“Nanohíbridos de puntos cuánticos de CdS y Polímeros ... · PDF...
120
1 CENTRO DE INVESTIGACIÓN EN QUÍMICA APLICADA TESIS “Nanohíbridos de puntos cuánticos de CdS y Polímeros Conjugados del tipo fenilenoetinileno” Presentada por: Marcos Joaquín Guillén Cruz Para obtener el grado de: Doctorado en Tecnología de Polímeros Asesores Dra. Ivana Moggio Dr. Ronald Francis Ziolo Saltillo, Coahuila Septiembre, 2014
Transcript of “Nanohíbridos de puntos cuánticos de CdS y Polímeros ... · PDF...

1
CENTRO DE INVESTIGACIÓN EN QUÍMICA APLICADA
TESIS
“Nanohíbridos de puntos cuánticos de CdS y Polímeros
Conjugados del tipo fenilenoetinileno”
Presentada por:
Marcos Joaquín Guillén Cruz
Para obtener el grado de:
Doctorado en Tecnología de Polímeros
Asesores
Dra. Ivana Moggio Dr. Ronald Francis Ziolo
Saltillo, Coahuila Septiembre, 2014

2

3

4

5
Dedicada a mis Padres (Joaquín Guillén y Reina Adelita Cruz)
y mis hermanos (Víctor Hugo Guillén y Reyna Victoria Guillén),
que siempre me han apoyado incondicionalmente

6
Mi más sincero agradecimiento a todas las personas que contribuyeron en el desarrollo de este
proyecto. En particular a:
La Dra. Ivana Moggio, el Dr. Ronald Francis Ziolo y el Dr. Eduardo Arias Marín por su dedicación
y asesoría en el desarrollo de esta tesis.
La L.C.Q. Gabriela Padrón Gamboa y el Dr. Antonio Ledezma Pérez por el apoyo técnico
constante y por su amistad.
La L.C.Q. María Guadalupe Méndez Padilla, MMC. Ma. Luisa López Quintanilla, M.C. Silvia
Torres Rincón, M.C. Jesús Ángel Cepeda Garza, M.C. Enrique Díaz Barriga Castro, Dr. José
Manuel Mata Padilla por la asistencia técnica para la caracterización Fisicoquímica y
Microscópica de los materiales.
El Dr. Darío Bueno Baques, Dr. Dámaso Navarro Rodríguez, Dr. Carlos Alberto Ávila
Orta por sus comentarios.
Mis queridos compañeros Lizeth García, Enrique Arias, Reyes, Diana, Laura Evelyn, Isela,
Marco Polo, Marlene Rodríguez, Pedro, Jessika González, Karla Gutiérrez, Miguel
Macías, Lidia, Arxel, Herlinda, Lydia Joanna, Denisse Azeneth, Anayantzi por esos
inolvidable momentos de alegría, consejos y porque siempre estuvieron ahí para apoyarme.
El Centro de Investigación en Química Aplicada, por facilitar la infraestructura que hizo
posible este trabajo.
El Consejo Nacional de Ciencia y Tecnología, por el apoyo económico a través de la beca
de doctorado No. 207893, el proyecto de Ciencia Básica SEP-CONACyT 98513-R, el
proyecto de colaboración CONACyT-American Air Force Office of Scientific Research
((1010/189/10C-286-10)-(FA5990-10-1-0236) y el proyecto 232753 Laboratorio Nacional
de Materiales Grafénicos.

7
Índice de contenido
Lista de figuras .............................................................................................................................. 10
Lista de tablas ............................................................................................................................... 14
Lista de acrónimos ........................................................................................................................ 14
Resumen ........................................................................................................................................ 16
Introducción .................................................................................................................................. 17
Capítulo 1.- Antecedentes ............................................................................................................. 19
1.1.- Celdas solares .................................................................................................................... 19
................................................................................................................................................ 21
1.1.1.- Celdas solares inorgánicas .......................................................................................... 22
1.1.2. Celdas solares orgánicas .................................................................................................. 23
1.1.3. Celdas solares híbridas con puntos cuánticos .............................................................. 30
1.2.- Nanocomposites de puntos cuánticos y polímeros ............................................................ 32
1.2.1.- Síntesis de puntos cuánticos ....................................................................................... 32
1.2.2.- Síntesis de polí(fenilenoetinileno)s ............................................................................. 34
1.2.3.- Síntesis de nanocomposites de puntos cuánticos con polímeros ................................ 38
1.3.- Estado del arte ................................................................................................................... 39
Capitulo 2: Enfoque de la tesis ..................................................................................................... 43
2.1.- Justificación ................................................................................................................... 43
2.2.- Hipótesis ............................................................................................................................ 48
2.3.- Objetivos ........................................................................................................................... 48
2.3.1. Objetivo particulares .................................................................................................... 48
Capitulo 3: Desarrollo experimental ............................................................................................. 49
3.1.- Síntesis de monómeros y polímeros .................................................................................. 49
3.1.1.- Reactivos ..................................................................................................................... 49
3.1.2.- Síntesis de monómeros halogenados .......................................................................... 51
3.1.2.1.- 1,4-bis(dodecanoxi)benceno(DOB)(2) ................................................................. 51
3.1.2.2.- 1,4-bis(dodecanoxi)-2-5diiodobenceno (DIDOB) (3) .......................................... 51
3.1.2.3.- (11-undecanol) 2,5-dibromobenzoato(5) .............................................................. 52
3.1.2.4.- (11-undecanol) 2,5-bis((trimetilsilil)etinilen)benzoato (6) ................................... 53

8
3.1.2.5.- (11-undecanol) 2,5-bis(etinil)benzoato (7) ........................................................... 54
3.1.2.6.- ácido 2-(3,3-dietil) triazenil)-5-iodobenzoico (9) ................................................. 54
3.1.2.7.- 11-hidroxilundecil 2-(3,3-dietil) triazenil)-5-iodobenzoato (10) .......................... 55
3.1.2.8.-(11-undecanol) 2-(3,3-dietil)triazenil)-5-((trimetilsilil)etinilen)benzoato (11) ..... 56
3.1.2.9.- (11-undecanol)-2-iodo-5-((trimetilsilil) etinilen)benzoato (12) ........................... 57
3.1.2.10.- (11-undecanol)-2-iodo-5-(etinil) benzoato (13) ................................................. 57
3.1.3. Síntesis de polímeros conjugados ................................................................................ 58
3.1.3.1.- co-polímero pPE(OC12-Co-BzC11OH) (14) ....................................................... 58
3.1.3.2.- Síntesis del homo-polímero conjugado pPE(BzC11OH)(15) .............................. 59
3.1.4.- Síntesis de puntos cuánticos ....................................................................................... 60
3.1.4.1.- Síntesis de puntos cuánticos de CdS(MPS) (17) ................................................. 60
3.1.5.- Síntesis de nanohíbrido ............................................................................................... 61
3.2.- Instrumentos y métodos de caracterización ...................................................................... 62
3.3- Celdas solares ..................................................................................................................... 65
3.3.1.-Materiales ..................................................................................................................... 65
4.1-Síntesis de monómeros ........................................................................................................ 67
4.1.1- Ruta de síntesis ............................................................................................................ 67
4.1.2. Mecanismos de reacción. ............................................................................................. 70
4.1.2.1. Mecanismos de reacción involucrados en la síntesis del 1,4-bis(dodecanoxi)-2,5-
diiodobenceno. .................................................................................................................... 70
4.1.2.2. Mecanismos de reacción involucrados en la síntesis del (11-undecanol) 2,5-
dietinilbenzoato. .................................................................................................................. 71
4.1.2.3. Mecanismos de reacción involucrados en la síntesis del monómero (11-undecanol)
2-[(3,3-dietil) trazenil]-5-etinil benzoato (13) .................................................................... 74
4.1.3.- Caracterización por resonancia magnética nuclear de 1H ........................................... 75
4.2.- Síntesis de polímeros ......................................................................................................... 80
4.2.1- Síntesis del co-polímero ............................................................................................... 80
4.2.1.1- Ruta de síntesis ...................................................................................................... 80
4.2.1.2- Mecanismo de reacción ......................................................................................... 80
4.2.2.- Caracterización por resonancia magnética nuclear ..................................................... 80
4.2.2.1.- Resonancia magnética nuclear de protón 1H ........................................................ 80

