programas para comparar las imágenes de 2D-PAGE y facilitar la identificación y cuantificación de manchas de proteínas entre diferentes muestras. La aplicación principal de la 2D-PAGE es la proteómica de expresión. En esta aproximación, la expresión de proteínas de dos muestras se puede comparar de forma cuantitativa. La aparición o desaparición de manchas proporciona información sobre la expresión diferenciadle proteínas y la intensidad de las manchas permite conocer los niveles de expresión. Para realizar estos estudios se pueden utilizar organismos completos, líneas celulares o fluidos biológicos. Se pueden comprar tejidos normales con tejidos enfermos, o células tratadas con drogas o diferentes estímulos. Otra aplicación importante de la proteómica del mapa celular, donde la 2D-PAGE se utiliza para hacer mapas de proteínas de microorganismos, orgánulos celulares y complejos de proteínas. También se puede utilizar para caracterizar proteínas en subproteomas que se han obtenido mediante alguna forma de purificación del proteoma. La 2D-PAGE también presenta muchas limitaciones. Es una técnica muy laboriosa que requiere bastante tiempo, y difícil de automatizar. La 2D-PAGE esta limitada por el número y el tipo de proteínas a resolver. Las proteínas muy grandes o hidrofóbicas no entran en el gel durante la primera dimensión y las proteínas muy ácidas o muy básicas no se resuelven bien. Algunos de estos problemas se pueden resolver mediante fraccionamiento, la utilización de IPGs con diferentes rangos de pH. Otra limitación importante de la 2D-PAGE es la detección de proteínas poco abundantes, algunas de ellas se consideran muy importantes (proteínas regulatorias, proteínas implicadas en la transducción de señales, receptores). En estos estudios es necesario realizar un fraccionamiento de la muestra para reducir la complejidad de los

5
extractos. Debido a estas limitaciones, la mayor aplicación de esta técnica en el futuro será el análisis y caracterización de subproteomas y de complejos proteicos.
Las limitaciones de la 2D-PAGE han impulsado el desarrollo de otras
metodologías. Una estrategia desarrollada consiste en digerir con tripsina una mezcla de proteínas para después purificar y analizar los péptidos mediante MS. Los péptidos se pueden purificar mediante cromatografía liquida, electroforesis capilar o mediante una combinación de técnicas como cromatografía de intercambio iónico y cromatografía de fase reversa. La ventaja de este procedimiento es que se dispone de una gran cantidad de proteínas. Sin embargo, el análisis de los datos requiere una enorme cantidad de tiempo y ordenadores muy potentes. Además se puede perder mucho tiempo y esfuerzo en el análisis de proteínas que no tienen interés. 1.1.2 Identificación y caracterización de proteínas En los proyectos proteómicos la identificación de proteínas es esencial. Es el primer paso para otros estudios que suponen en última instancia la caracterización funcional. Además, en el caso de los geles bidimensionales, la identificación de las manchas conduce a la creación de mapas de referencia, que definen las proteínas expresadas por unos organismos o tejido en unas condiciones determinadas. Las proteínas pueden ser identificadas por diversos procedimientos, entre los que se incluyen la secuenciación del extremo N-terminal, detección con anticuerpos específicos, composición de aminoácidos, co-migración con proteínas conocidas, y sobre-expresión y deleción de genes. Todos estos métodos generalmente son lentos, laboriosos, o caros, y por tanto no resultan apropiados para su utilización como estrategias a gran escala. Debido a su rapidez y elevada sensibilidad, la espectrometría de masas se ha convertido en el método de detección para la identificación de proteínas a gran escala, el primer paso para el estudio de proteoma de distintos organismos. También permite la caracterización de modificaciones post-traduccionales que presentan relevancia fisiológica, tales como la glicosilación y la fosforilación. La estrategia general empleada para la identificación a gran escala de proteínas mediante espectrometría de masas se muestra en la Fig. 3.
El análisis de las proteínas mediante espectrometría de masas ha sido posible gracias al desarrollo de varios métodos de ionización suave para convertir biomoléculas grandes, polares y no volátiles en iones en fase gaseosa.
Los espectrómetros de masa están formados al menos por una fuente de iones,
un analizador de masas y un detector que mide la relación masa/carga (m/z) de los iones en fase gaseosa. Esta técnica tan robusta implica: 1.- La conversión de los péptidos en iones en fase gaseosa mediante técnicas de ionización suave, como ionización desorción con láser asistida con matriz (MALDI) a partir de una muestra en estado sólido, o la ionización mediante electrospray (ESI) de una muestra en solución.

6
2.- Separación de los iones según su m/z en un analizador de masas (por ejemplo un analizador tipo ToF (Time of Flight), cuadrupolo, trampa iónica, etc.) 3.- Fragmentación opcional de los iones peptídicos. 4.- Medida de las masas en un detector obteniendo un espectro de masas que refleja la abundancia de los iones frente a su valor m/z.
Fig. 3 Estrategia para la identificación de proteínas mediante espectrometría de masas (Pitarch et al. 2003).
Recientemente se han desarrollado también fuentes de MALDI que se acopan a un analizador QToF o a dos analizadores ToF en tándem (MALDI-ToF/ToF).
Para la identificación de proteínas se han desarrollado diversas estrategias, de
las cuales, dos se encuentran representadas esquemáticamente en la Fig. 4:
• Identificación mediante huella peptídica (PMF: peptide mass fingerprinting) o mapeo peptídico utilizando un espectrómetro tipo MALDI-TOF.
• Identificación mediante fragmentación de péptidos obteniendo la secuencia
total o parcial de los aminoácidos (etiqueta de secuencia) utilizando un espectrómetro de masas en tándem.

7
Fig. 4 Dos nuevas aproximaciones para la detección y cuantificación diferencial de proteínas. A. ICAT (isotope coded affinity tag). B. DIGE (Differential gel electrophoresis). (Pitarch et al. 2003).
En esta revisión sobre tecnología proteómica pretende solamente resaltar algunos aspectos de interés, especialmente de la separación de proteínas mediante electroforesis y del análisis de éstas mediante espectrometría de masas (MS) señalando las aproximaciones para la identificación y caracterización. 2. FUNDAMENTOS DE LA ELECTROFORESIS
La electroforesis es la migración de solutos iónicos bajo la influencia de un campo eléctrico; estas partículas migran hacia el cátodo o ánodo (electrodos - y +), en dependencia de una combinación de su carga, peso molecular y estructura tridimensional.
Es de destacar que a escala analítica, los métodos electroforéticos son de alta sensibilidad, poder de resolución y versatilidad, y sirven como método de separación de mezclas complejas de ácidos nucleicos, proteínas y otras biomoléculas, donde aportan un potente criterio de pureza. Se pueden conocer también mediante estas técnicas, las características ácido-básicas de las proteínas presentes en un extracto crudo, lo que da la información necesaria si se pretende realizar una separación cromatográfica basada en diferencias de carga. Es útil además para determinar otros parámetros como peso molecular, punto isoeléctrico y número de cadenas polipeptídicas de las proteínas.

8
La velocidad de migración (v) de la partícula es directamente proporcional al producto de su carga efectiva (q) y el gradiente de potencial eléctrico (E) e inversamente proporcional al coeficiente de fricción (f) relativo a la talla y forma de la molécula, o sea, a la resistencia que le ofrece el medio.
V = q E / f
La movilidad electroforética (Me) es un caso particular de la velocidad de migración de un ión, cuando se aplica un campo eléctrico de 1 V/cm. Su signo es igual al de la carga de la partícula.
La velocidad de migración electroforética depende de la densidad de carga de la molécula (relación carga / peso), del voltaje aplicado y de la porosidad del gel de electroforesis. El voltaje no se puede incrementar indiscriminadamente porque se genera un excesivo calor. En contraste, al aplicar bajo voltaje puede ocurrir una pobre separación, por causa de la difusión por tiempo muy prolongado de la corrida electroforética.
La mayoría de las biomacromoléculas (proteínas, ácidos nucleicos y polisacáridos) poseen determinada carga eléctrica con grupos aniónicos y catiónicos capaces de disociarse. La carga neta de la partícula está dada por el pH del medio y puede ser modificada por la interacción con pequeñas moléculas de iones u otras macromoléculas. De lo anterior se deduce que el pH influye sobre la velocidad de migración de las moléculas.2 En el punto isoeléctrico de la biomolécula, pH al cual su carga neta es 0, esta no migra. Por debajo del punto isoeléctrico tiene carga neta positiva y migra hacia el cátodo, y por encima del punto isoeléctrico tienen carga neta negativa y migra hacia el ánodo.
3. TIPOS DE ELECTROFORESIS
Existen dos modalidades de electrofóresis:
3.1 Electrofóresis libre
El campo eléctrico se aplica a disoluciones o suspensiones (Fig. 5). Históricamente, es el origen de la electroforesis tal y como hoy la conocemos y fue desarrollada por Arne Tiselius en 1937. Actualmente está en desuso, ya que al ser un fluido el medio en el que se desarrolla, tiene poco poder de resolución.

9
Fig. 5 Las moléculas cargadas migran hacia los electrodos, aunque no bien separadas por tratarse de una disolución.
3.2 Electroforesis zonal
Es una de las técnicas más utilizadas en bioquímica y biología molecular por su elevada resolución. El campo eléctrico se aplica a un medio o soporte estabilizante (en vez de a un fluido) y la muestra se deposita en una reducida zona del mismo (Fig. 6).
Fig. 6 Electroforesis de zona sobre un soporte.
Son útiles para lograr la separación de componentes de mezclas complejas. Se aplican pequeñas cantidades de la disolución de proteínas a un soporte sólido, que se impregna con una solución de tampón. Los soportes son en general polímeros y forman un gel poroso que restringe el movimiento de las moléculas a través del medio durante la electroforesis y disminuyen los flujos de convección del solvente. Como soportes han sido utilizados papel (celulosa), almidón, poliacrilamida, agarosa, acetato de celulosa, etcétera.
Este método tiene gran poder resolutivo porque se aplica una cantidad pequeña de proteína a una zona estrecha, mientras que la longitud del trayecto es mucho mayor que la zona de aplicación. El equipamiento requerido es simple (fuente de poder y cubeta vertical u horizontal donde se coloca el soporte y 2 electrodos). Con el desarrollo de la ciencia se han diseñado equipos automatizados de electroforesis como el PhastSystem.
3.2.1 Equipamiento de la electrofóresis
Para llevar a cabo una separación electroforética zonal se necesitan los elementos mostrados en la Fig. 7:
• Fuente de alimentación. Proporciona el campo eléctrico mediante dos electrodos, uno positivo (ánodo) y otro negativo (cátodo) entre los que se establece una diferencia de potencial.
• Cubeta. Recipiente en cuyos extremos se sitúan los electrodos.
• Soporte electroforético.

10
Fig. 7 Equipo electroforético. La cubeta tiene en sus extremos los reservorios para el tampón de electroforesis. En el centro se aloja el soporte (gel) con la muestra aplicada en su zona central.
3.3 Tipos de soporte
El elemento fundamental es el soporte. Aunque todas las electroforesis responden a un mismo principio, las de zona pueden ser de distintos tipos en función del soporte que se utilice. En todos los casos, la muestra se dispone inicialmente en una pequeña zona del soporte, y posteriormente lo va recorriendo impulsado por el campo eléctrico. Para que esta situación sea estable, tanto al comienzo como a lo largo del proceso de separación, es necesario contrarrestar el efecto de la difusión y las posibles corrientes de convección. Para ello se utilizan reticulados moleculares (el soporte de la electroforesis), que forman un entramado tridimensional que impide o reduce dicha difusión y permite la entrada de agua en sus poros; el tamaño de estos poros debe permitir el paso de moléculas voluminosas como son los ácidos nucleicos y las proteínas. Además, el soporte debe ser inerte, es decir, no debe tener cargas que interfieran con la migración de las moléculas de la muestra.
Los diferentes tipos de soportes se clasifican en dos grupos:
3.3.1 Soportes no restrictivos tipo I
Tamaño de poro no opone impedimento al paso de las moléculas. El agar es el ejemplo más representativo y se obtiene a partir de algas marinas y esta formado por dos tipos de polisacáridos: la agarosa y la agaropectina; ésta última posee varios grupos cargados (grupos carboxilo y sulfato), lo que la hacen inadecuada como soporte electroforético por el efecto electroendosmótico que produce. La agarosa (Fig. 8) se utiliza ampliamente sobre todo para la separación de ácidos nucleicos y para el análisis de proteínas. Los diferentes tipos de agarosas se caracterizan por su punto de fusión (entre 35-95°C), su resistencia física y el grado de electroendósmosis.
Fig. 8 Estructura química de la agarosa, polisacárido lineal formado por la repetición de la estructura
básica (agarobiosa), con un peso molecular medio de 120kDa (que corresponde a unas 400 unidades). Las características de la agarosa se ven modificadas dependiendo de la naturaleza de los diferentes radicales
R.