9
4.2.2.2.- Resonancia magnética nuclear de carbono 13C ..................................................... 82
4.2.2- Síntesis del homopolímero ........................................................................................... 83
4.2.2.1- Ruta de síntesis del homopolímero ........................................................................ 83
4.2.1.2- Mecanismo de reacción ......................................................................................... 84
4.2.3.- Caracterización por resonancia magnética nuclear de 1H ........................................... 84
Capitulo 5: Resultados y discusiones-síntesis y caracterización de puntos cuánticos CdS(MPS) 85
5.1.- Síntesis .............................................................................................................................. 85
5.2.- Caracterización .................................................................................................................. 87
5.2.1.- Espectroscopia de resonancia magnética nuclear de estado sólido (SS-NMR de las
siglas en ingles) ...................................................................................................................... 87
5.2.2.- Espectroscopia fotoelectrónica de rayos x (XPS) ....................................................... 88
5.2.3.- Análisis termogravimétrico (TGA) ............................................................................. 92
5.2.4.- Microscopia electrónica de transmisión...................................................................... 94
5.4.- Propiedades ópticas ........................................................................................................... 95
Capitulo 6: Resultados y discusiones-síntesis y caracterización de nanohíbrido de puntos cuánticos
con el co-polímero ........................................................................................................................ 97
6.1.- Síntesis .............................................................................................................................. 97
6.2.- Caracterización .................................................................................................................. 97
6.2.1.- Resonancia magnética nuclear de estado sólido (SS-NMR)....................................... 97
6.3.- Propiedades electroquímicas ........................................................................................... 104
6.4.- Propiedades fotofísícas .................................................................................................... 106
Capítulo 7: Resultados y discusiones-celdas solares .................................................................. 110
7.1.- Consideraciones finales ................................................................................................... 113
8. - Conclusiones ......................................................................................................................... 116
Trabajo a futuro ........................................................................................................................... 117
Esta tesis es la primera del grupo en investigar la posibilidad de utilizar nanohíbridos de puntos
cuánticos directamente funcionalizados con polímeros conjugados, por lo que se abren varías
líneas futuras a desarrollar; ......................................................................................................... 117
Referencias .................................................................................................................................. 118

10
Lista de figuras
Figura 1.- Irradiancia solar diaria en México ................................................................................ 19
Figura 2.- Esquema de mecanismo de fotogeneración en celdas solares ..................................... 20
Figura 3.- Espectro de irradiancia del sol ..................................................................................... 21
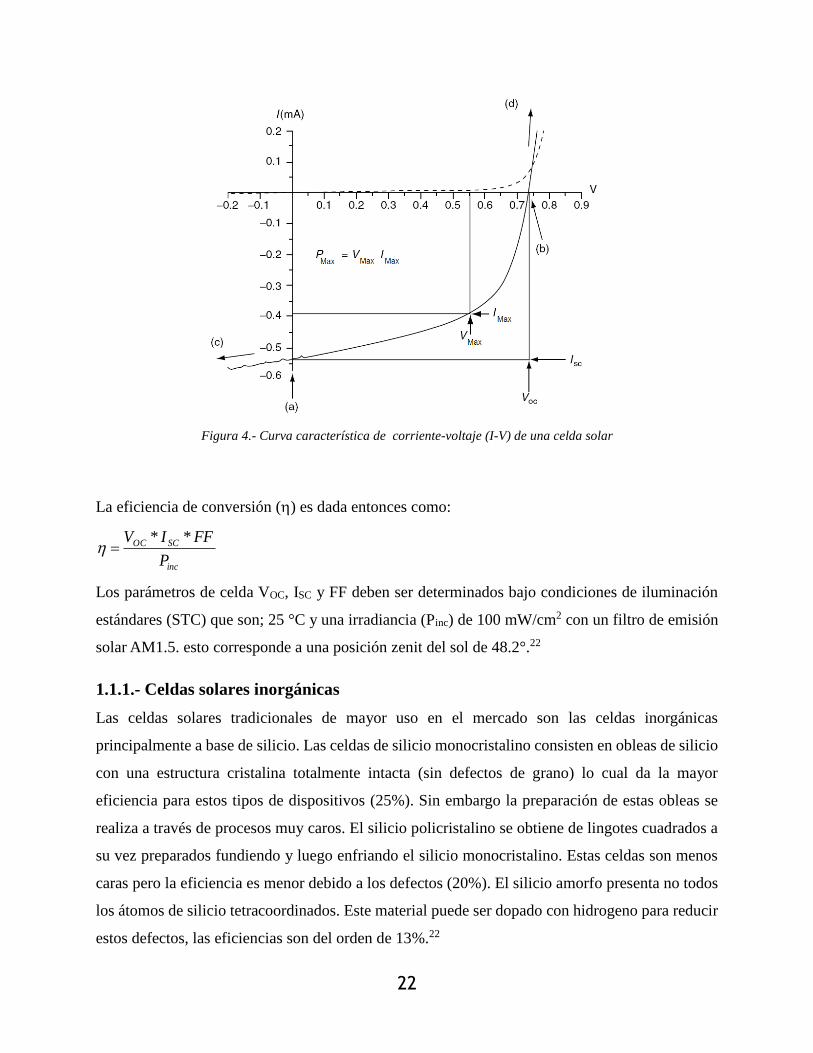
Figura 4.- Curva característica de corriente-voltaje (I-V) de una celda solar .............................. 22
Figura 5.- Esquema de proceso de transferencia de carga en celdas inorgánicas (a) y orgánicas (b)23
................................................................................................................................................ 23
Figura 6.- Esquema de un excitón para el caso del poli(fenilenovinilideno) ................................ 24
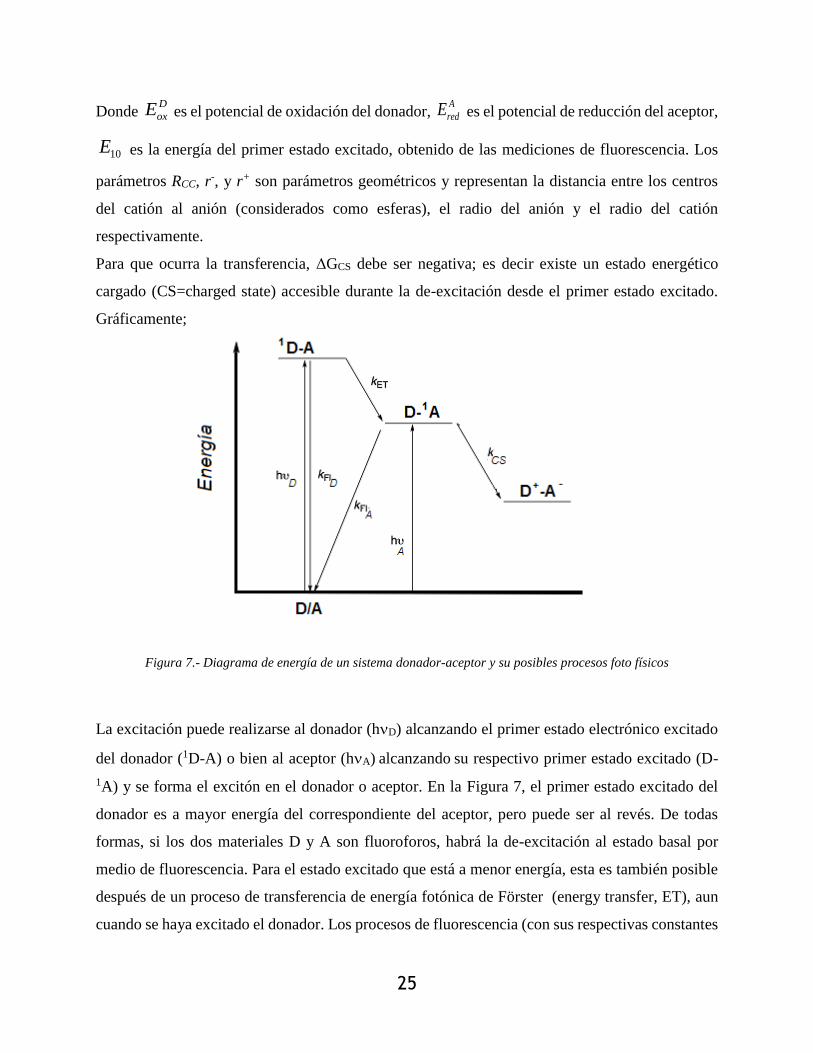
Figura 7.- Diagrama de energía de un sistema donador-aceptor y su posibles procesos foto físicos
................................................................................................................................................ 25
Figura 8.- Configuraciones de una celda solar orgánica ............................................................... 27
Figura 9.- Estructura de algunos de los materiales más utilizados en celdas solares orgánicas ... 28
Figura 10.- Proceso de transferencia de energía en celdas solares híbridas de polímeros conjugados
y puntos cuánticos (QD de quantum dots en la figura). Adaptado de ref.42........................... 31
Figura 11.- Niveles energéticos de varios materiales utilizados en celdas solares híbridas.
Adaptado de ref.42 .................................................................................................................. 31
Figura 12.- Esquema de la formación de nanocristales según el modelo de LaMer y Dinegar.
Adaptado de ref.45 .................................................................................................................. 32
Figura 13.-Especies producidas durante la primera etapa descrita por Amatore. ......................... 35
Figura 14.- Adición oxidativa de la segunda etapa descrita por Amatore. ................................... 35
Figura 15.-Ciclo catalítico del Paladio propuesto por Amatore para el acoplamiento de halogenuros
de arilo y acetilenos. ............................................................................................................... 36
Figura 16.- Esquema de la formación de nanocristales funcionalizados con PPV, adaptada de ref.3
................................................................................................................................................ 39
Figura 17.- Celda solar tandem orgánica60 ................................................................................... 41
Figura 18.- Eficiencias reportadas para varias celdas base orgánico. Adaptado de ref.71 UCSB:
Universidad de California, Santa Barbara; UCSC: Universidad de California, Santa Cruz; UC
Berkeley: Universidad de California, Berkeley; UCLA: Universidad de California, Los
Angeles; TU/e: Technische Universiteit Eindhoven (Alemania), Universidad de Linz,

11
Austria.Konarka: empresa localizada en Massachussets; Wake Forest: universidad en
California; Cambridge: Inglaterra; ECN: empresa de Holanda. ............................................ 44
Figura 19.- Esquema de trabajo necesario para llegar a una celda solar eficiente ........................ 45
Figura 20.- Materiales de estudio en esta tesis ............................................................................. 47
Figura 21.- Esquema de obtención de DOB ................................................................................. 51
Figura 22.- Esquema de obtención de DIDOB(3) ........................................................................ 52
Figura 23.- Esquema de obtención de (5) ..................................................................................... 53
Figura 24.- Esquema de obtención de (6) ..................................................................................... 53
Figura 25.- Esquema de obtención de (7) ..................................................................................... 54
Figura 26.- Esquema de obtención de (9) ..................................................................................... 55
Figura 27.- .- Esquema de obtención de (10) ................................................................................ 55
Figura 28.- Esquema de obtención de (11) ................................................................................... 56
Figura 29.- Esquema de obtención de (12) ................................................................................... 57
Figura 30.- Esquema de obtención de (13) ................................................................................... 57
Figura 31.- Esquema de obtención de (14) ................................................................................... 58
Figura 32.- Esquema de obtención de (15) ................................................................................... 59
Figura 33.- Esquema de obtención de (17) ................................................................................... 60
Figura 34.- Esquema de la Síntesis de los diferentes composite .................................................. 61
Figura 35.- Configuración de las celdas solares ........................................................................... 65
Figura 36.- Ruta de síntesis para obtener el monómero halogenado (3) ...................................... 68
Figura 37.- Ruta de síntesis para obtener el monómero (11-undecanol) 2,5-
bis((trimetilsilil)etinilen)benzoato (7) .................................................................................... 68
Figura 38.- Ruta de síntesis para obtener el monómero bifuncional (13). .................................... 69
Figura 39.- Mecanismos de reacción en la alquilación de Williamson. ........................................ 70
Figura 40.- Mecanismo de reacción para la obtención del (3) ...................................................... 71
Figura 41.- Mecanismo de esterificación mediante una amidina (DBU) ..................................... 71
Figura 42.- Ciclo catalítico de reacción de Sonogashira para acoplar un acetileno con un
halogenuro de arilo. ................................................................................................................ 72
Figura 43.- Mecanismo de reacción involucrado en la desprotección del grupo acetileno. ......... 73
Figura 44.- Mecanismo de reacción para la síntesis del monómero triazeno (9). ........................ 74
Figura 45.- Mecanismo de reacción involucrado en la sustitución del triazeno por el iodo ......... 75

12
Figura 46.- 1H RMN del precursor para formar el 2,5-bis(dodecanoxi)-1,4-diiodobenzeno en
CDCl3. Arriba; espectro de 2 y debajo de 3. .......................................................................... 76
Figura 47.- 1H RMN de los intermediarios para la formación del (11-undecanol)-2,5-dietilbenzoato
(7) en CDCl3. Desde arriba hacia abajo; 5,6 y 7. ................................................................... 77
Figura 48.- 1H RMN de los intermediarios para la formación del (11-undecanol)-2-yodo-5) etinil)
benzoato (13) en CDCl3. Desde arriba hacia abajo; 9-13. ..................................................... 79
Figura 49.- Ruta de síntesis de Sonogashira para la preparación del co-polímero (14) ............... 80
Figura 50.- 1H RMN del co-polímero de estudio en este trabajo en CDCl3. ................................ 81
Figura 51. -13C RMN del co-polímero de estudio en este trabajo en CDCl3. ............................... 83
Figura 52.- Ruta de síntesis de Sonogashira para la preparación del homopolímero (15) ........... 83
Figura 53.- 1H RMN del homopolímero (15) en CDCl3. .............................................................. 84
Figura 54.- Representación esquemática de las posibles estructuras de CdS empleando (3-
mercaptopropil) trimetoxisilano (MPS) como ligante. R=CH3/H. ......................................... 86
Figura 55.- Espectros de Izquierda: 13C SSNMR de CdS(MPS) (12064 barridos). *=DMF.
R=CH3/H. Las estructuras insertadas corresponden a algunas de la Figura 54, a fin de mejor
visualizar la asignación de los picos. ..................................................................................... 88
Figura 56.- Espectro XPS deconvolucionado de C 1s para CdS(MPS) ........................................ 89
Figura 57.- Espectro XPS deconvolucionado de O 1s para CdS(MPS) ....................................... 89
Figura 58.- Espectro XPS deconvolucionado de Si 2p para CdS(MPS)....................................... 90
Figura 59.- Espectro XPS deconvolucionado de S 2p para CdS(MPS) ........................................ 91
Figura 60.- Espectro XPS deconvolucionado de Cd 3d para CdS(MPS) ..................................... 91
Figura 61.- Termograma de CdS(MPS). ....................................................................................... 93
Figura 62.- Imagen TEM del CdS(MPS) y su distribución de tamaño (insertada). ...................... 94
Figura 63.- Imagen de difracción de electrones del CdS(MPS) anillo 1, 2, 3 para los planos (100),
(200), (220) respectivamente.................................................................................................. 95
Figura 64.- Espectro de UV-Vis (línea negra), y fluorescencia (línea azul) de CdS(MPS) en DMF.
Solución con absorbancia a la longitud de onda de excitación<0.1. Figura insertada: zoom del
espectro UV-Vis de la región entre 300-500 nm.................................................................... 96
Figura 65.- Espectro de 13C CP/MAS SSNMR (a) CdS(MPS) (scans=12064), (b) pPE(OC12-Co-
BzC11OH) (scans=3832), (c) CdS(pPE)(H+) (scans 1062CdS(pPE) (OH-) (scans=4400). .. 98