11
3.3.2 Soportes restrictivos tipo II
El tamaño de poro opone resistencia al paso de las moléculas. El ejemplo característico son los geles de poliacrilamida, que no sólo evitan la convección y minimizan la difusión, sino que además participan en el proceso de separación. Estos geles son medios porosos en los que el tamaño de poro es similar al de las proteínas, de tal modo que existe un efecto de tamizado molecular y la separación electroforética depende de la densidad de carga de las moléculas y de su tamaño. 3.4 Factores que afectan la electrofóresis
El resultado de una electroforesis depende de numerosos factores y es necesario conocerlos para lograr la máxima resolución. 3.4.1 Campo eléctrico
Es un gradiente de potencial que posibilita que haya electroforesis. A su vez, depende de diferentes parámetros:
• Diferencia de potencial (V). Se mide en voltios y es lo que define el campo eléctrico. Cuanto mayor sea, mayor será la velocidad de migración. Las electroforesis se consideran de bajo voltaje si se realizan entre 10-500 voltios (la mayoría) y de alto voltaje si se realizan entre 500-10,000 voltios.
• Intensidad (I). Se mide en amperios y cuantifica el flujo de carga eléctrica. Esta
relacionada con la diferencia de potencial por la Ley de Ohm: V=IR
y para una diferencia de potencial dada, su valor (normalmente entre 5-50mA) viene determinado por la resistencia del soporte, por lo que, en última instancia, da cuenta de la distancia recorrida por las moléculas.
• Resistencia (R). Se mide en ohmios y cuanto mayor sea la resistencia del soporte, menor será la movilidad electroforética (ya que la intensidad ha de ser menor para que se cumpla la Ley de Ohm). Depende de la naturaleza del soporte, sus dimensiones (anchura, longitud y sección) y de la concentración del tampón de electroforesis.
• Temperatura. Por el efecto Joule, el paso de una corriente eléctrica produce
calor, y este efecto será tanto mayor cuanto mayor sea la diferencia de potencial y la resistencia del sistema. Debe controlarse de forma estricta, puesto que puede afectar a la muestra (desnaturalizándola), al soporte e incluso al equipo electroforético. Todo esto justifica, por sí solo, la necesidad de un sistema de refrigeración en las cubetas de electroforesis.
3.4.2 Muestra
La movilidad electroforética de una molécula es tanto mayor cuanto mayor sea su carga y disminuye según aumenta su tamaño, ya que mayor es la fricción de la molécula con el medio y menor es su carga por unidad de superficie. Por esta razón, las moléculas globulares se mueven con mayor facilidad que las elongadas, ya que la esfera presenta la mínima superficie a igualdad de volumen. Así, moléculas del mismo

12
tamaño y carga, pero de diferente forma, pueden tener una movilidad electroforética diferente y ser, por tanto, separables mediante una electroforesis. 3.4.3 Tampón
Durante una electroforesis, se produce una electrolisis del agua por acción del campo eléctrico, lo cual genera protones en la proximidad del ánodo e iones hidroxilo en el cátodo. El tampón es, por esta razón, esencial durante la electroforesis para evitar que el ánodo se acidifique y el cátodo se haga más básico, pero no debe afectar a las moléculas a separar.
Además, la fuerza iónica condiciona la movilidad electroforética de las substancias
a separar, ya que influye en su esfera de solvatación y en la conductividad de todo el sistema, por lo tanto, es necesaria una fuerza iónica suficiente para mantener el pH y la conductividad durante todo el proceso, pero no muy elevada, ya que llegaría a interferir en el sistema.
Normalmente, se emplean tampones con fuerzas iónicas moderadas (0.05-0.10M),
siendo el pH la característica que debe ser más controlada, puesto que determina la carga de la muestra.
Conviene señalar también que atendiendo a la distribución del tampón, los
sistemas se clasifican en continuos cuando se emplea un único tampón en el proceso, y discontinuos cuando se emplean, al menos, dos tampones de pH y composición diferentes. 3.4.4 Soporte La simple presencia del soporte en una electroforesis hace que se presenten una serie de fenómenos de elevada trascendencia para el resultado del proceso:
• Adsorción. Es una retención inespecífica de las moléculas de la muestra sobre el soporte, por lo que afecta negativamente a la resolución, ensanchando las bandas de migración.
• Electroendósmosis (EEO). Es un fenómeno que se da en algunos soportes
(sobre todo en los no reductivos tipo I), en los que aparecen cargas negativas, debido a grupos carboxilo o sulfato, al estar en contacto con una disolución iónica. Al aplicar el campo eléctrico, los iones y las moléculas se desplazan según su carga, pero en su migración se encuentran con un flujo de cationes desplazándose sobre la superficie del soporte y otro de aniones haciéndolo por el centro de los poros (Fig. 9). Este flujo orientado se llama flujo electroendosmótico. El EEO va en contra de la resolución de una electroforesis, ya que las bandas de la muestra se ensanchan y se distorsionan; sin embargo, la EEO contribuirá a una mejor resolución en las inmunoelectroforesis y en la electroforesis.

13
Fig. 9 Efecto electroendosmótico en la superficie de un soporte cargado negativamente. Los aniones migran desplazándose por el centro del poro, mientras que los cationes lo hacen por la superficie.
• Tamizado molecular. Es un efecto debido al mayor o menor reticulado de los
soportes; cuanto más tupida sea la red de fibras, menores serán los poros. El movimiento de las moléculas a través del soporte es más o menos fácil en función de su tamaño respecto al de los poros; aquellas moléculas cuyo tamaño se aproxime al del poro, verá impedido su avance respecto a aquellas cuyo tamaño sea muy inferior. Por esta razón, el soporte actúa como un cedazo que separa o tamiza las moléculas en función de su tamaño.
3.5 Tipos de electrofóresis
Las proteínas son moléculas cuya carga neta depende del contenido de una serie de aminoácidos (fundamentalmente ácido glutámico, ácido aspártico, lisina, arginina e histidina) y del grado de ionización de éstos al pH considerado (Fig. 10 ).
Fig. 10 Principales aminoácidos responsables de la carga neta de una proteína, dependiendo del pH.
En los soportes no restrictivos (en los que el entramado no interfiere en la
migración, ya que el tamaño de las proteínas es muy pequeño en comparación con el tamaño de poro del soporte), la separación depende de la densidad de carga de las moléculas y, así, cuanto mayor sea la densidad, mayor será la velocidad de migración en un campo eléctrico hacia el polo que determine su carga neta.
La primera etapa del proceso es la aplicación de la muestra. En papel o acetato
de celulosa, esto se puede efectuar de forma puntual (permite el análisis de varias muestras simultáneamente en pequeñas cantidades) o longitudinalmente (permite el estudio de una sola muestra pero en mayor cantidad). La muestra se aplica disuelta en el tampón de electroforesis, del que está impregnado el soporte y que se encuentra en los reservorios de la cubeta, y se deposita en una pequeña zona, lo más estrecha posible, en el centro o en un extremo del soporte (si se conoce cuál va a ser la dirección

14
de desplazamiento de la misma) y de forma perpendicular a la dirección del campo eléctrico. Conviene evitar la proximidad de los bordes del papel, ya que allí el campo eléctrico no es homogéneo y se distorsionan las bandas según avanzan. El volumen que se aplica suele ser inferior a 10 µL y si se necesitan mayores volúmenes porque la muestra se encuentre muy diluida, pueden hacerse aplicaciones sucesivas sobre la misma zona, dejando secar entre aplicación y aplicación. Es necesario humedecer el soporte (papel o acetato de celulosa) para que sea conductor; para ello, se impregna el soporte de tampón de electroforesis por ambos extremos y de forma simultánea para que por capilaridad se desplace hacia la zona de aplicación de la muestra.
En los geles de agarosa, se perfora el gel con ayuda de un troquel o una pipeta
de diámetro adecuado y se elimina esa porción por succión. La perforación puede ser cilíndrica o rectangular, dependiendo del número y cantidad de muestra a analizar y la muestra se deposita en el orificio practicado sin que llegue a rebosar. En este caso, el tampón está embebido en el soporte (agarosa).
En todos los casos, la zona de aplicación debe ser lo más estrecha posible, para
aumentar la resolución y evitar solapamientos entre bandas que migren en posiciones cercanas.
La electroforesis termina cuando se haya producido la máxima separación de
los componentes de la muestra, pero sin sobrepasar los límites del soporte. Para evitar que se sobrepasen estos límites, se utilizan los marcadores electroforéticos que son moléculas coloreadas con una movilidad electroforética superior a cualquier componente de la muestra (poseen elevada carga respecto a su tamaño). Entre los marcadores más utilizados se encuentran la dinitrofenil-lisina (DNP-Lys) y el azul de bromofenol.
Una vez acabada la electroforesis, se procede a la etapa de tinción. El soporte se
retira de la cubeta y se sumerge en un recipiente que contiene una disolución de un colorante que se une de manera específica a los componentes de la muestra y que precipita a los componentes separados en la posición en la que se encuentren. Las disoluciones más utilizadas en el caso de proteínas son el Negro Amidón al 1% (p/v) en ácido acético y el Azul de Coomassie al 0.1% (p/v) en metanol/agua/ácido acético (9:9:2 v/v/v).
Si la electroforesis se pretende desarrollar a escala preparativa, ha de utilizarse un
papel de mayor grosor (175 gr/cm2), en lugar del utilizado en la electroforesis analítica (85-100 gr/cm2; 0.15mm de espesor), lo que permite la aplicación de una mayor cantidad de muestra (50-100µL).
3.5.1 Inmunoelectrofóresis
Es una combinación de una electroforesis en geles de agarosa seguida de una inmunodifusión (Fig. 11). En la primera parte, se realiza una aplicación puntual de la muestra, por lo que al final las distintas proteínas estarán distribuidas a lo largo del gel según el eje de migración. Acabada la electroforesis, y sin efectuar la tinción, se practica en el gel un canal (“trinchera”) paralelo a la dirección de migración con un bisturí de doble hoja (la separación de las hojas determina la anchura de la trinchera) en el que se deposita un antisuero que contenga anticuerpos específicos frente a una o varias de las proteínas separadas en la electroforesis.

15
Los geles se mantienen así a temperatura ambiente (20-22°C) durante 24-48 horas.
Los anticuerpos y las proteínas (que actúan como antígenos) difunden, y si se encuentran en la proporción adecuada, producen complejos antígeno-anticuerpo insolubles que precipitan y aparecen en el gel como bandas elipsoidales.
Estos complejos solamente se producen cuando ambos compuestos se hallan en lo
que se conoce como relación de equivalencia y que no son idénticas para todas las proteínas presentes en la muestra; por eso, deben determinarse en experimentos preliminares las cantidades idóneas de antisuero y de la muestra, así como las distancias entre el pocillo de aplicación de la muestra y la trinchera, ya que si las distancias son muy pequeñas, el antígeno (las proteínas) difunde rápidamente y puede no formarse el precipitado, y si la distancia es mayor de la idónea, hay que emplear mayor cantidad de antisuero y la duración del proceso se alarga.
Una vez formadas las bandas de precipitación, el gel puede lavarse para eliminar
las proteínas que no hayan inmunoprecipitado y, posteriormente, teñirse como se ha descrito anteriormente. Cada proteína de la muestra produce una banda de precipitación independiente, por lo que es posible identificar el número de constituyentes de la muestra que son reconocidos por los anticuerpos.
La gran especificidad y sensibilidad de las reacciones de precipitación, permite
diferenciar substancias con movilidades electroforéticas idénticas y detectar componentes que se encuentren en concentraciones mínimas (µg).
Fig. 11 Fases de una inmunoelectroforesis (IEF). El número de bandas observado en una IEF debe entenderse como el número
mínimo de componentes presentes, es decir, puede haber más componentes que no sean detectados por el antisuero empleado o que no hayan alcanzado la relación de equivalencia antígeno-anticuerpo adecuada.
La IEF es una técnica principalmente cualitativa, que se desarrolla en condiciones
nativas y es especialmente apropiada para el análisis de líquidos fisiológicos y extractos celulares. Permite identificar substancias en mezclas muy complejas y en concentraciones muy bajas. Sin embargo, requiere que dichas substancias sean inmunogénicas y depende de los anticuerpos utilizados, por lo que es difícil de estandarizar.