13
Figura 66.- Espectro de 29Si CP/MAS SSNMR (a) CdS(MPS) (scans=2800), (b) CdS(pPE)(H+)
(scans=27000) (a) CdS(pPE) (OH-) (scans=26696). .............................................................. 99
Figura 67.- Imagen STEM CdS(pPE)(H+) .................................................................................. 100
Figura 68.- Imagen STEM de arriba hacia abajo: CdS(pPE)(OH-) y CdS(pPE) neutro. ............ 101
Figura 69.- Imagen TEM de CdS(pPE)(H+) a mayor magnificación, detalle de una superpartícula.
.............................................................................................................................................. 101
Figura 70.- Imagen STEM de, desde arriba hacia abajo: CdS(MPS), pPE(OC12-Co-BzC11OH)
(DMF:THF) y CdS(pPEOC12). ........................................................................................... 103
Figura 71.- Voltagramas en solución de CdS(MPS) pPE(OC12-Co-BzC11OH), y CdS(pPE). 104
Figura 72.- Comparación de los niveles energéticos HOMO-LUMO del pPE(OC12-Co-
BzC11OH) y CdS(MPS). ..................................................................................................... 105
Figura 73.- Espectros UV-Vis del pPE(OC12-Co-BzC11OH) (THF, línea negra), nanohíbrido en
THF:DMF (línea verde) y CdS(MPS) en DMF (línea roja) ................................................ 106
Figura 74.- Espectro de fluorescencia de CdS(pPE) en THF:DMF ............................................ 107
Figura 75.- Espectro de excitación de CdS(pPE) en THF:DMF y del co-polímero en THF ..... 107
Figura 76. Decaimiento de fluorescencia del co-polímero, puntos cuánticos y nanohíbrido. En la
leyenda se reporta la longitud de onda de emisión al cual se obtuvieron las diferentes curvas.
.............................................................................................................................................. 108
Figura 77.- Curva densidad de corriente vs Voltaje de una celda de CdS(pPE) ....................... 110
Figura 78. Curvas densidad de corriente vs Voltaje para celdas de co-polímero (puntos rojos) y
nanohíbrido (cuadrados negros) con PCBM 1:3. ................................................................. 111
Figura 79. Diagrama energético de celdas del co-polímero y CdS(pPE) con PCBM 1:3. ....... 112

14
Lista de tablas
Tabla 1.- Porcentaje en átomo de C,S,Si,O y C obtenidos a partir de la integración de los picos
XPS correspondientes ............................................................................................................ 92
Tabla 2.- Propiedades electroquímicas en solución de los materiales: partículas de CdS, co-
polímero, CdS(pPE) por Voltametría Cíclica. ..................................................................... 105
Tabla 3.- Propiedades fotofisícas de CdS(pPE) y sus precursores, CdS(MPS) y co-polímero
pPE(OC12-Co-BzC11OH) ................................................................................................... 108
Tabla 4.-Parámetros fotovoltaicos de celdas de co-polímero y CdS(pPE) con PCBM 1:3 ........ 111
Lista de acrónimos
Acrónimo Significado
DEDOB 1,4-dietinil-2,5-bis(dodecanoxi)benceno
RMN 1H Resonancia magnética nuclear de protón
GPC Cromatografía de permeación en gel
UV-Vis Espectroscopia ultravioleta-visible
pPE(OC12-Co-BzC11OH) Co-polímero de estudio
MPS 3-(mercaptopropil)-trimetoxisilano
SS-NMR Resonancia magnética nuclear en estado sólido (de
las siglas en ingles)
STEM Microscopía electrónica de transmisión y barrido
VCS Voltametría cíclica en solución
LUMO Lowest Unoccupied Molecular Orbital
CdS(MPS) Nanopartículas de sulfuro de cadmio estabilizadas
con el agente MPS

15
CdS(pPE) Composite obtenido por la funcionalización de
Nanopartículas CdS(MPS) y el polímero conjugado
pPE(OC12-Co-BzC11OH)
TOPO oxido de tri-n-octilfosfina
PC Polímeros conjugados
PT poli(tiofeno)
PPV poli(parafenilenovinilidenos)
PPE poli(parafenilenoetinilenos)
HOMO Highest Occupied Molecular Orbital
QD Quantum dots
P3HT poli(3-hexiltiofeno)
DOPO-Br Oxido de [(4-bromofenil)metil]dioctilfosfino
PAE poli(arilenoetinilenos)
PPF poli(p-fenilenos)
PFV poli(fenilenovinilidenos)
C60 Molécula de fulereno
PCPDTBT poli[2,6- (4,4-bis(2-ethilhexil)-4H-cyclopenta[2,1-
b;3,4-b′]-dithiofeno)-alt-4,7- (2,1,3-benzotiadiazolo)]
PCBM fenil C61 ácido butírico metil éster
DBU 1,8,diazabiciclo[5.4.0]unde-7-eno
NEt3 Trietilamina
THF Tetrahidrofurano

16
Resumen
En este proyecto de tesis se realizó la síntesis de puntos cuánticos de Sulfuro de Cadmio
estabilizados con el 3-(mercaptopropil)-trimetoxisilano (MPS) para su sucesiva funcionalización
con un polímero fenilenoetinileno de nueva síntesis por medio de una reacción de condensación
entre los grupos hidroxilos de la cadena lateral del polímero y los grupos metoxi terminales del
MPS. Los puntos cuánticos se caracterizaron por TEM y HRTEM, XPS, TGA, resonancia
magnética nuclear en estado sólido (SS-NMR), rayos X, UV y fluorescencia. El tamaño promedio
obtenido por TEM fue de 3.02 ± 0.46 nm con estructura cristalográfica cubica zinc blenda. La
formación del composite fue corroborada al observarse el desplazamiento perteneciente al enlace
Si-O-CH2- en SS-NMR. Superestructuras de tipo vesicular de 200-600 nm fueron observados
mediante microscopía electrónica de transmisión y barrido (STEM). Los resultados obtenidos por
voltametría cíclica en solución muestran que el nivel LUMO (Lowest Unoccupied Molecular
Orbital) del polímero es 0.5911 eV superior al de las nanopartículas de sulfuro de cadmio
sugiriendo que el composite cumple con los requerimientos energéticos para poder funcionar en
un dispositivo fotovoltaico. Sin embargo el estudio fotofísico reveló que las propiedades de
fluorescencia del co-polímero quedan prácticamente iguales en el composite, y el rendimiento
cuántico del co-polímero no disminuye. Esto puede explicase por la formación de las
superestructuras la cual origina una distancia grande entre co-polímero y puntos cuánticos. Como
consecuencia de esto, la celda del composite no dio corriente fotogenerada, sin embargo se obtuvo
un incremento en el valor de voltaje de circuito abierto VOC y por ende de la eficiencia al mezclar
el composite con el derivado del fulereno PCBM con respecto a la celda del puro co-polímero con
PCBM, debido a la disminución del potencial de oxidación y por ende menor valor de HOMO.
NFBU4 Fluoruro de tetrabutil amonio
DOB 1,4-bis(dodecanoxi)benceno
DIDOB 1,4-bis(dodecanoxi)-2-5diidobenceno
Et2NH dietilamina
PEDOT:PSS poli (3,4-etilendioxi-2,4-tiofeno)-poliestirensulfonato

17
Introducción
En la actualidad la mayoría de las celdas solares comerciales son fabricadas de materiales
semiconductores inorgánicos como el silicio altamente puro, lo que eleva considerablemente su
costo así como el del proceso de fabricación. En la búsqueda de nuevas alternativas han surgido
materiales con características que los hacen aptos en la aplicación como dispositivos fotovoltaicos.
Entre éstos materiales se encuentran los semiconductores orgánicos, un ejemplo de ellos son los
polímeros conjugados(PC)1-3, los cuales poseen en su estructura principal una combinación de
compuestos insaturados como de anillos aromáticos con dobles y triples enlaces conjugados, dando
lugar a una elevada deslocalización electrónica y por consiguiente estos exhiben propiedades
semiconductoras4 de interés.
Un ejemplo de polímeros conjugados utilizados son los de tipo poli(tiofeno) (PT),
poli(parafenilenovinilidenos) (PPV) y poli(parafenilenoetinilenos) (PPE)5 los cuales presentan la
habilidad para ser electrón-donadores, sin embargo los PC presentan una menor eficiencia con
respecto a la de los semiconductores inorgánicos, ya que absorben en una región limitada del
espectro de emisión solar y además requieren de una morfología controlada de la superficie activa
de la celda solar.
Por otro lado un factor importante de los PC es la capacidad de generar excitones, la cual está
relacionada con su coeficiente de absorción molar, el rango de absorción en el visible, el
rendimiento cuántico, la distancia de difusión del excitón que se ha calculado ser menor a 100 nm
para los PC, además de que presentan viabilidad de procesamiento6 así como fabricación de celdas
fotovoltaicas con técnicas de bajo costo. Por lo que es necesario el uso de compuestos que
incrementen su eficiencia aprovechando las propiedades de los PC.
Un requisito en la elaboración de celdas solares orgánicas es la presencia de un grupo electrón-
donador y un grupo electrón-aceptor, por lo que se requiere el uso de compuestos con alta
capacidad de ser electrón-aceptores. Una eficiente separación de cargas está relacionada con el
hecho de que la diferencia de energía entre los niveles HOMO-HOMO (Highest Occupied
Molecular Orbital) y LUMO-LUMO del electrón-donador y el electrón-aceptor sobrepase la
energía de enlace coloumbica que une el excitón.

18
Para superar estas limitaciones y aprovechar las propiedades de los PC antes mencionadas, en los
últimos años emergieron los llamados materiales híbridos, los cuales unen de manera sinérgica las
propiedades de los semiconductores orgánicos e inorgánicos. De particular interés son los
obtenidos a partir de PC y puntos cuánticos (Quantum Dots (QD) por sus siglas en inglés) estos
semiconductores inorgánicos nanométricos poseen buena fotoconductividad7-9, así como la
posibilidad de modular sus propiedades ópticas10 según el tamaño de la nanopartícula obtenidas y
además son materiales electrón-aceptores. Entre los QD11 con mayor aplicación se encuentran, de
CdS12, PbS13, CdTe14, ZnS15, InP16, CdSe17.
Existen diferentes métodos de síntesis de materiales híbridos PC/QD siendo las principales: mezcla
física6 o elaboración de bi- o múltiples capas18. Sin embargo en ambas metodologías es difícil de
controlar la morfología provocando que solo una pequeña fracción de excitones sea capaz de
difundir por la interfase PC/QD dando bajas eficiencias en las celdas. En la actualidad se ha
encontrado que al unir químicamente un QD con PC se obtiene una buena dispersión de las
nanopartículas, contribuyendo a una mayor difusión de los excitones lo cual es crucial en celdas
fotovoltaicas.
Una clara demostración del efecto del método de síntesis de materiales híbridos PC/QD en la
eficiencia de las celdas lo reporta Xu y col.19 preparan dos nanocomposites de poli(3-hexiltiofeno)
(P3HT) con puntos cuánticos de CdSe por mezcla física y unión química. En la mezcla física se
observó claramente la separación de fases entre ambos componentes y una mala dispersión de las
nanopartículas. En la unión química el QD fue funcionalizado con [oxido(4-
bromofenil)metil]dioctilfosfino (DOPO-Br) y posteriormente acoplado con la terminación vinil
del P3HT. El material híbrido da lugar a películas con una buena dispersión de las nanopartículas
de CdSe y una mejora en el desempeño del dispositivo.
Basado en lo anterior, en este trabajo se reporta la síntesis y caracterización de un composite
híbrido a partir de la unión química entre un co-polímero conjugado del tipo fenilenoetinileno
portador de cadenas laterales que terminan con grupos hidroxilos y puntos cuánticos de CdS
estabilizadas con MPS, el estudio fisicoquímico del composite y de superestructuras que se
generan del mismo y su evaluación para la aplicación en celdas fotovoltaicas.

19
Capítulo 1.- Antecedentes
1.1.- Celdas solares
Frente a la creciente demanda de energía que ya no puede ser satisfecha por los combustibles
fósiles, como gas, carbono y petróleo así como los efectos indeseados de estos en el ambiente, se
ha incrementado la investigación en recursos de energía alternativos y renovables.20 Entre ellos, la
energía solar es la mejor alternativa ya que no produce contaminantes (a diferencia por ejemplo de
la energía nuclear), su fuente es inagotable a escala humana, dando casi 104 veces la energía
requerida para todo el planeta21. México se encuentra en una zona (ver Figura 1), que le permite
obtener una capacidad de generación eléctrica equivalente a la capacidad instalada actual de la
CFE (39,270MW). El problema radica en que a los costos actuales de la energía fotovoltaica,
(<1$US/Wp para la tecnología de película delgada) esta capacidad instalada constaría casi 39,
270MD.
Figura 1.- Irradiancia solar diaria en México
Bajo este antecedente, el gobierno está impulsando la investigación en esta área a través de varios
programas y proyectos.
Las celdas solares son dispositivos que convierten la energía solar en energía eléctrica usando el
efecto fotovoltaico. De forma general, el efecto fotovoltaico procede de la siguiente manera. (1)
El material semiconductor absorbe la luz solar lo cual provoca su excitación (2) pasando un

20
electrón (e) de la banda de valencia (si es inorgánico) o del orbital molecular ocupado de mayor
energía (HOMO de sus siglas en inglés; Highest Occupied Molecular Orbital, si es orgánico) a la
banda de conducción (inorgánico) o al orbital desocupado de menor energía (LUMO de Lowest
Unoccupied Molecular Orbital, si es orgánico) dejando una vacancia o hueco (h) en la banda de
valencia (o HOMO). 3) El electrón se transfiere al cátodo y luego 4) fluye a través del circuito
externo hasta combinarse con el hueco que pasó al ánodo. Generando de esta forma una corriente.
En la Figura 2 se muestra el mecanismo para el caso de celdas solares orgánicas. Ver también
Figura 5 más adelante.
Figura 2.- Esquema de mecanismo de fotogeneración en celdas solares
La eficiencia de una celda depende de la capacidad del material de absorber la luz solar. Teniendo
la luz solar el rango del UV-Visible y cercano IR, los materiales más prometedores son los que
presentan una brecha energética en este rango es decir deben ser materiales semiconductores.
Particularmente si hacemos referencia al espectro de emisión solar (Figura 3), podemos distinguir
su máximo alrededor de 500 nm, por lo cual los candidatos óptimos para celdas deberían de tener
un alto coeficiente de extinción molar alrededor de esta longitud o bien de 2.5 eV y que a esta
energía corresponda la transición HOMO-LUMO. Una vez excitado, el material debe poder
transferir los electrones al cátodo y los huecos al ánodo lo cual involucra las barreras de energías
entre LUMO-función de trabajo del cátodo y HOMO-función del trabajo del ánodo. Finalmente
también debe haber una alta movilidad de cargas para que los electrones puedan moverse en el
circuito.

21
Figura 3.- Espectro de irradiancia del sol
Para caracterizar una celda solar, se realizan mediciones de corriente contra voltaje bajo
iluminación, obteniendo curvas I-V como la de Figura 4. En estas curvas, los puntos relevantes
son; potencial en circuito abierto VOC que representa la máxima fuerza electromotriz que puede ser
desarrollada, y la corriente en corto circuito ISH, que es la máxima fotocorriente que puede ser
obtenida. En oscuro (línea punteada) no hay corriente fluyendo a través del dispositivo, hasta que
los contactos empiezan a inyectar una fuerza electromotriz de polarización directa mayor que el
voltaje en circuito abierto. Cuando el dispositivo se irradia, la corriente fluye en dirección opuesta
a la corriente inyectada. La máxima fotocorriente generada fluye en condiciones de cortocircuito
en (a); y la fotocorriente es balanceada a cero obteniéndose la condición de banda plana. Entre (a)
y (b) en el cuarto cuadrante, el dispositivo genera energía. Hay un cierto punto denominado punto
de potencia máxima (Pmax) que es el producto de la corriente máxima IMax y el voltaje máximo
VMax. Si se colectara la totalidad de portadores de carga disociados en los electrodos, entonces la
potencia máxima sería max OC SHP V I . Las pérdidas después de la disociación pueden ser
reflejadas en el factor de llenado (FF), el cual representa la fracción del producto OC SHV I que
puede ser utilizada como fuente energética (OCSC
MaxMax
VI
VIFF
*
* ).
22
Figura 4.- Curva característica de corriente-voltaje (I-V) de una celda solar
La eficiencia de conversión () es dada entonces como:
inc
SCOC
P
FFIV **
Los parámetros de celda VOC, ISC y FF deben ser determinados bajo condiciones de iluminación
estándares (STC) que son; 25 °C y una irradiancia (Pinc) de 100 mW/cm2 con un filtro de emisión
solar AM1.5. esto corresponde a una posición zenit del sol de 48.2°.22
1.1.1.- Celdas solares inorgánicas
Las celdas solares tradicionales de mayor uso en el mercado son las celdas inorgánicas
principalmente a base de silicio. Las celdas de silicio monocristalino consisten en obleas de silicio
con una estructura cristalina totalmente intacta (sin defectos de grano) lo cual da la mayor
eficiencia para estos tipos de dispositivos (25%). Sin embargo la preparación de estas obleas se
realiza a través de procesos muy caros. El silicio policristalino se obtiene de lingotes cuadrados a
su vez preparados fundiendo y luego enfriando el silicio monocristalino. Estas celdas son menos
caras pero la eficiencia es menor debido a los defectos (20%). El silicio amorfo presenta no todos
los átomos de silicio tetracoordinados. Este material puede ser dopado con hidrogeno para reducir
estos defectos, las eficiencias son del orden de 13%.22