16
3.5.2 Electrofóresis en gel de poliacrilamida (PAGE)
La poliacrilamida es un soporte empleado frecuentemente en electroforesis en gel, es químicamente inerte, de propiedades uniformes, capaz de ser preparado de forma rápida y reproducible. Forma, además, geles transparentes con estabilidad mecánica, insolubles en agua, relativamente no iónicos y que permiten buena visualización de las bandas durante tiempo prolongado. Además tiene la ventaja de que variando la concentración de polímeros, se puede modificar de manera controlada el tamaño del poro.3 3.5.2.1 Formación del gel de poliacrilamida
Los geles de poliacrilamida se forman por la polimerización vinílica del monómero acrilamida y del monómero entrecruzador N, N'-metilen-bis-acrilamida (Fig. 12). La polimerización se inicia con la formación de radicales libres del monómero, que se producen por radicales libres de oxígeno, por causa de la acción de iones persulfato. Las aminas terciarias como el N, N, N, N´-tetrametilen-diamina (TEMED) se emplean como catalizadores de esta reacción, porque causan la formación de radicales libres del persulfato. Esta reacción es fuertemente inhibida por altos niveles de oxígeno, por lo que la solución debe ser desgasificada para lograr una formación de gel reproducible. Hay muchos factores que desempeñan una función importante en la separación electroforética, como son pH, fuerza iónica, gradiente de potencial, tiempo de corrida, concentraciones de acrilamida y bis-acrilamida, etc. Las condiciones óptimas de una buena separación, en la práctica se determinan experimentalmente, con el análisis de cómo influyen los diferentes factores en la electroforesis en cuestión. La naturaleza de la muestra sirve de guía para alcanzar las condiciones en la que se deben obtener los mejores resultados.
Durante la polimerización del gel a temperaturas inferiores a 17 ºC, la iniciación del control catalítico y el tiempo de elongación de la cadena comienzan a ser diferencialmente controlables.
Fig. 12 Estructura de la acrilamida, bisacrilamida y poliacrilamida
La concentración de acrilamida (% T) representa el porcentaje en peso del
monómero total empleado (acrilamida + entrecruzador en gramos por 100 mL) y determina la longitud promedio de la cadena del polímero. La concentración de bis-

17
acrilamida (% C) representa el porcentaje de este monómero en el gel y determina el grado de entrecruzamiento.
La estructura del gel comienza a ser evidente a 4 % T; 0,2-5 % C. Los factores que
gobiernan la talla del poro del gel son complicados, pero en general el tamaño del poro disminuye con el incremento del % T. Las proporciones en que ambas se encuentran determinan las propiedades físicas del gel como su densidad, elasticidad, resistencia mecánica y el tamaño del poro. En general se recomiendan valores de T de 5 a 15 % y de C de 2 a 4 %. Para separar moléculas por tamaño, es necesario tomar en cuenta la relación entre el poro efectivo del medio y el tamaño de la molécula que se pretende separar. Si el poro del medio es significativamente mayor que la molécula, la separación electroforética será sobre la base de diferencias de carga y el tamizaje molecular será mínimo. Al aplicar muestras biológicas en un gel con gradiente de acrilamida, todas las macromoléculas comienzan su migración a bajo porcentaje y en la medida en que avanzan se encuentran con poros más pequeños que retardan su movimiento. Las moléculas menores comienzan a tener alta fricción cuando los poros son más pequeños, mientras que las grandes comienzan la fricción en los poros de mayor tamaño.
Al polimerizar la acrilamida, adquiere la forma del recipiente, por lo que es
necesario un molde para preparar el gel; los hay de dos tipos: • Tubo. Consiste en un tubo de vidrio de dimensiones variables, aunque
normalmente es de 15-20 cm de largo × 0.5-0.8 cm de diámetro interno, abierto por los extremos. Se tapa el orificio inferior y se rellena con la disolución de polimerización sobre la que se deposita una pequeña cantidad de butanol (con esto se evita que al polimerizar el gel forme menisco y se excluye el oxígeno que interfiere en la polimerización). Una vez formado el gel, se elimina el butanol y se retira la tapa inferior del tubo. En la actualidad, la utilización de PAGE en tubo ha quedado restringida a la primera dimensión en electroforesis bidimensionales, PAGE preparativa y algunos casos muy específicos de análisis de proteínas radioactivas.
• Placa. Es la forma más habitual en la que se desarrolla la PAGE. Puede llevarse
a cabo en posición horizontal o vertical (dependiendo del tipo de cubeta empleada), aunque lo más habitual (sobre todo para la separación de proteínas) es el modo vertical (Fig. 13). Si se aumenta la longitud del gel, aumenta la resolución de la electroforesis, aunque también se incrementa su duración. Asimismo, cuanto mayor sea la anchura del gel, mayor será el número de pocillos y, por lo tanto, el de muestras que se pueden analizar simultáneamente y en idénticas condiciones. El espesor del gel y las dimensiones de los pocillos viene determinado por el grosor de los espaciadores y el tamaño del peine determina la cantidad de muestra que puede aplicarse.
Una modalidad de la electroforesis vertical es la realizada con geles en gradiente en
los que la concentración de acrilamida (%T) aumenta progresivamente desde la parte superior del gel hasta la inferior, es decir, el tamaño de poro decrece de forma inversa. Este gradiente de concentración de acrilamida puede ser lineal o no (cóncavo, en pasos, etc.), aunque los primeros son los más utilizados. Con estos geles el intervalo de tamaños de proteínas que pueden separarse se incrementa considerablemente frente a los geles homogéneos y presentan una mayor resolución. Además, las moléculas de la

18
parte delantera de la banda se frenan más (encuentran antes los poros más pequeños) que las moléculas de la parte trasera, con lo que las bandas se estrechan, concentrándose sobre su parte delantera, produciendo bandas muy finas y con una gran resolución.
En la cubeta, el tampón del reservorio superior cubre el gel y los pocillos en los que
se ha de cargar la muestra. Con el fin de que ésta, disuelta en el mismo tampón, no difunda al depositarla en los pocillos, se le puede añadir un compuesto que aumente su densidad (normalmente, glicerol o sacarosa). El volumen de muestra que puede aplicarse a cada pocillo depende del tamaño de la propia muestra y del pocillo, pero oscila entre 10- 100µL con un máximo de 200µL. En el tampón de la muestra, también se añade el marcador electroforético (el más utilizado es el azul de bromofenol) para poder determinar el final del proceso.
Fig. 13 Preparación del gel para una electroforesis en placa. Elementos para ensamblar el molde. Los espaciadores (sujetados con pinzas de presion) colocados sobre los cristales forman el molde. Adición de la
solución de monómeros. Colocación del peine en la parte superior. El gel formado presenta los pocillos donde aplicar la muestra. Las dimensiones habituales de los geles son: 10-20 cm (L) x 10-15 cm (A) x 0.5-1.5
mm (E).
3.5.2.2 Tipos de monómeros
La bis-acrilamida es el agente entrecruzador más comúnmente empleado en este tipo de electroforesis. O Gaal et al. (1984), plantearon que la bis-acrilamida puede actuar como terminador de la cadena en el proceso de polimerización y concentraciones altas pueden disminuir el tamaño de poro máximo de gel.
Se han reportado otros agentes entrecruzadores como: la N, N´-diallitartar diamina
(DATD) que da un tamaño de poro mayor que el obtenido con bis-acrilamida; la N, NC-bisacrililcistamina (BAC) cuyo gel resultante se une por los grupos bisulfuro, lo que hace que pueda disolverse en soluciones de reactivos tiólicos yel N,N´-(1,2- dihidroxietileno) bis-acrilamida o DHEBA forma geles de poros altamente hidrofílicos. 3.5.2.3 Catalizadores empleados en la formación del gel de poliacrilamida
La polimerización se lleva a cabo con la utilización de sistemas catalíticos redox como por ejemplo:
• Persulfato de amonio (agente oxidante) y TEMED como agente reductor.

19
• Persulfato de amonio y 3-dimetilamino propio-nitrilo (DMAPN). • Peróxido de hidrógeno, sulfato de hierro y ácido ascórbico.
La polimerización no ocurre a bajo pH, ya que requiere radicales de monómeros
libres formados por catálisis básica de radicales oxígeno del persulfato. La fotopolimerización con riboflavina no es inhibida por el oxígeno; sin embargo,
en ambos casos el TEMED actúa como agente reductor suave y acelera la polimerización. 3.5.2.4 Sistemas de tampón que se emplean en la electroforesis en geles de
poliacrilamida
La fuerza iónica (m) determina el potencial electrocinético ya que reduce la carga neta de los grupos con cargas efectivas. Se ha determinado que la movilidad de las partículas cargadas es inversamente proporcional a la raíz cuadrada de la fuerza iónica, a baja m se eleva la velocidad de migración y es menor la difusión, de manera que la zona de separación es más ancha y mayor la resolución.1 Con el aumento de la m, se incrementa el desprendimiento de calor y la evaporación. En la electroforesis, los efectos de absorción de agua, no-homogeneidad del material, intercambio iónico con grupos cargados del soporte y electroendósmosis, pueden influir sobre la movilidad y la calidad de la separación. La electroendósmosis generalmente ocurre porque grupos cargados del soporte se ionizan en soluciones tampón neutras o ácidas y los contraiones libres hidratados.
(H 3O+) migran hacia el cátodo, esto provoca un movimiento relativo del solvente
respecto al soporte sólido y que las moléculas no cargadas sean transportadas hacia el cátodo a pesar de no tener grupos ionizados.
La electroforesis en geles de poliacrilamida puede realizarse en el rango de pH de 2
a 11, sin embargo la desaminación de las proteínas o las reacciones hidrolíticas pueden ser severas a valores de pH inferiores a 3 y superiores de 10. La mayoría de las proteínas con puntos isoeléctricos comprendidos entre pH 4 y 7,5, son separadas en geles con pH entre 8 y 9.
La fuerza iónica del sistema tampón debe mantenerse a un nivel adecuado para
garantizar la solubilidad de la muestra y suficiente capacidad amortiguadora. Es usual utilizar concentraciones de tampón en un rango entre 0,025 y 0,1 mol/L, pero pueden emplearse concentraciones sobre 0,5 mol/L en sistemas fuertementes ácidos, pues a bajos valores de pH muchos ácidos débiles (que son los más empleados) están débilmente ionizados y es necesaria una alta concentración para obtener la adecuada capacidad amortiguadora. 3.5.2.5 Formatos de la PAGE
Existen 3 formas diferentes de realizar electroforesis en geles de poliacrilamida: lámina vertical,varilla cilíndrica y lámina horizontal.
Los geles más finos son más económicos porque emplean menos reactivos y
pueden ser eficientemente refrigerados durante la corrida, por lo tanto pueden ser corridos a mayor voltaje, lográndose así una alta resolución en corto tiempo. Los

20
tiempos de tinción y destinción son menores también porque la difusión es muy rápida; sin embargo como el gel es tan fino es más propenso a perturbaciones y problemas en la polimerización.
Se ha planteado que la resolución de las moléculas generalmente aumenta con la
disminución del grosor del gel. 3.5.2.6 PAGE en condiciones no desnaturalizantes
La modalidad más sencilla es la descrita en apartados anteriores en las que la muestra se carga directamente en el gel en el cual se va a producir la separación, por lo tanto, la concentración del gel es uniforme y hay un único tampón. La PAGE se desarrolla en condiciones en las que no se altera la conformación nativa de las proteínas separadas, por lo que finalizada la electroforesis, pueden realizarse estudios de funcionalidad de dichas proteínas separadas (actividad enzimática, capacidad de unión de anticuerpos, unión a receptores, etc.).
Otra modalidad de electroforesis no desnaturalizante es aquella en la que el
tampón presente en los reservorios es diferente al que impregna el gel, el cual tampoco es homogéneo, ya que, en realidad, hay dos geles diferentes en uno solo: una zona de resolución (donde tiene lugar la separación de las proteínas) que se polimeriza sin colocar el peine (por lo que la parte superior presenta un frente plano) y otra pequeña zona de concentración de 1-3 cm, que se polimeriza sobre la anterior (quedan fundidas), que contiene los pocillos de aplicación de la muestra y cuya finalidad es concentrar la muestra en la zona de contacto con la zona de resolución (Fig 14).
Este hecho es debido a que la zona de concentración tiene un tamaño de poro muy
grande (2.5-3.0%T), por lo que no es restrictiva frente al de la zona de resolución cuyo tamaño de poro sí es restrictivo.
Fig. 14 PAGE en condiciones no desnaturalizantes en sistemas de tampón discontinuo.
3.5.2.7 PAGE en condiciones desnaturalizantes
En presencia de algunos compuestos químicos, las proteínas pierden su estructura nativa; tales compuestos, llamados agentes desnaturalizantes, producen el desplegamiento de la proteína que queda, así, sin la organización tridimensional característica de su funcionalidad biológica.
En algunas proteínas hay puentes disulfuro entre los residuos de Cys (para formar cistinas) de una misma cadena polipeptídica (puentes intracatenarios) o de diferentes cadenas (puentes intercatenarios) de algunas proteínas con estructura cuaternaria.

21
Estos puentes se pueden romper mediante tratamiento con determinados agentes reductores, como por ejemplo, el ß-mercaptoetanol (ß-ME) o el ditiotreitol (DTT).
Otros agentes desnaturalizantes de proteínas, son la urea y determinados
detergentes. La urea interfiere con las reacciones hidrofóbicas de las proteínas y debe incluirse en el tampón de la muestra y en todos los que se empleen para la preparación de los geles; las principales aplicaciones de la urea es en la separación de las histonas, así como de proteínas nucleares y ribosomales (ya que tienen mucha tendencia a agregar y son insolubles) y para el análisis conformacional de proteínas (mediante geles con gradientes transversales de urea).
Los detergentes afectan a la estructura nativa de las proteínas y a las interacciones
con otras moléculas (proteínas, lípidos, etc.), ya que las interacciones hidrofóbicas de las proteínas son sustituidas por interacciones detergentes-proteína. Existen tres tipos principales de detergentes:
• Detergentes no iónicos. Son débilmente desnaturalizantes y no alteran la carga
de las proteínas a las que se unen; se pueden utilizar en geles con urea y en electroenfoque. Los más empleados son el Triton X-100 y el Octilglucósido, particularmente para solubilizar proteínas de membrana. Se utilizan a concentraciones de 0.1-3.0% (p/v) y deben incluirse en el tampón de la muestra y en el del gel.
• Detergentes iónicos. Pueden tener carga positiva (catiónicos) que se usan para
la separación de proteínas muy ácidas o muy básicas; el más utilizado es el cetiltrimetilamonio (CTAB). Otros poseen carga negativa y un fuerte carácter desnaturalizante; el más empleado es el dodecilsulfato sódico o lauril sulfato sódico (SDS).
• Detergentes anfóteros. Son débilmente desnaturalizantes y no afectan a la carga de las proteínas; algunos, como el 3-(cloroamido propil)-dimetilamonio-1-propanosulfato (CHAPS) solubilizan muy bien las proteínas de membrana. Son compatibles con la utilización de urea y su uso es frecuente como alternativa a los detergentes no iónicos.
3.5.2.8 PAGE-SDS
Permite el cálculo de parámetros moleculares (al contrario que el resto de los tipos de electroforesis), pues los complejos SDSproteína se separan estrictamente según su tamaño molecular. El SDS interacciona con las proteínas formando complejos de características comunes independientemente de las de cada proteína.
Las proteínas unen una molécula de SDS por cada dos aminoácidos, lo que implica
que las cargas propias de las proteínas quedan enmascaradas o anuladas; asimismo, la molécula de SDS proporciona una carga negativa, por lo que los complejos SDS-proteína están cargados negativamente de forma uniforme (la carga por unidad de masa es prácticamente constante para todos los complejos).
Como ya se ha comentado, la movilidad electroforética en una PAGE es función del
tamaño y de la carga por unidad de masa; como ésta es constante para todos los