23
1.1.2. Celdas solares orgánicas
Las celdas solares orgánicas son una alternativa más económica a las celdas de silicio. Utilizan
oligómeros y polímeros conjugados para obtener el efecto fotovoltaico. Estos materiales son
semiconductores orgánicos con una estructura química caracterizada por la alternancia de dobles
o triples enlaces con enlaces sencillos. Presentan brecha energética entre 1 y 3.5 eV, altos
coeficientes de absorción y buena procesabilidad que permite la fabricación de los dispositivos en
películas delgadas (100-150 nm) con técnicas de depositación sencillas y cubriendo áreas grandes
inclusive sobre substratos flexibles. Sin embargo, el efecto fotovoltaico en estos materiales
procede de una forma un poco diferente con respecto a los materiales inorgánicos como se
representa en la Figura 5.
Figura 5.- Esquema de proceso de transferencia de carga en celdas inorgánicas (a) y orgánicas (b)23
En ambos materiales, la excitación da lugar a pares de electrón-hueco (llamados excitones) pero
en los compuestos inorgánicos (Figura 5a) estos son débilmente ligados, la energía térmica a
temperatura ambiente (menor a 0.05 KeV) siendo suficiente a romperlos de manera que el electrón
pasa de la banda de valencia (Valence Band=VB) a la de conducción (Conduction Band=CB)
dejando una vacancia o hueco () en la VB. Las cargas libres (electrones y huecos) pueden
entonces difundirse a los respectivos electrodos bajo el efecto del campo eléctrico generado por la
diferencia en la función de trabajo de los mismos. En los semiconductores orgánicos (Figura 5b)
la energía de atracción coloumbica de las cargas en el excitón (llamada en inglés exciton binding

24
energy) se encuentra típicamente entre 0.4-1 eV.24 En la Figura 6 se muestra la representación de
un excitón para un polímero conjugado, el poli(fenilenovinilideno).
Figura 6.- Esquema de un excitón para el caso del poli(fenilenovinilideno)
En este caso, entonces para romper el excitón es necesario crear un campo eléctrico local utilizando
dos materiales con diferente afinidad electrónica y energía de ionización. El material con mayor
afinidad electrónica y mayor potencial de ionización funciona como un electrón aceptor mientras
el otro como un electrón donador. El excitón difunde a la interface entre los dos materiales por una
distancia determinada por su tiempo de vida (entre nano y picosegundos lo cual da una distancia
de difusión típicamente de 10-20 nm), por un proceso de transferencia de energía conocido como
transferencia de carga fotoinducida. Tomando en cuenta el diagrama de energía de la Figura 5b, si
el excitón se forma en el electrón donador, entonces el proceso de transferencia de carga ocurre
del orbital molecular no ocupado de menor energía del donador (D-LUMO) al del aceptor (A-
LUMO). Este es el caso más frecuente en celdas de moléculas orgánicas conjugadas. Por el
contario, si el excitón se forma del lado del aceptor, la transferencia ocurre del orbital molecular
ocupado de mayor energía del aceptor (A-HOMO) al del donador (D-HOMO). En algunos casos,
pueden ocurrir ambos procesos.
Sin embargo, el proceso de transferencia fotoinducida es factible si es energéticamente favorable
ya que de forma paralela habrá la emisión del electrón donador y/o aceptor. De esta manera, del
punto de vista energético, el proceso de transferencia de energía es dictaminado por la energía libre
de Gibbs ΔGCS.25,26 Esta se puede calcular mediante la ecuación de Weller:
2 2
10
0 0
1 1 1 1( )
4 8
D A
CS ox red
s cc r s
e eG e E E E
R r r
25
Donde D
oxE es el potencial de oxidación del donador, A
redE es el potencial de reducción del aceptor,
10E es la energía del primer estado excitado, obtenido de las mediciones de fluorescencia. Los
parámetros RCC, r-, y r+ son parámetros geométricos y representan la distancia entre los centros
del catión al anión (considerados como esferas), el radio del anión y el radio del catión
respectivamente.
Para que ocurra la transferencia, ΔGCS debe ser negativa; es decir existe un estado energético
cargado (CS=charged state) accesible durante la de-excitación desde el primer estado excitado.
Gráficamente;
Figura 7.- Diagrama de energía de un sistema donador-aceptor y su posibles procesos foto físicos
La excitación puede realizarse al donador (hD) alcanzando el primer estado electrónico excitado
del donador (1D-A) o bien al aceptor (hA) alcanzando su respectivo primer estado excitado (D-
1A) y se forma el excitón en el donador o aceptor. En la Figura 7, el primer estado excitado del
donador es a mayor energía del correspondiente del aceptor, pero puede ser al revés. De todas
formas, si los dos materiales D y A son fluoroforos, habrá la de-excitación al estado basal por
medio de fluorescencia. Para el estado excitado que está a menor energía, esta es también posible
después de un proceso de transferencia de energía fotónica de Förster (energy transfer, ET), aun
cuando se haya excitado el donador. Los procesos de fluorescencia (con sus respectivas constantes

26
kFLD y kFLA) compiten con el proceso de transferencia fotoinducida (con su constante kCS) el cual
es posible si de acuerdo a la ecuación de Weller, ΔGCS<0 es decir si el estado excitado
correspondiente a la separación de cargas del excitón (D+-A-) es a menor energía de ambos estados
excitados de singulete (o primeros estados). La posición del estado CS se obtiene de la ecuación
de Weller y está relacionada con los potenciales de óxido-reducción de los materiales (obtenidos
experimentalmente por voltametría). La posición de los primeros estados excitados y las constantes
se obtienen de estudios foto físicos.
La transferencia de energía fotoinducida puede ser intra o intermolecular. La primera se da en una
molécula que presenta los dos grupos electrón donador y aceptor en la estructura, y la segunda se
da entre moléculas vecinas. Una forma de obtener un cálculo cuantitativo de la energía libre del
proceso de transferencia electrónica intermolecular es empleando la ecuación de Weller en donde
se considere que debido a que el donador y el aceptor no están enlazados, CCR , y así,
2
0/ (4 ) 0s CCe R .
1.1.2.1. Arquitecturas de celdas solares orgánicas
La configuración más sencilla (Figura 8) de una celda orgánica consiste de una capa de un
oligómero o polímero conjugado depositado entre los dos electrodos. Normalmente el ánodo es
una capa de óxido de indio y estaño (ITO) mientras que el cátodo puede ser un metal, como Al,
Mg o Ca (evaporado en vacío). Más recientemente se han encontrado buenos resultados también
con metales fundidos como el Woods metal o Fields metal así como cátodos líquidos como
suspensiones coloidales de plata.27-29
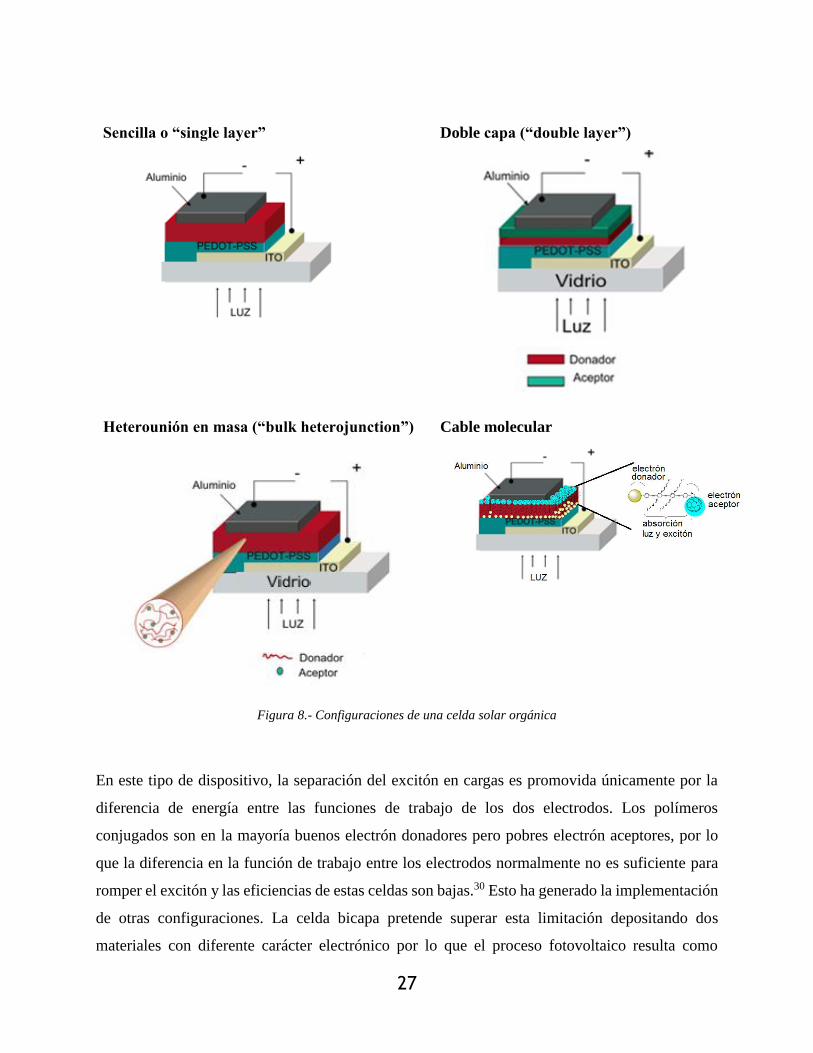
27
Sencilla o “single layer” Doble capa (“double layer”)
Heterounión en masa (“bulk heterojunction”) Cable molecular
Figura 8.- Configuraciones de una celda solar orgánica
En este tipo de dispositivo, la separación del excitón en cargas es promovida únicamente por la
diferencia de energía entre las funciones de trabajo de los dos electrodos. Los polímeros
conjugados son en la mayoría buenos electrón donadores pero pobres electrón aceptores, por lo
que la diferencia en la función de trabajo entre los electrodos normalmente no es suficiente para
romper el excitón y las eficiencias de estas celdas son bajas.30 Esto ha generado la implementación
de otras configuraciones. La celda bicapa pretende superar esta limitación depositando dos
materiales con diferente carácter electrónico por lo que el proceso fotovoltaico resulta como

28
consecuencia de la transferencia de energía fotoinducida. Como se ha mencionado anteriormente
los polímeros conjugados son generalmente electrón donadores. Mientras que como materiales
electrón aceptores comúnmente se utilizan derivados solubles del fulereno. Recientemente se han
reportado buenas eficiencias también con otros alotropos del carbono como los nanotubos31-35 y
grafenos36,37 así como con algunas moléculas orgánicas pequeñas como los perilenos.38 Otra
posibilidad es utilizar materiales electrón aceptores inorgánicos dando lugar a celdas híbridas (ver
siguiente sección). En la Figura 9 se muestran las estructuras de algunos materiales más utilizados
en celdas solares orgánicas.
Figura 9.- Estructura de algunos de los materiales más utilizados en celdas solares orgánicas
Como se ha deducido de la ecuación de Weller, la transferencia de energía fotoinducida (en este
caso solo puede ser intermolecular), se obtiene por CCR es decir cuando la distancia los dos
grupos es alta lo cual permite evitar la recombinación de las cargas; en términos de interfase,
cuando el área de la interface es grande. Además los materiales deben tener una elevada movilidad

29
de carga para que los electrones y huecos se transporten rápidamente a los electrodos. Si no es así,
la recombinación de las cargas provoca calentamiento de la celda y por ende bajas eficiencias o
cortos. Comparada con la configuración de una sola capa, la de bicapa permite incrementar la
eficiencia de hasta un orden de magnitud. Por ejemplo se ha reportado una eficiencia de 0.1% para
una celda de capa sencilla de poli(2-methoxi,5-(2’-etil-hexiloxi)-p-fenilenovinilideno) o MEH-
PPV39 (Figura 9) y de 1% para una de bicapa, MEH-PPV con fulereno C60.40
La configuración de heterounión en masa o bulk heterojunction permite alcanzar mayores
eficiencias gracias a que la capa activa está formada por una mezcla física del electrón aceptor y
electrón donador lo cual permite la formación de una red. Sin embargo siendo una mezcla física,
es necesario lograr una buena miscibilidad entre la solución del electrón donador y la del electrón
aceptor y que una vez evaporado el solvente la separación de fase entre estos dos materiales sea
menor de la distancia de difusión del excitón. Por esta razón la morfología de la capa activa afecta
en gran medida la eficiencia de estas celdas.
Una posible solución consiste en diseñar moléculas que presenten en la misma estructura grupos
electrón donadores y electrón aceptores separados por un sistema conjugado que genere los
excitones. Estas estructuras denominadas “push-pull” por la presencia de los dos polos (electrón
negativo y positivo) o D--A (donador-conjugado-aceptor) permiten el desarrollo de celdas de
cable molecular. En este caso, la transferencia de energía debería ser más efectiva ya que la
interface entre electrón donador y aceptor está constituida únicamente por los grupos funcionales
requeridos para el enlace covalente. Además para esta configuración es factible no solamente la
transferencia intermolecular (como en las demás) sino también la intramolecular. Finalmente, la
posibilidad de inferir una orientación preferencial de las moléculas con respecto a los electrodos
por medio de técnicas de depositación específicas (por ejemplo Langmuir-Blodgett) abre la
perspectiva de llegar a mayores eficiencias.41
Como conclusión, y tomando en cuenta el esquema de funcionamiento del efecto fotovoltaico
(Figura 2) una molécula orgánica puede ser una buena candidata para aplicaciones en celdas
solares si presenta las siguientes características;
1.- Absorbe la mayor parte de la región espectral correspondiente a la emisión de la luz solar.
2.- Presenta altos rendimientos cuánticos los cuales se reflejen en una alta posibilidad de formar
excitones de singuletes.