22
complejos SDS-proteína (que, además, tienen la misma forma elipsoide), esta movilidad es solamente función de la masa molecular, es decir, cuanto menor sea la masa molecular de la proteína, mayor será la movilidad de la misma y viceversa. En resumen, la SDS-PAGE es la electroforesis más utilizada para el análisis de proteínas debido a:
• La gran mayoría de las proteínas son solubles en SDS.
• Todos los complejos SDS-proteína tienen carga negativa y migran, por lo tanto, en el mismo sentido.
• Su densidad de carga es muy elevada, por lo que su velocidad de migración
también lo es y las electroforesis son muy rápidas.
• La separación depende de un parámetro físico-químico, como es la masa molecular, que se puede calcular.
• Los complejos SDS-proteína se tiñen fácilmente.
La preparación de la muestra es de la máxima importancia para asegurar que todo
lo comentado anteriormente se cumple. Al tampón en el que va disuelta la muestra (Tris HCl; 0.05M, pH = 6.8) se añade un exceso de SDS (2% p/v); para facilitar la unión del SDS a las proteínas, la muestra se calienta a 100°C durante al menos 3-5 minutos y, opcionalmente, se añade ß-ME (5% v/v) ó DTT (20 mM) si se requieren condiciones reductoras. Para incrementar la densidad de la disolución de la muestra, se añade glicerol o sacarosa al 10% (p/v) y como marcador azul de bromofenol al 0.001% (p/v). La cantidad de muestra que se añade depende de la sensibilidad de la tinción posterior que se vaya a utilizar, pero como regla general y para geles de 20×20 cm de 1.5mm de espesor y tinción con azul de Coomassie, se suele añadir entre 1-10µg de muestra (más de 100µg de proteína supone una sobrecarga del gel).
El SDS debe incluirse en el tampón de los reservorios, en los de los geles (de concentración y de resolución) y en el de la muestra para mantener las condiciones desnaturalizantes. La PAGE-SDS puede realizarse en condiciones reductoras o no reductoras; la ausencia de reductores se traduce en que si las proteínas poseen puentes disulfuro, el SDS sólo producirá una desorganización parcial de la estructura (Fig. 15). Si son dos o más las cadenas unidas por puentes disulfuro intercatenarios, en presencia de SDS, quedarán más o menos desplegados en función de la posición de los puentes disulfuros (Fig. 16).
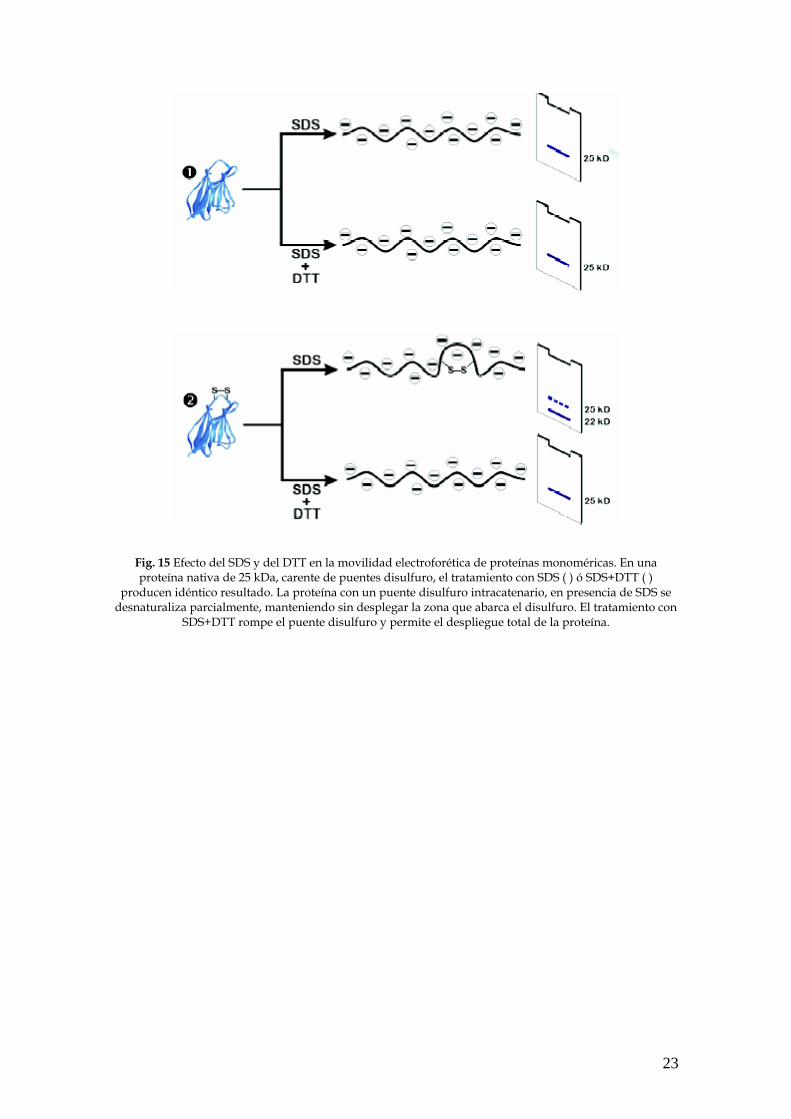
23
Fig. 15 Efecto del SDS y del DTT en la movilidad electroforética de proteínas monoméricas. En una proteína nativa de 25 kDa, carente de puentes disulfuro, el tratamiento con SDS ( ) ó SDS+DTT ( )
producen idéntico resultado. La proteína con un puente disulfuro intracatenario, en presencia de SDS se desnaturaliza parcialmente, manteniendo sin desplegar la zona que abarca el disulfuro. El tratamiento con
SDS+DTT rompe el puente disulfuro y permite el despliegue total de la proteína.

24
Fig. 16 Efecto del SDS y del DTT en la movilidad electroforética de proteínas oligoméricas.
Una vez finalizada la PAGE, se separa el gel de las placas de vidrio haciendo
palanca con una espátula y se visualiza con un colorante; el más utilizado es el azul de Coomassie con el que se pueden visualizar 0.1-0.5 µg de proteína por banda. Otro método de tinción es con sales de plata, que tiene como principal ventaja su elevada sensibilidad (hasta 0.1ng de proteína por banda), aunque tiene como desventajas que es muy laboriosa y cara, presenta elevado background, baja reproducibilidad, algunas proteínas no se tiñen y, sobre todo, no es totalmente específico de proteínas, ya que algunos lipopolisacáridos, ácidos nucleicos y polisacáridos también se tiñen.
Un método de sensibilidad comparable a la tinción con sales de plata, emplea
compuestos fluorescentes que se unen específicamente a las proteínas (la unión puede realizarse antes o después de la electroforesis), como el cloruro de dansilo (detecta hasta 10ng de proteína por banda), la fluorescamina (detecta hasta 3-5ng de proteína por banda) y el MDPF (2-metoxi-2,4-difenil-3(2H) furanona; detecta hasta 1ng de proteína por banda).
Una vez teñido el gel de poliacrilamida, la imagen que se obtiene es un conjunto de
bandas coloreadas sobre un soporte transparente. Su análisis permite identificar el

25
número mínimo de componentes (bandas) de cada muestra. Cada banda se caracteriza por su movilidad electroforética relativa (“Ur”).
Ur = Lmuestra/Lmarcador
donde: Lmuestra = Longitud recorrida por la muestra Lmarcador = Longitud recorrida por el marcador
La PAGE es un criterio de pureza negativo, es decir, permite saber si una muestra no es homogénea (porque aparecen varias bandas en un gel) pero no permite establecer que sea homogénea (puede haber varias proteínas que tengan la misma movilidad electroforética y aparecerían como una sola banda).
En las condiciones de la SDS-PAGE, si se representan los valores de logaritmo del
peso molecular (logM) frente a la movilidad electroforética (Ur) de un conjunto de proteínas, se obtiene una relación lineal (Fig. 17). Normalmente, se utiliza un conjunto de proteínas de peso molecular conocido (marcadores de peso molecular) que se someten a SDS-PAGE en el mismo gel que se analizan las proteínas cuyos peso molecular se desea conocer.
Fig. 17 Cálculo de pesos moleculares de proteínas por SDS-PAGE
Es importante señalar que la relación lineal entre logM y Ur se mantiene para una
concentración dada de poliacrilamida (%T) y sólo en un corto intervalo de peso molecular. De forma general, en geles del 15%T la relación lineal es válida para proteínas comprendidas entre 12 y 45 kDa; en geles del 10%T, el intervalo lineal va entre 15 y 70 kDa, y en los del 5%T, entre 25 y 200 kDa.
Hay proteínas con un comportamiento anómalo en SDS-PAGE; en las glicoproteínas y las lipoproteínas, que llevan unidos azúcares o lípidos, la parte no proteica no une SDS y puede impedir el acceso del mismo a la proteína. Por ello, las glicoproteínas y lipoproteínas unen menos SDS que la mayoría de las proteínas; esto hace que la relación carga/tamaño sea menor y, por lo tanto, tengan menor movilidad electroforética y se comporten como si tuvieran mayor tamaño.
3.5.3 Electrofóresis en geles de gradientes
El uso de geles de poliacrilamida que tienen un gradiente creciente de concentración (Fig. 18) de acrilamida + bisacrilamida, y en consecuencia un gradiente decreciente en el tamaño del poro, pueden tener ventajas sobre los geles de concentraciones uniformes de acrilamida. En un gel en gradiente la proteína migra hasta alcanzar una zona donde el tamaño de poro impida cualquier avance. Una vez se alcanza el limite del poro no se produce una migración apreciable aunque no se detiene completamente. Una de las ventajas de este tipo de geles es que resuelve mejor las

26
bandas pues las concentra en regiones mas estrechas, además de incrementar el rango de pesos moleculares que se pueden resolver en un mismo gel comparado con los de una concentración fija.
Fig. 18 Electroforesis en gel con gradiente.
3.5.4 Electroforesis en geles de agarosa
La agarosa es un polisacárido (originalmente obtenido de algas, como el agar-agar, pero de composición homogénea), cuyas disoluciones (típicamente de 0.5 a 2 %) poseen la propiedad de permanecer liquidas por encima de 50 °C y formar un gel, semisólido al enfriarse. Este gel esta constituido por una matriz o trama tridimensional de fibras poliméricas embebida en gran cantidad de medio líquido, que retarda el paso de las moléculas, se usa usualmente para separar moléculas grandes de alrededor 20.000 nucleótidos.
3.5.5 Electrofóresis capilar
La electroforesis capilar (Fig. 19) se basa en los mismos principios de las técnicas electroforeticas convencionales, pero utiliza condiciones y tecnología distinta que nos permiten obtener una serie de ventajas al respecto. Esta separación de péptidos realizada sobre un capilar de silica fundida a potenciales elevados 20 a 30 Kv en un campo de 400 a 500 v/cm refrigerados por aire. La corriente electroendosmótica (FEO) generada por los grupos silanol de la superficie interna del capilar da como resultado una corriente plana del frente del líquido que contrasta con el frente parabólico de la cromatografía líquida de alta resolución. La ventaja de esta técnica es que el capilar de silica fundida que generalmente se cubre con una capa de polimina para darle mayor rigidez y resistencia, tiene una ventaja a través de ella que permite el pasaje de la luz UV de tal manera que la visualización es on-line. Con esta técnica descrita es posible separar cationes, aniones, proteínas, macromoléculas y sustancias no cargadas en forma simultánea.

27
Fig. 19 Electroforesis capilar 3.5.6 Enfoque isoeléctrico
Es un tipo particular de electroforesis en la que los compuestos anfóteros se separan según su punto isoeléctrico (pI; pH al cual la carga neta de la proteína es nula) en un gradiente continuo de pH.
Fig. 20 Curva de titulación de una proteína. Representación de la carga neta (Q) de una proteína en
función del pH.
En todos los métodos electroforéticos descritos anteriormente, la difusión es un fenómeno con efectos negativos, que tiende a ensanchar las bandas, disminuyendo la resolución. En el EEF el efecto de la difusión es mínimo y por ello posee una enorme resolución, pudiendo llegar a separar proteínas que difieran en 0.001 unidades de pI. Esto se debe a que si una proteína focalizada en su pI se desplazase por difusión, adquiriría carga (positiva o negativa según hacia el polo que difundiera) y por efecto del campo eléctrico volvería a la zona de su pI; esto se llama efecto focalizante del EEF y es el responsable de su enorme resolución. Además, el EEF aumenta su resolución al disminuir el intervalo de gradiente de pH.
Por todo esto, el disponer de un gradiente estable de pH es lo esencial del EEF;
para ello, se emplean compuestos anfóteros de bajo peso molecular llamados anfolitos portadores, que son pequeñas poliaminas (alrededor de 750 kDa) con abundantes
La carga neta de una proteína es función del pH:
pH>pI Carga nula negativa pH=pI Carga neta nula (Fig. 20) pH<pI Carga nula positiva

28
grupos ácidos y básicos, con un pI que abarca desde 2.5 hasta 11.0, con gran capacidad de tamponación cerca de su pI y con una gran solubilidad y conductividad.
Una disolución de una mezcla de estos anfolitos tendrán un pH próximo al
promedio de los pIs. Si esta disolución se emplea para la preparación de un gel de poliacrilamida, al aplicar una diferencia de potencial cada molécula de anfolito migrará hacia uno u otro polo en función de su carga, hasta alcanzar la zona de su pI. Cuando se alcanza el equilibrio queda formado un gradiente de pH a lo largo del gel. Si, debido a la difusión, los anfolitos abandonasen la zona de su pI, adquirirían carga y experimentarían el efecto focalizante mencionado anteriormente.
Los gradientes de pH generados en algunos soportes, como los geles de
poliacrilamida, muestran el fenómeno denominado deriva catódica, que consiste en que el gradiente se hace inestable con el tiempo y se desplaza, junto con las proteínas, hacia el cátodo; esto se debe, fundamentalmente, al flujo electroendosmótico. La deriva catódica puede evitarse variando la naturaleza química del gel (se usan derivados de acrilamida, como la 3-dimetilaminopropil metacrilamida) o utilizando gradientes de pH inmovilizados (que se consigue copolimerizando con la acrilamida los compuestos químicos que establecerán el gradiente, por lo que las moléculas que forman el gradiente están fijas sobre las propias fibras de poliacrilamida).
La variedad más utilizada de EEF es la analítica. El gel se prepara a partir de
una disolución de acrilamida (3-5%T para facilitar la movilidad) y bisacrilamida en agua, a la que se añaden los anfolitos. La preparación del gel es totalmente análoga a la de PAGE, aunque también puede llevarse a cabo en cubetas horizontales, en cuyo caso los geles son de muy poco grosor (<0.5 mm) y se polimerizan sobre láminas de plástico tratadas especialmente para que se adhieran al gel.
El EEF puede llevarse a cabo en condiciones que mantengan la estructura nativa
de las proteínas o en condiciones desnaturalizantes, incorporando, además de los anfolitos, urea (8M) o detergentes ni iónicos o anfóteros (pero nunca detergentes iónicos como el SDS).
Si el EEF se va a desarrollar en un gel con un gradiente de pH inmovilizado, se
utiliza un formador de gradiente (sistema de vasos comunicantes) y dos disoluciones con distinta densidad (una más densa de carácter ácido, por lo que quedará en la parte inferior del gel y otra más básica de menor densidad que quedará en la parte superior del gel).
El desarrollo del EEF es más complejo que el de la PAGE, ya que se utilizan
voltajes variables (ya que según se va formando el gradiente, los anfolitos van perdiendo carga hasta llegar a su pI, con lo que se reduce la conductividad del medio); en el EEF se necesitan fuentes de alimentación capaces de generar 3.000V, para proporcionar intensidades constantes del orden de 50-150mA. Estos voltajes tan elevados lleva asociado un importante incremento de temperatura, por lo que el soporte debe estar refrigerado a 4°C o alrededor de 10°C si se utilizan geles con urea (ya que la urea precipita a 4°C).
Una de las aplicaciones más importantes del EEF es el cálculo de pI (Fig. 21); en
el pocillo I se aplican los marcadores de pI, y en el II la muestra (X) cuyo pI se desea conocer. El carril III (sombreado) se emplea para determinar el gradiente de pH. Una

29
vez corrido el gel, se trocea el carril III y cada segmento (de 1-2 mm) se introduce en 1mL de agua, determinándose su pH. Finalmente, se representa el pH de cada fracción, frente a su posición en gel. La distancia migrada por la muestra (X) se interpola en la recta obtenida, determinándose así su pH.
Fig. 21 Determinación del punto isoeléctrico de una muestra.
Hay que volver a destacar, una vez más, que el EEF es un criterio de pureza
negativo, es decir, varias bandas indican que la muestra es heterogénea, pero la aparición de una única banda no permite afirmar que la muestra sea homogénea, ya que pueden coexistir proteínas diferentes con igual pI; no obstante, cuanto menor sea el gradiente de pH utilizado, mayor será la probabilidad de que al aparecer una banda, la muestra sea homogénea. 3.5.7 Electrofóresis bidimensional
Es una sucesión de dos electroforesis distintas (o en distintas condiciones) realizadas sobre una misma muestra. En la primera de ellas (primera dimensión) se separan los componentes de la muestra según un criterio (carga, tamaño o pI), y en la segunda (segunda dimensión) según un parámetro distinto del anterior. De esta manera, se combinan dos modos de separación diferentes y se consigue el máximo de resolución posible mediante técnicas electroforéticas.
La primera dimensión puede llevarse a cabo, por ejemplo, en condiciones no
desnaturalizantes y la segunda en condiciones desnaturalizantes; las proteínas ribosomales se separan en un gel discontinuo en la primera dimensión, seguido por otro continuo en la segunda, y ambos en presencia de urea; las histonas se separan en geles con urea y ácido en la primera dimensión y utilizando tritón/ácido/urea en la segunda.

30
Sin embargo, normalmente, la idea de electroforesis bidimensional suele referirse a la combinación de los dos tipos de electroforesis más resolutivos: electroenfoque (primera dimensión) y SDS-PAGE (segunda dimensión) (Fig. 22).
La electroforesis bidimensional puede considerarse como un criterio de pureza
positivo debido a su gran poder de resolución, aceptándose que la aparición de una sola mancha indica una muestra homogénea.
Fig. 22 Electroforesis bidimensional. Primera dimensión (SDS-PAGE). En el pocillo I se aplica el marcador de pesos moleculares y en los pocillos II y III la muestra. El carril del pocillo III (sombreado) se corta y el
resto del gel se tiñe. El carril III se coloca sobre un nuevo gel que se ha polimerizado sin pocillos, donde se desarrolla la segunda dimensión (EEF) perpendicularmente a la primera. Obsérvese que algunas bandas
producen más de una mancha en la segunda dimensión.
4. INFORMACIÓN SOBRE LA ESTRUCTURA DE LAS PROTEÍNAS. 4.1 Fragmentación de Proteínas.
Las estrategias más utilizadas en la identificación de proteínas por espectrometría de masas (MS) requieren que la muestra sea fragmentada, ya sea por métodos químicos o enzimáticos, en sus constituyentes peptídicos.
En general las proteínas que poseen enlaces disulfuro en su estructura no se digieren, y aquellas que son digeridas producen mezclas de péptidos muy complejas. Por esta razón resulta necesario reducir y alquilar los residuos de cisteína en las proteínas que han sido resueltas por electroforesis antes de llevar a cabo la digestión. Este proceso comprende 2 pasos: primero se reduce el enlace disulfuro y posteriormente se alquilan los grupos tiol de los residuos de cisteína. De esta manera la proteína se encuentra desplegada de forma tal que mejora el rendimiento durante el proceso de digestión. 4.1.1 Proteólisis.
La fragmentación enzimática se ha convertido en el método más utilizado en proteómica para el rompimiento de proteínas, teniendo a su disposición una gran variedad de enzimas. Algunas de las ventajas que ofrece este método son la alta
31
especificidad y eficiencia en el rompimiento de las proteínas, así como un mínimo de reacciones laterales.
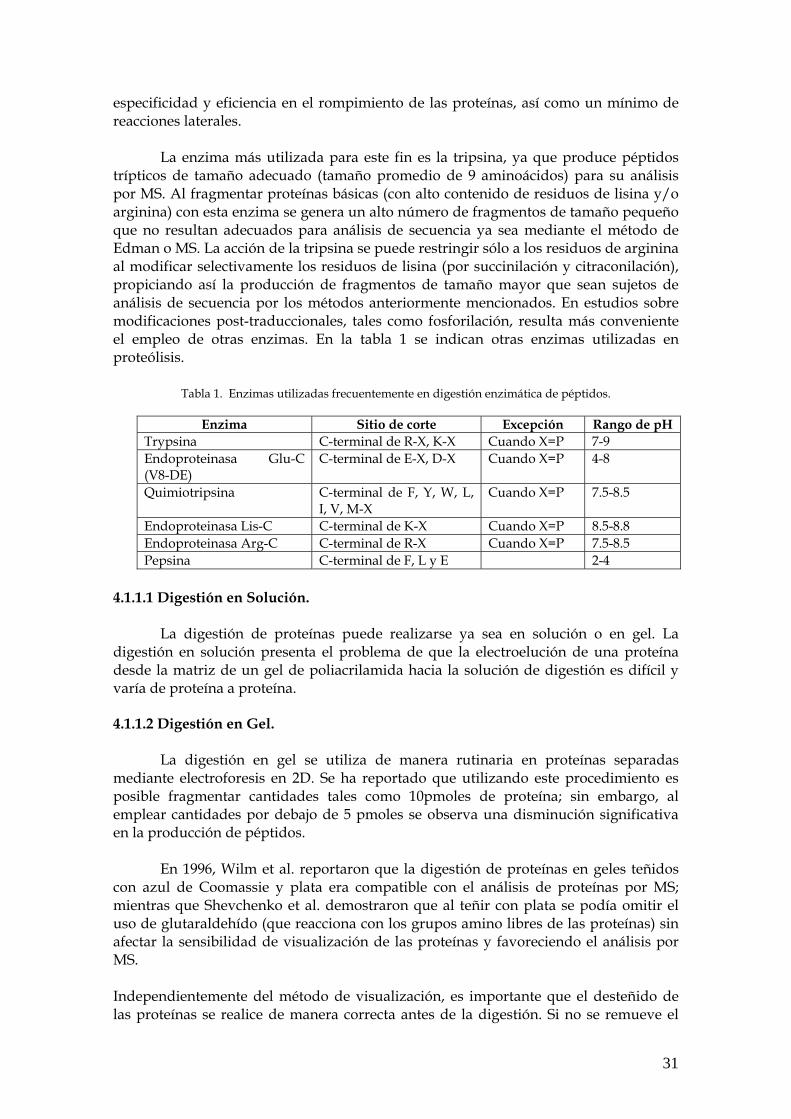
La enzima más utilizada para este fin es la tripsina, ya que produce péptidos trípticos de tamaño adecuado (tamaño promedio de 9 aminoácidos) para su análisis por MS. Al fragmentar proteínas básicas (con alto contenido de residuos de lisina y/o arginina) con esta enzima se genera un alto número de fragmentos de tamaño pequeño que no resultan adecuados para análisis de secuencia ya sea mediante el método de Edman o MS. La acción de la tripsina se puede restringir sólo a los residuos de arginina al modificar selectivamente los residuos de lisina (por succinilación y citraconilación), propiciando así la producción de fragmentos de tamaño mayor que sean sujetos de análisis de secuencia por los métodos anteriormente mencionados. En estudios sobre modificaciones post-traduccionales, tales como fosforilación, resulta más conveniente el empleo de otras enzimas. En la tabla 1 se indican otras enzimas utilizadas en proteólisis.
Tabla 1. Enzimas utilizadas frecuentemente en digestión enzimática de péptidos.
Enzima Sitio de corte Excepción Rango de pH Trypsina C-terminal de R-X, K-X Cuando X=P 7-9 Endoproteinasa Glu-C (V8-DE)
C-terminal de E-X, D-X Cuando X=P 4-8
Quimiotripsina C-terminal de F, Y, W, L, I, V, M-X
Cuando X=P 7.5-8.5
Endoproteinasa Lis-C C-terminal de K-X Cuando X=P 8.5-8.8 Endoproteinasa Arg-C C-terminal de R-X Cuando X=P 7.5-8.5 Pepsina C-terminal de F, L y E 2-4
4.1.1.1 Digestión en Solución.
La digestión de proteínas puede realizarse ya sea en solución o en gel. La digestión en solución presenta el problema de que la electroelución de una proteína desde la matriz de un gel de poliacrilamida hacia la solución de digestión es difícil y varía de proteína a proteína. 4.1.1.2 Digestión en Gel.
La digestión en gel se utiliza de manera rutinaria en proteínas separadas mediante electroforesis en 2D. Se ha reportado que utilizando este procedimiento es posible fragmentar cantidades tales como 10pmoles de proteína; sin embargo, al emplear cantidades por debajo de 5 pmoles se observa una disminución significativa en la producción de péptidos.
En 1996, Wilm et al. reportaron que la digestión de proteínas en geles teñidos con azul de Coomassie y plata era compatible con el análisis de proteínas por MS; mientras que Shevchenko et al. demostraron que al teñir con plata se podía omitir el uso de glutaraldehído (que reacciona con los grupos amino libres de las proteínas) sin afectar la sensibilidad de visualización de las proteínas y favoreciendo el análisis por MS. Independientemente del método de visualización, es importante que el desteñido de las proteínas se realice de manera correcta antes de la digestión. Si no se remueve el

32
azul de Coomassie de la muestra se afectará la eficiencia de digestión de la tripsina ya que éste se une a los residuos básicos de las proteínas. La presencia de iones de plata, de azul de Coomassie, y de detergentes como el SDS (que se ioniza fácilmente) puede afectar también el análisis de MS. La proteína de interés se recupera del gel cortando la banda manualmente, aunque existen también sistemas automatizados. Antes de añadir la enzima al fragmento de gel con la proteína de interés, éste se deshidrata mediante lavados sucesivos con acetonitrilo y posteriormente se seca al vacío. Este paso favorece que la enzima se incorpore eficientemente por difusión cuando el gel es rehidratado con el buffer de digestión. Una vez llevada a cabo la reacción enzimática los péptidos producidos se extraen de la matriz del gel por difusión utilizando solventes orgánicos y ácidos, entre ellos el ácido trifluoroacético y el acetonitrilo. Para eliminar eficientemente sales, buffers y contaminantes de los péptidos extraídos de la matriz del gel se utilizan columnas de cromatografía en fase reversa (RP-HPLC). Una vez eliminados los contaminantes, las muestras están listas para ser analizadas por MS si se encuentran en la concentración requerida por el método.
Las proteínas resueltas en geles de poliacrilamida pueden también ser transferidas por electroblotting a membranas con tripsina inmovilizada donde se lleva a cabo su fragmentación. 4.1.2 Rompimiento químico. Los métodos químicos son complementarios a la proteólisis, y aunque no siempre se utilizan tienen aplicaciones específicas en casos donde la proteólisis no es apropiada o no se cuenta con la enzima adecuada. El rompimiento químico presenta varias desventajas, entre ellas: baja eficiencia de corte, inespecificidad en el corte con la mayoría de los métodos y reacciones laterales indeseables. Un método que presenta una excelente especificidad de corte es el rompimiento con bromuro de cianógeno (CNBr), que se utiliza para cortar proteínas insolubles o de membrana generando péptidos relativamente largos. En la siguiente tabla se muestran los métodos químicos empleados más comúnmente para digerir proteínas.
Tabla 2. Agentes químicos utilizados frecuentemente en fragmentación de péptidos. Agente químico Sitio de corte Observaciones Bromuro de cianógeno (CNBr)
C- terminal de Metionina Excelente especificidad y rendimiento de corte. Exceso de CNBr puede causar rompimiento en residuos de Y y W.
Hidrólisis ácida (ácido fórmico)
Residuos de Aspartil Rompimiento específico.
Hidroxilamina Enlaces Asn-Gly Algunos enlaces Asn-X se pueden romper.
BNPS-skatole Triptofano Inestable en condiciones ácidas.

33
4.2 Análisis de Secuencias Amino y Carboxilo terminal 4.2.1 Secuenciación del extremo amino terminal.
Uno de los primeros métodos utilizados en la identificación de proteínas fue el desarrollado por Pehr Victor Edman para obtener la secuencia amino terminal.
El método de degradación de Edman se divide en 3 etapas: - Acoplamiento: Se lleva a cabo en un tiempo de 15-30 min a pH 9 y 40-55°C. En la
reacción de acoplamiento el fenilisotiocianato (PITC) modifica químicamente al grupo α-amino del amino terminal libre de un polipéptido para formar un feniltiocarbamil (PTC) polipéptido. Esta reacción se inhibe por modificaciones en el amino terminal, tales como formilación, acetilación, acilación y ciclización de residuos de glutamina.
- Rompimiento: En presencia de ácido anhidro el residuo PTC amino terminal se
libera rápidamente del polipéptido. Esto genera un aminoácido anilinotiazolinona (ATZ) y un polipéptido. El ATZ aminoácido generado es hidrofóbico, por lo que puede ser extraído de la solución empleando solventes como el etilacetato. El polipéptido liberado tiene un aminoácido menos y posee un extremo amino terminal libre capaz de reaccionar con el PITC.
- Conversión: En este paso, mediante la reacción con ácido acuoso el ATZ
aminoácido que es muy inestable se convierte en un feniltiohidantoin aminoácido (PTH) más estable. El PTH es posteriormente analizado por HPLC para definir la identidad del aminoácido.
Si bien el método de secuenciación de Edman está perdiendo terreno en el campo
de la proteómica, aún sigue siendo una alternativa viable para la obtención de secuencias en caso de no contar con un espectrómetro de masas. Hoy en día se utiliza en la técnica de secuenciación de mezcla de péptidos. En esta técnica la mezcla de proteínas resuelta por electroforesis en 2-D se transfiere a una membrana inerte por electroblotting. Las proteínas son visualizadas en la superficie de la membrana, escindidas y fragmentadas en 3 ó 5 péptidos largos con CNBr o skatole. El fragmento de la membrana se coloca directamente en un secuenciador de Edman automatizado. Esta metodología sirve para analizar proteínas con modificaciones en el extremo N-terminal que no son susceptibles de ser secuenciadas por el método de degradación tradicional.

34
Figura 23. Método de degradación de Edman.
4.2.1.1 Proteínas con modificaciones en el amino terminal.
Estimaciones empíricas sugieren que cerca del 40-50% de las proteínas que se
resuelven por electroforesis en 2-D están bloqueadas en el extremo N-terminal por grupos formil, acetil y piroglutamil (por ciclización de residuos de glutamina en condiciones ácidas). Si la proteína de interés no está bloqueada naturalmente aún puede sufrir un bloqueo de su N-terminal durante la preparación de la muestra, en especial durante la electroforesis. Para evitar el bloqueo del extremo N- terminal en las proteínas de interés se han implementado algunas medidas tales como: adicionar glicerol grado HPLC al buffer de muestra o correr la electroforesis con sólo el gel de corrida y antes de colocar el gel empacador para evitar que los monómeros de poliacrilamida reaccionen con los N- terminales de las proteínas. Existen también diversos métodos para desbloquear proteínas, aunque no han probado ser muy eficientes; es por ello que no se recomienda su empleo para proteínas poco abundantes.

35
4.2.2 Secuenciación del extremo carboxilo terminal. Las técnicas de secuenciación del carboxilo terminal se han desarrollado poco en
comparación con la metodología empleada para secuenciar el amino terminal. La mayoría de las secuencias del extremo carboxilo terminal reportadas en los últimos años se han obtenido utilizando MALDI en espectrometría de masas; para ello los péptidos son digeridos con carboxipepsidasas.
5. ESPECTROMETRÍA DE MASAS Mediante espectrometría de masas es posible obtener información estructural de
las proteínas tal como secuencia de aminoácidos y la masa de los péptidos. Esta información puede utilizarse para identificar proteínas comparando los resultados con bases de datos. La espectrometría de masas también resulta útil para identificar y localizar modificaciones post-traduccionales en las proteínas. La recolección de información por medio de MS se podría dividir en las 3 etapas que se mencionan a continuación.
5.1 Preparación de la muestra
Como se mencionó previamente, la proteína se resuelve de una mezcla proteíca empleando generalmente técnicas electroforéticas. Debido a que la conversión de la proteína en sus constituyentes peptídicos proporciona mayor información que la obtenida con la proteína completa es necesario llevar a cabo una fragmentación de la misma por los métodos que ya se describieron en este trabajo. Una vez obtenidos los constituyentes peptídicos estos pueden ser purificados por RP-HPLC, utilizando ZipTips (Millipore) o material de perfusión Poros R2 (PerSeptive Biosystems, Framingham, Mass). (Fig. 3)
5.2 Ionización de la muestra
Para el análisis de muestras biológicas por MS es necesario que las moléculas estén cargadas eléctricamente y secas. Este requisito se cumple convirtiendo las moléculas en iones desolvatados. Han sido varias las técnicas que se han desarrollado con este fin, las primeras de ellas se basaron en el impacto de electrones y en la ionización química. Estas técnicas resultaron efectivas para ionizar moléculas pequeñas pero no para la ionización de moléculas de alto peso molecular, como las biomoléculas. La espectrometría de masas se revolucionó en 1981 gracias a la introducción del bombardeo rápido de átomos (FAB, fast atom bombardment) por Barber. Esta técnica posibilitó la ionización y detección de biomoléculas con relativamente buena sensibilidad. Hoy en día, los métodos más comúnmente empleados para generar iones desolvatados son el MALDI (matrix-assisted laser desorption/ionization) y el ESI (electrospray ionization), estos han reemplazado por completo al FAB. Tanto el MALDI como el ESI son métodos de ionización suaves donde se mantiene relativamente la integridad de la muestra.
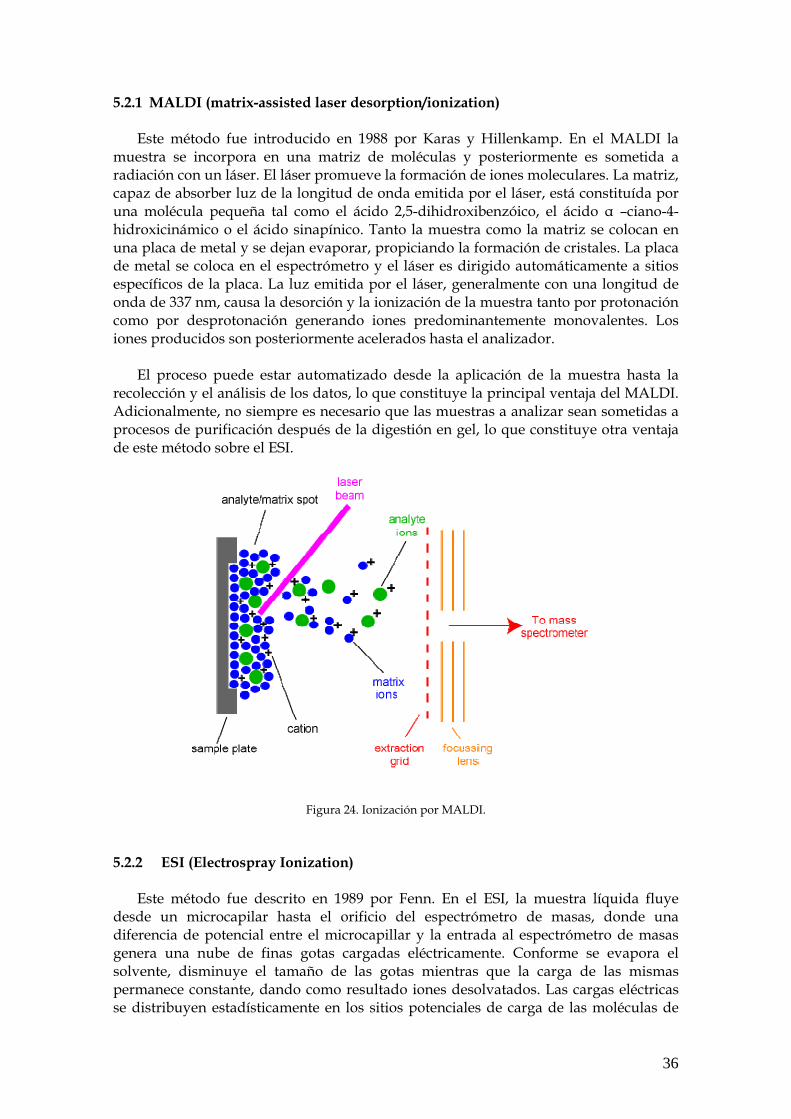
36
5.2.1 MALDI (matrix-assisted laser desorption/ionization)
Este método fue introducido en 1988 por Karas y Hillenkamp. En el MALDI la muestra se incorpora en una matriz de moléculas y posteriormente es sometida a radiación con un láser. El láser promueve la formación de iones moleculares. La matriz, capaz de absorber luz de la longitud de onda emitida por el láser, está constituída por una molécula pequeña tal como el ácido 2,5-dihidroxibenzóico, el ácido α –ciano-4-hidroxicinámico o el ácido sinapínico. Tanto la muestra como la matriz se colocan en una placa de metal y se dejan evaporar, propiciando la formación de cristales. La placa de metal se coloca en el espectrómetro y el láser es dirigido automáticamente a sitios específicos de la placa. La luz emitida por el láser, generalmente con una longitud de onda de 337 nm, causa la desorción y la ionización de la muestra tanto por protonación como por desprotonación generando iones predominantemente monovalentes. Los iones producidos son posteriormente acelerados hasta el analizador. El proceso puede estar automatizado desde la aplicación de la muestra hasta la recolección y el análisis de los datos, lo que constituye la principal ventaja del MALDI. Adicionalmente, no siempre es necesario que las muestras a analizar sean sometidas a procesos de purificación después de la digestión en gel, lo que constituye otra ventaja de este método sobre el ESI.
Figura 24. Ionización por MALDI.
5.2.2 ESI (Electrospray Ionization)
Este método fue descrito en 1989 por Fenn. En el ESI, la muestra líquida fluye desde un microcapilar hasta el orificio del espectrómetro de masas, donde una diferencia de potencial entre el microcapillar y la entrada al espectrómetro de masas genera una nube de finas gotas cargadas eléctricamente. Conforme se evapora el solvente, disminuye el tamaño de las gotas mientras que la carga de las mismas permanece constante, dando como resultado iones desolvatados. Las cargas eléctricas se distribuyen estadísticamente en los sitios potenciales de carga de las moléculas de

37
analito, dando como resultado iones multivalentes. A cada ión multivalente se le denomina un estado de carga, y es común obtener una distribución de estados de carga al analizar macromoléculas por ESI. Las proteínas también generan iones multivalentes, sin embrago generalmente la resolución de cada ión multivalente no es suficiente para determinar el número de cargas que posee el ión, con el cual se podría determinar su masa real. Por lo tanto se utilizan las masas de al menos 2 iones multivalentes para calcular la masa real de la proteína mediante algoritmos de deconvolución. La obtención de iones multivalentes es una característica única del método de ESI; esto permite utilizar una menor cantidad de masa del analito, lo que constituye una ventaja para el análisis de macromoléculas. La técnica de electrospray genera flujos que van de 10 a 100 µL/min. Este flujo resulta problemático para el análisis de proteínas, específicamente debido a que la muestra se consume rápidamente y a que el tiempo de análisis es muy corto. Por ello, una mejora importante a este método de ionización ha sido el desarrollo de la ionización por nanospray, donde se utilizan microcapilares con diámetro interno de 1 a 2 µm y velocidades de flujo de 5 a 10 nL/min. Estos flujos permiten reducir la cantidad de muestra consumida e incrementar el tiempo disponible para el análisis. Existen diversas maneras de hacer llegar la muestra al espectrómetro. La más simple es utilizando microcapilares individuales para cada muestra, de esta manera se evita la contaminación cruzada entre muestras. La purificación de la muestras posterior a la digestión en gel es un requisito para utilizar el método de ESI, y puede llevarse a cabo directamente en los tubos microcapilares. Es debido a este pre-requisito que algunas fuentes de electrospray están conectadas a sistemas de cromatografía que de manera automática purifican y dirigen la muestra al espectrómetro de masas.
Figura 25. Ionización por ESI En la siguiente tabla se enlistan las ventajas y desventajas de los 2 métodos descritos previamente.

38
Tabla 3. Ventajas y desventajas de los métodos de ionización MALDI y ESI
Ventajas Desventajas MALDI Límite de tamaño 300,000 Da Sensibilidad en el orden de fentomoles a picomoles. Ionización suave con baja o nula fragmentación de los péptidos. Apropiado para análisis de mezclas complejas. Las muestras no requiren preparación previa.
Ruido de fondo generado por la matriz, problemas en compuestos de menos de 1,000 Da. Imposible estudiar interacciones no covalentes. Capacidad mínima para secuenciación (MS/MS) y determinación de modificaciones post-traduccionales a menos que se equipe con un reflectrón (post-source decay).
ESI Límite de tamaño 70,000 Da Sensibilidad en el orden de fentomoles a picomoles. Ionización suave. Capaz de observar interacciones no covalentes nativas. No hay interferencia de alguna matriz. Fácil de adaptar a análisis de tripe cuadrupolo. Formación de iones multivalentes: mayor precisión en el cálculo de masas, análisis de iones de alto peso con un relativamente bajo rango m/z del instrumento. Excelente para determinar modificaciones post-traduccionales. Capacidad para secuenciación de péptidos excelente.
Baja tolerancia a sales. Necesario utilizar RP-HPLC. Alta sensibilidad a contaminantes. Baja tolerancia a mezclas. La pureza de la muestra es muy importante. Iones multivalentes. Puede ocasionar confusión especialmente al analizar mezclas.
5.3 Análisis de masa.
Después de la conversión de proteínas o péptidos en iones moleculares, estos son acelerados desde la fuente de iones hacia el analizador de masa. En el analizador de masa los iones son separados de acuerdo a su relación carga-masa (m/z) en el vacío. 5.3.1 Analizadores de Cuadrupolo. Uno de los analizadores de masa más comúnes es el analizador de cuadrupolo. En él los iones son conducidos a través de un campo eléctrico creado por un arreglo de 4 barras metálicas paralelas, el cuadrupolo. Un cuadrupolo puede transmitir todos los iones, o bien actuar como un filtro másico que permita la transmisión de de iones con una cierta relación m/z. Un solo analizador de cuadrupolo no brinda información útil en análisis de proteómica, sin embargo al combinar tres cuadupolos en serie (figura 27) y acoplarlos a un sistema de ESI es posible obtener información acerca de la secuencia de amino ácidos de un péptido. Empleando esta configuración, el primer y tercer cuadrupolo actúan como filtros de masa, mientras que el segundo funciona como una celda de colisión en la cual se produce la información sobre la estructura proteíca. A esta configuración se le denomina “tandem space” o MS/MS.

39
Figura 26. Analizador de iones de cuadrupolo.
Figura 27. Configuración de un analizador de cuadrupolo triple.
Utilizando un analizador de cuadrupolo triple es posible realizar tres tipos de experimentos en espectrometría MS/MS, estos experimentos se denominan: product ion scan, precursor ion scan y neutral loss scan. (ver figura 28) En product ion scan, se selecciona el ión de interés en el primer cuadrupolo, que posteriormente es dirigido al segundo cuadrupolo, que actúa como una celda de colisón. En esta celda el ión se fragmenta para producir una distribución de fragmentos de ión, o iones producto. Estos iones se resuelven en el tercer cuadrupolo, que actúa como un filtro de masas. En precursor ion scan, el primer cuadrupolo permite que todos los componentes de la mezcla pasen a la celda de colisión (segundo cuadrupolo). El tercer cuadrupolo, que actúa como filtro de masas, está fijo en un valor de masa, de manera que sólo los péptidos que hayan generado fragmentos con la masa especificada por el filtro podrán ser detectados en el analizador de masas. En neutral loss scan, el primer y tercer cuadrupolos se barren sincronizadamente con un offset de valor m/z específico. Toda la mezcla de iones pasa al segundo cuadrupolo, que nuevamente es la celda de colisiones, pero sólo las especies que generan fragmentos de masa igual a la del offset son observadas en el detector.

40
Figura 28. Modos en que se emplea un cuadrupolo triple. 1) Product ion scan, 2) Precursor ion scan, 3) Neutral loss scan.
5.3.2 Analizador de tiempo de vuelo (ToF). Éste es uno de los analizadores más simples. Mide la relación m/z de un ión determinando el tiempo requerido para recorrer la longitud de un tubo de vuelo desde que el ión abandona la fuente de iones; el ión es impulsado con una velocidad inicial, la cual depende directamente de su masa. El tiempo de vuelo del ión es proporcional a la raíz cuadrada de su relación m/z dada una aceleración constante provocada por el voltaje.
Tiempo de vuelo= k √(m/z)
Algunos analizadores de masa de ToF incluyen un espejo de iones o reflectrón al final del tubo de vuelo, el cual repele los iones a través de este tubo y los dirige al detector. De esta forma, se ve incrementada la longitud del tubo de vuelo con lo que mejora la resolución. El reflectrón sirve también para corregir las pequeñas diferencias en la energía cinética (Ec) que existe entre iones de la misma masa. Estas diferencias se deben a la posición que guarda cada ión en la fuente de iones al momento de que son acelerados al aplicar una diferencia de potencial. Las diferencias en la energía cinética de los iones se reduce debido a que los iones con Ec mayor viajan más lejos en el reflectrón que los iones con Ec menor. Esto ocasiona que los iones con la misma masa se enfoquen mejor en el detector mejorando considerablemente la resolución y la precisión del cálculo de masas.

41
Figura 29. Analizador de tiempo de vuelo. Los espectrómetros de masa con analizadores de tiempo de vuelo acoplados a sistemas de ionización MALDI se denominan espectrómetros MALDI-ToF. Estos se utilizan principalmente en fingerprinting de péptidos y se usan como herramienta primaria en la identificación de proteínas debido su velocidad. Un sistema MALDI-ToF sirve para obtener la secuencia de péptidos empleando la técnica de decaimiento post -fuente (post source decay, PSD). 5.3.3 Analizador de trampa de iones. La función de estos analizadores es atrapar iones moleculares en un campo eléctrico tridimensional. En contraste con un analizador de cuadrupolo, en el cual los iones son descartados antes de iniciar el análisis, el analizador de trampa de iones posee la capacidad de almacenar los iones y liberarlos de manera selectiva de la trampa. Esto incrementa la sensibilidad.
Figura 30. Analizador de Trampa de iones. 5.3.4 Analizadores híbridos. Los analizadores híbridos se han convertido en una herramienta muy útil para la secuenciación de proteínas. Combinando un cuadrupolo que actúe como filtro de masa y una celda de colisión con un analizador ToF con reflectrón, es posible obtener información con alta precisión, resolución y sensibilidad. Los analizadores híbridos usualmente están acoplados a un sistema de HPLC.

42
5.3.5 Analizadores ToF/ToF. Este tipo de analizador, acoplado a una fuente de iones MALDI, se utiliza para secuenciación de péptidos y huella de masa de péptidos. Dos analizadores ToF se encuentran separados por una celda de colisión. El primer analizador ToF se emplea como selector de iones. En la celda de colisión se producen choques de alta energía entre los iones. Y el segundo analizador ToF resuelve los iones. 5.3.6 FT-ICR (Fourier transform-ion cyclotron resonance). El analizador de masas FT-ICR se utiliza para el análisis de biomoléculas acoplado a una fuente de iones MALDI o ESI. Se ha empleado para resolver mezclas complejas, estudiar interacciones de proteínas, así como para elucidar la conformación de proteínas. El FT-ICR, es una trampa de iones magnética. En este analizador los iones son atrapados en una celda que está bajo un campo magnético fuerte. La celda está compuesta por tres pares de placas (figura 31): placas de confinamiento (perpendiculares al campo magnético), transmisoras y receptoras. Dentro de la trampa, los iones se mueven en órbita circular, perpendicular al campo magnético. Los iones se mantienen en la celda debido al potencial eléctrico aplicado a las placas de confinamiento. Cuando se aplica un potencial eléctrico de radio-frecuencia en las placas transmisoras los iones atrapados se excitan. Una frecuencia dada excita iones con una relación m/z particular. La excitación de los iones provoca que giren en órbitas mayores. Cuando los iones excitados pasan cerca de las placas receptoras se detecta su frecuencia de rotación (que es inversamente proporcional a su masa) y se induce una corriente en las placas. A esta corriente se le denomina corriente de imagen. Mediante las transformadas de Fourier es posible medir simultáneamente todas las frecuencias de rotación al mismo tiempo.
Figura 31. Analizador de masas FT-ICR. A) Placas de confinamiento, atropamiento de iones por campo magnético. B) Placas transmisoras, excitación de los iones. C) Placas receptoras, generación de corriente de
imagen.

43
5.3.7 Fragmentación de Iones.
Una vez que los iones son dirigidos a la cámara de colisión del espectrómetro de masas, estos interactúan con el gas de colisión (N2 o Ar) y sufren una fragmentación primaria. Los fragmentos que se generan pueden ser muy variados (Figura 32). Si el después de la fragmentación la carga se mantiene en el extremo N-terminal, el ión es designado como “ión -b”; en cambio, si la carga se mantiene en el extremo C-terminal, el ión es denominado un “ión -y”. La diferencia en masa entre un ión -b y un ión -y corresponde a un aminoácido. Esta diferencia de masa entre los iones se utiliza para identificar los aminoácidos, y con ello, la secuencia peptídica. Para distinguir entre los aminoácidos leucina e isoleucina, que poseen la misma masa molecular, es necesario que la fragmentación se lleve a cabo con energías de colisión muy altas, de manera que se propicie la fragmentación de cadenas laterales de los aminoácidos. Tanto los iones -b como los -y pueden perder NH3 (principalmente de los residuos básicos como arginina y lisina), H2O (principalmente del alcohol contenido en residuos de serina y treonina; y de los residuos que contienen grupos carboxilo libres como el ácido glutámico y el aspártico) y CO. Cuando un ión –b pierde CO se forma un ión-a.
Figura 32. Fragmentación de péptidos. Además de la formación de iones – y y b, se indica la formación de
otros iones. 6. IDENTIFICACIÓN DE PROTEÍNAS UTILIZANDO BASES DE DATOS. Los datos generados en un espectrómetro de masas, son visualizados en forma de picos; a la representación gráfica de los datos se le denomina espectro de masas. Cada uno de los picos de un espectro de masas representa valores de m/z y la intensidad de cada señal. Definir la identidad o secuencia de una proteína a partir de su espectro de masa es un procedimiento común en estudios de proteómica. Utilizando la información contenida en bases de datos es posible la identificar rápidamente un gran número de proteínas con gran precisión. En la tabla 4 se muestran algunas de las bases de datos más utilizadas en estudios de proteómica.

44
Tabla 4. Bases de datos utilizadas para identificación de proteínas. Nombre del sitio URL Comentarios ProFound http://prowl.rockefeller.edu/cgi-
bin/ProFound Peptide mass fingerprinting y secuenciación. Considera propiedades de la proteína, y preparación de la muestra. Calcula peso molecular, pI, etc.
MOWSE http://srs.hgmp.mrc.ac.uk/cgi-bin/mowse Peptide mass fingerprinting y secuenciación.
MASCOT http://matrixscience.com/ Mapeo de masa de péptidos y secuenciación (uninterpreted MS/MS).
SEQUEST http://fields. Scripps.edu/sequest/ Secuenciación (uninterpreted MS/MS).
FindMod http://www.expasy.ch/tools/findmod/ Modificaciones post-traduccionales.
FASTA http://fasta.bioch.virgina.edu/ Base de datos de proteínas y nucleótidos.
La identificación exitosa de proteínas depende de la calidad de los datos generados en el espectrómetro de masas, de los datos contenidos en la base de datos y, del método empleado en la búsqueda de datos. En general, existen diversas estrategias para el manejo de datos obtenidos por MS, algunas de ellas se describen a continuación. 6.1 Huella de masa del péptido (peptide mass fingerprinting, PMF). Esta técnica comprende los siguientes pasos: digestión de la proteína, análisis por MALDI-ToF y empleo de algoritmos de búsqueda en bases de datos. Para llevar a cabo el análisis de datos, cada proteína en la base de datos es digerida teóricamente empleando el mismo agente utilizado para fragmentar la proteína problema. Posteriormente se compara el espectro obtenido por MS (la huella de masa del péptido) con los espectros teóricos generados en la base de datos; se calcula y asigna una “puntuación”, la cual refleja la paridad entre los datos experimentales y los teóricos obtenidos por la base de datos. La proteína identificada como la más probable es aquella con la puntuación más alta. Esta técnica posee la ventaja de ser bastante rápida, sin embargo no es buena cuando se trabaja con mezclas de proteínas a pesar de que se han desarrollado algunos programas para tal fin. La presencia de un alto número de modificaciones post-traduccionales en la proteína problema puede generar un espectro de masa distinto a los predichos por las bases de datos, lo que representa un problema para el análisis. La técnica PMF puede ofrecer mejores resultados si la búsqueda en la base de datos es suplementada con una secuencia parcial. En este caso sí es posible identificar proteínas en una mezcla.

45
6.2 Secuenciación utilizando datos generados con la técnica product ion scan en MS/MS. Como se mencionó previamente, la fragmentación de péptidos utilizando la técnica ion product scan o de decaimiento post-fuente de masas proporciona información acerca de la secuencia. La información obtenida a partir de una secuencia es indudablemente más precisa que la proporcionada por la masa molecular. Por tanto, la secuencia de un péptido suficientemente largo puede ser suficiente para identificar una proteína que ha sido caracterizada previamente. Es posible llevar a cabo un análisis manual del espectro de masa para determinar la secuencia tomando como base los iones (de la serie b y y) generados. Los datos también pueden interpretarse en bases de datos, a este tipo de búsqueda se le denomina MS/MS no interpretado (uninterpreted MS/MS). Y generalmente basta con obtener la secuencia de 2 péptidos para identificar una proteína de un genoma conocido. Son muchas las bases de datos disponibles para realizar búsquedas de este tipo, sin embargo presentan problemas en el análisis cuando existen modificaciones y polimorfismo en las proteínas. 6.3 Secuenciación de novo.
El objetivo de esta aproximación es obtener secuencias completas de aminoácidos que se deduzcan de novo de los espectros MS/MS (bien mediante interpretación manual o con ayuda de algoritmos) y buscar homologías frente a proteínas presentes en bases de datos. La secuenciación de novo es necesaria cuando el análisis por MFP o MS/MS no interpretado no produce resultados confiables. Esto es común en casos en que el genoma del organismo del cual procede la proteína no ha sido caracterizado completamente. Aunque la proteína de interés pueda no ser identificada o sea una proteína desconocida, la información de la secuencia puede ser utilizada para clonar el gen correspondiente. En condiciones de baja energía, el ión precursor se fragmenta para producir iones –b y –y de longitudes variadas. La secuencia de los iones de ambas series es complementaria, de manera que es posible obtener una secuencia completa aún cuando existan gaps en la secuencia de una u otra serie. Cuando los péptidos son obtenidos por digestión con tripsina, el extremo C- terminal de los mismos es arginina o lisina. En un espectro MS/MS, el residuo C-terminal se puede observar en el fragmento correspondiente al ión-y1. Por tanto, el punto de inicio de la serie y siempre será conocido. A la masa observada del ión-y1 hay que añadir la masa de una molécula de agua y de un protón. Si la masa del ión– y1 es de 175 Da, entonces el residuo es arginina; en cambio, si la masa es de 147 Da el residuo es lisina. Una vez identificado el ión– y1 en el espectro, la secuencia de la serie puede iniciarse a partir de ese punto.
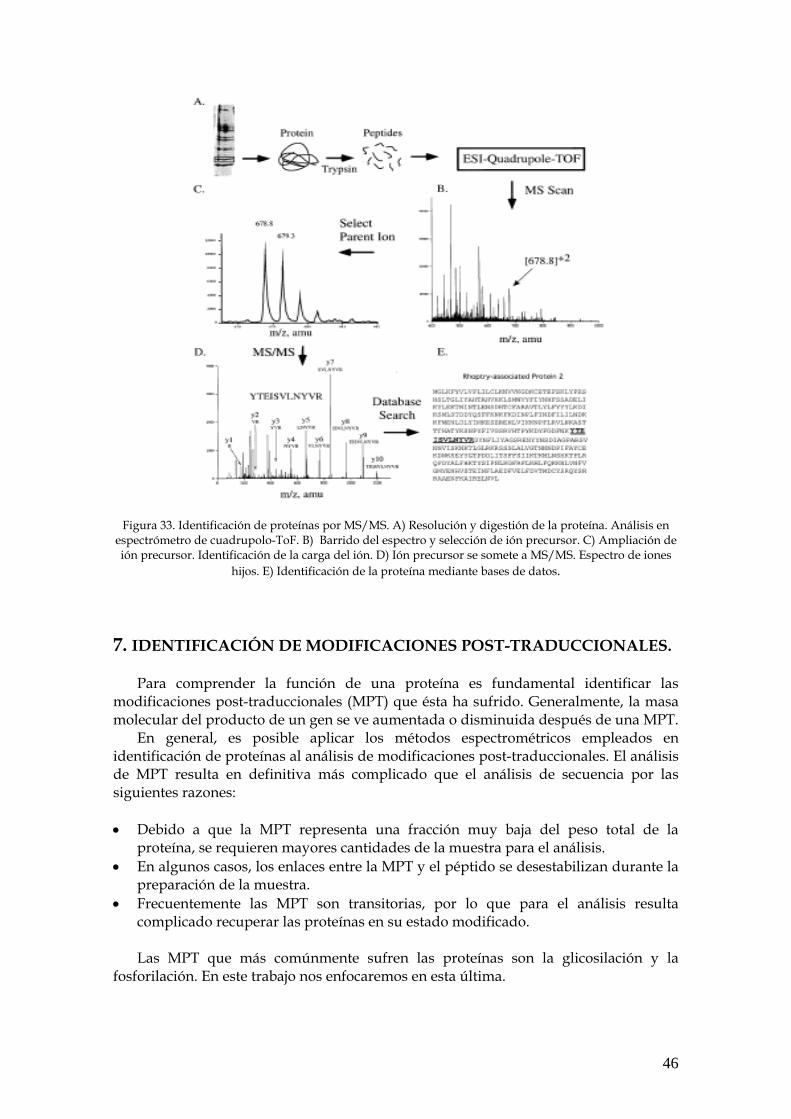
46
Figura 33. Identificación de proteínas por MS/MS. A) Resolución y digestión de la proteína. Análisis en espectrómetro de cuadrupolo-ToF. B) Barrido del espectro y selección de ión precursor. C) Ampliación de ión precursor. Identificación de la carga del ión. D) Ión precursor se somete a MS/MS. Espectro de iones
hijos. E) Identificación de la proteína mediante bases de datos. 7. IDENTIFICACIÓN DE MODIFICACIONES POST-TRADUCCIONALES. Para comprender la función de una proteína es fundamental identificar las modificaciones post-traduccionales (MPT) que ésta ha sufrido. Generalmente, la masa molecular del producto de un gen se ve aumentada o disminuida después de una MPT. En general, es posible aplicar los métodos espectrométricos empleados en identificación de proteínas al análisis de modificaciones post-traduccionales. El análisis de MPT resulta en definitiva más complicado que el análisis de secuencia por las siguientes razones: • Debido a que la MPT representa una fracción muy baja del peso total de la
proteína, se requieren mayores cantidades de la muestra para el análisis. • En algunos casos, los enlaces entre la MPT y el péptido se desestabilizan durante la
preparación de la muestra. • Frecuentemente las MPT son transitorias, por lo que para el análisis resulta
complicado recuperar las proteínas en su estado modificado. Las MPT que más comúnmente sufren las proteínas son la glicosilación y la fosforilación. En este trabajo nos enfocaremos en esta última.

47
7.1 Identificación de sitios de fosforilación. Identificar los sitios de fosforilación en una proteína proporciona información acerca de los mecanismos de regulación enzimática, así como de las enzimas involucradas (cinasas y fosfatasas). Para estudiar la fosforilación de proteínas in vivo se utiliza un marcaje de las fosfoproteínas con 32P inorgánico. Se puede determinar los sitios de fosforilación de una proteína utilizando el método de degradación de Edman o espectrometría de masas. 7.1.1 Enriquecimiento de fosfoproteínas. El enriquecimiento de fosfoproteínas en una célula favorece la identificación de proteínas fosforiladas que se encuentran en baja abundancia y que de otra manera no serían detectadas. Para enriquecer fosfoproteínas se emplean anticuerpos que reconocen proteínas con tirosina fosforilada en baja abundancia, estos anticuerpos pueden utilizarse en ensayos de inmunoprecipitación. Otros métodos de enriquecimiento involucran la conversión de residuos de fosfoserina a residuos biotinilados; las proteínas derivatizadas pueden purificarse por cromatografía de afinidad utilizando avidina, por lo que utilizando esta técnica es posible aislar es fosfoserin-proteoma de una célula. Esta técnica no modifica residuos de fosfotirosina y fosfotreonina. Empleando Fe, Al y Ga en columnas de cromatografía de afinidad, también es posible llevar a cabo el enriquecimiento de proteínas debido a la alta afinidad que presenta el grupo fosfato por estos metales. 7.1.2 Determinación de sitios de fosforilación mediante la degradación de Edman. El método de degradación de Edman se utiliza ampliamente para determinar sitios de fosforilación en proteínas marcadas con 32P. En este método las proteínas son digeridas y los fosfopéptidos generados se purifican por RP-HPLC. Los péptidos purificados se fijan a membranas inertes en su extremo C-terminal. La membrana se somete a varios ciclos de degradación de Edman. El 32P liberado es cuantificado en un contador de centelleo. Este método posiciona el fosfoamino en la secuencia del fosfopéptido. 7.1.3. Determinación de sitios de fosforilación mediante espectrometría de masas. La espectrometría de masas permite la secuenciación directa de fosfopéptidos y la identificación de los sitios de fosforilación. Debido a que una MPT puede alterar el patrón de fragmentación de una proteína, el tratamiento de la muestra previo al análisis de los sitios de fosforilación por MS puede ser muy diferente del que se aplica en análisis de secuencias también por este método. De la mezcla de péptidos, producto de la fragmentación enzimática o química de la proteína, se pueden identificar los fosfopéptidos utilizando el método de precursor ion scan. Para este fin, el tercer cuadrupolo se fija en el valor de masa del ión reportero del

48
grupo fosfato (PO3- ) con una relación carga/masa de 79. La mezcla de péptidos se rocía bajo condiciones básicas o neutras, y sólo se detectan los péptidos que al fragmentarse generan un ión con m/z igual a 79. Una vez que se detecta un fosfopéptido, la mezcla se rocía bajo condiciones ácidas y el análisis de la secuencia se realiza por MS/MS convencional. Durante la fragmentación del péptido los residuos modificados pueden ser identificados por la formación de productos específicos de la β–eliminación del fosfoaminoácido. La fosfoserina produce deshidroalanina (69 Da), mientras la fosfotreonina produce deshidroamino-2 ácido butírico. Utilizando la técnica de neutral loss scan, también es posible identificar sitios de fosforilación. En esta técnica, el tercer cuadrupolo está fijo en un offset de 49; este valor corresponde a la masa de ácido fosfórico en un ión con doble carga. Durante la colisión de los péptidos, los residuos de serina y treonina pierden ácido fosfórico fácilmente, aún en colisiones de baja energía. Por tanto, cualquier péptido con doble carga que pierda 49 Da es identificado como fosfopéptido.

49
8. BIBLIOGRAFÍA Gaal, O., Medgyesi, G.A. and Vereczkey, L. (1984). Electrophoresis in the separation of biological macromolecules. Akademiai Kiado Budapest and Jhon Wiley and Sons. García, P.H.M. (2000). Electroforesis en geles de poliacrilamida: Fundamentos, actualidad e importancia. Universo Diagnóstico 1(12):31-41. Gil, C. (2003). La metodología proteómica, una herramienta para la búsqueda de función. Actualidad SEM (35): 12-20. Graves, R. P. and Haystead, A.J.T. (2002). Molecular Biologist´s Guide to Proteomics. Microbiology and Molecular Reviews 66(1): 39-63. Pitarch, A., Sánchez, M., Nombela, C. and Gil, C. (2003). Analysis of the Candida albicans proteome I. Strategies and applications. Journal of Chromatography B787: 101-128. Rabilloud, T. (2000). Proteome Research: Two-Dimensional Gel Electrophoresis and Identification Metods. Springer. Germany. Simpson, R.J. (2003). Proteins and Proteomics A Laboratory Manual. Cold Spring Harbor, New York. USA. Westermeier, R. And Naven, T. (2002). Proteomics in Practice. A laboratory manual of proteome analysis. Wiley-VCH Verlag-Gmbh (eds). Weinheim, Germany. Sitios Web: www.abrf.org/ABRFNews/1997/June1997/jun97lennon.html www.chm.bris.ac.uk/ms/theory/tandem-ms.html www.cultek.com/index.asp?p=Soluciones-Electroforesis-Principal www.chm.bris.ac.uk/ms/theory/maldi-ionisation.html www.javeriana.edu.co/Facultades/Ciencias/neurobioquimica/libros/celular/electroforesis.html www.waters.com/watersdivision/ContentD.asp?watersit=EGOO-66MNYR&WT.svl=1


