30
3.- Es procesable por medio de técnicas sencillas en forma de películas homogéneas.
4.- Presenta en su estructura un grupo electrón aceptor y un grupo electrón donador o bien varias
moléculas, cada una con efecto electrónico especifico, pueden mezclarse o ensamblarse para
conllevar a un sistema del tipo electrón donador/sistema conjugado excitónico/electrón aceptor
con una interface adecuada es decir donde la distancia entre el sistema electrón donador y el
electrón aceptor está adentro de la distancia de difusión del excitón.
6.- Presenta una buena movilidad de cargas para permitir el transporte de estas hacia los electrodos.
La mayor parte de los materiales orgánicos no cumplen con todas estas características, por esta
razón se han desarrollados dispositivos híbridos.
1.1.3. Celdas solares híbridas con puntos cuánticos
Los dispositivos híbridos orgánico/puntos cuánticos son celdas de heterounión que utilizan
moléculas conjugadas como materiales electrón donadores y nanopartículas semiconductoras
inorgánicas como electrón aceptores. Ambos materiales absorben luz y presentan normalmente
altos rendimientos cuánticos de manera que se incrementa el número de excitones formados con
respecto a las celdas orgánicas. Además, debido a que las propiedades ópticas de los puntos
cuánticos pueden ser moduladas según su tamaño, es posible modificarlas de manera que el sistema
híbrido absorba regiones del espectro electromagnético que el material orgánico solo no absorbe.
Comparativamente al dispositivo totalmente inorgánico, la incorporación del material orgánico
ofrece la posibilidad de usar técnicas de fabricación de menor costo.
El proceso fotovoltaico en los dispositivos híbridos orgánico/punto cuántico es similar al descrito
anteriormente para las celdas de heterounión orgánicas, sin embargo debido a que como antes
mencionado, ambos materiales pueden formar excitones, la transferencia de energía fotoinducida
puede ocurrir ya sea del lado del polímero (transferencia de electrones del LUMO del polímero a
la banda de conducción del punto cuántico) que del lado del punto cuántico (transferencia de
huecos de la valencia del punto cuántico al HOMO del polímero) siempre que haya una buena
alineación entre los diferentes orbitales moleculares (ver Figura 10).
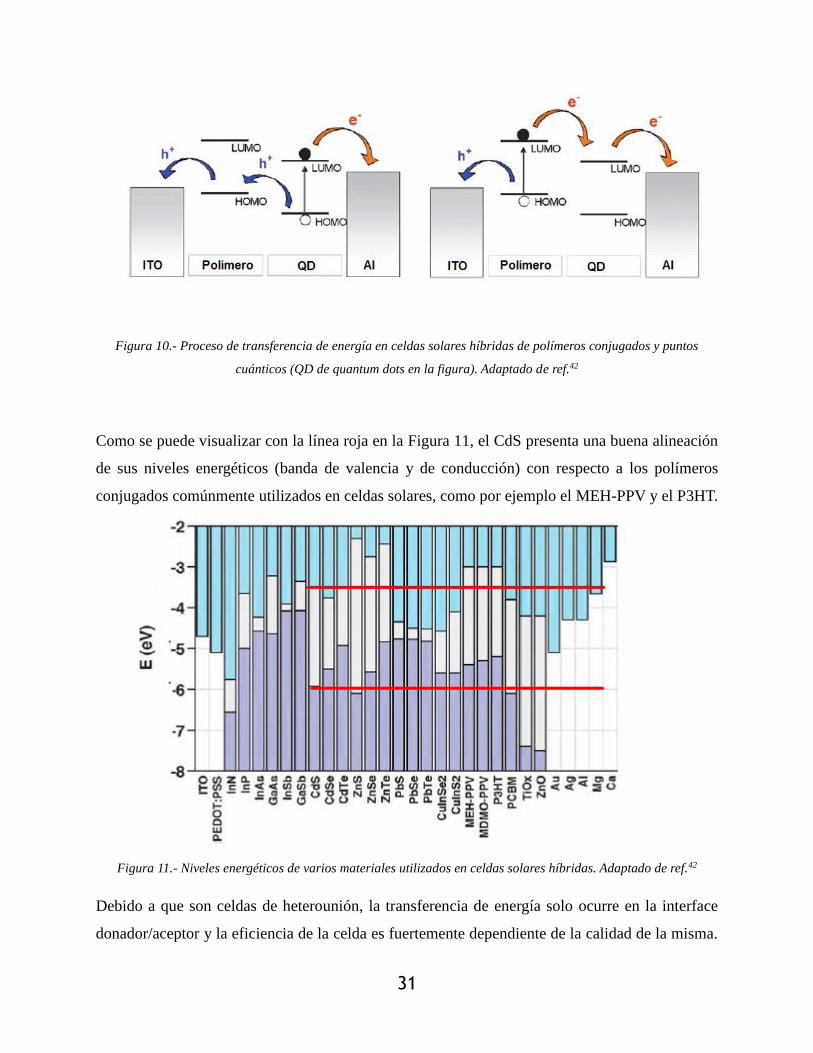
31
Figura 10.- Proceso de transferencia de energía en celdas solares híbridas de polímeros conjugados y puntos
cuánticos (QD de quantum dots en la figura). Adaptado de ref.42
Como se puede visualizar con la línea roja en la Figura 11, el CdS presenta una buena alineación
de sus niveles energéticos (banda de valencia y de conducción) con respecto a los polímeros
conjugados comúnmente utilizados en celdas solares, como por ejemplo el MEH-PPV y el P3HT.
Figura 11.- Niveles energéticos de varios materiales utilizados en celdas solares híbridas. Adaptado de ref.42
Debido a que son celdas de heterounión, la transferencia de energía solo ocurre en la interface
donador/aceptor y la eficiencia de la celda es fuertemente dependiente de la calidad de la misma.

32
En este aspecto, el método de síntesis del composite es un primer punto a considerarse ya que las
condiciones de síntesis del punto cuántico y el tipo de pasivante pueden afectar el tamaño de la
nanopartícula (a menor tamaño, mayor área superficial de contacto entre punto cuántico y
polímero, es decir mayor interfase), su morfología, la eficacia en el transporte de cargas de la
interface punto cuántico/polímero así como la procesabilidad del composite y por ende la
formación de microdominios en la película de la capa activa. En la sección siguiente se reportan
los métodos más utilizados para la obtención de nanocomposites de puntos cuánticos con
polímeros conjugados.
1.2.- Nanocomposites de puntos cuánticos y polímeros
1.2.1.- Síntesis de puntos cuánticos
De forma general para que una nanopartícula semiconductora se pueda definir como un punto
cuántico debe satisfacer el tamaño (2 y 10 nm) y monodispersidad. El término de monodispersidad
corresponde a que las partículas se puedan identificar de forma aislada y con un solo tamaño, sin
embargo en la práctica se consideran como nanopartículas monodispersas, las que presentan una
desviación estándar en la determinación de su diámetro menor de 5%43. Según estudios por LaMer
y Dinegar44, la producción de coloides monodispersos requiere de la formación de unos pocos
núcleos cristalinos seguidos por un lento crecimiento de los núcleos existentes (Figura 12).
Figura 12.- Esquema de la formación de nanocristales según el modelo de LaMer y Dinegar. Adaptado de ref.45

33
Si los reactivos vienen adicionados de forma rápida, la concentración de los precursores supera la
concentración mínima de núcleos (umbral de nucleación) es decir estamos en un régimen de
supersaturación. Si el consumo de los precursores a través de la nucleación es más rápido de la
adición subsiguiente de los reactivos, no se formarán nuevos núcleos y por lo tanto se tiene el
crecimiento de los núcleos existentes. Puesto que el crecimiento de cada nanocristal es igual al de
otro, la distribución inicial de tamaños es determinada en gran medida por el tiempo durante el
cual los núcleos se forman y empiezan a crecer. Si el crecimiento de cristales durante la nucleación
es mínimo el tamaño de los nanocristales será más uniforme a medida que pasa el tiempo. Algunos
sistemas tienen un segundo periodo de crecimiento que se llama “maduración de Ostwald” durante
el cual los cristales más pequeños se disuelven en la mezcla de reacción y nuevo material se
deposita sobre los cristales más grandes es decir el tamaño de cristal crece a dependiendo del
número de cristales. Este modelo fue comprobado sacando en tiempo de reacción diferentes
alícuotas de mezcla de reacción (ver Figura 12).
A pesar de que el crecimiento de cristales puede ser regulado de acuerdo con los tiempos de
inyección de los reactivos según el modelo antes descrito, los nanocristales por su gran área
superficial presentan la tendencia a aglomerarse por diferentes posible interacciones (Van der
Waals, estéricas, electroestáticas etc.)46. Para evitar este proceso, durante la síntesis se agrega un
agente estabilizante (o pasivante) el cual tiene la función de formar una capa en la superficie de la
nanopartícula la cual a su vez crea una interface entre una partícula y otra. Los pasivantes pueden
ser moléculas orgánicas pequeñas o polímeros en donde una parte tenga afinidad con el elemento
constituyente el nanocristal y otra parte quede al exterior creando fuerzas repulsivas entre una
partícula y otra.47 La pasivación con estos ligantes orgánicos puede alterar las propiedades de
dispersión de los puntos cuánticos en matrices poliméricas. Por otra parte estos actúan también
como barreras para la transferencia de carga de una partícula a la otra por lo que tienen que ser
removidos para permitir el contacto eléctrico entre partículas. Para la aplicación de puntos
cuánticos en dispositivos electrónicos u optoelectrónicos donde es necesaria la continuidad
eléctrica entre partículas, se requiere buscar ligantes que no sean aislantes. De allí ha surgido el
interés en sintetizar nanocomposites de puntos cuánticos con sistemas conjugados.

34
1.2.2.- Síntesis de polí(fenilenoetinileno)s
Como se ha mencionado anteriormente, los polímeros conjugados presentan en su estructura
enlaces sencillos alternados con enlaces dobles y/o triples lo cual les confiere una deslocalización
electrónica muy extendida y por consecuencia propiedades ópticas y eléctricas interesantes para
aplicaciones en optoelectrónica. Entre los diferentes polímeros conjugados, en esta tesis se
sintetizaron poli(fenilenoetinilenos) debido a su alto rendimiento cuántico de fluorescencia,
estabilidad térmica y fotoquímica mayor a la de otros polímeros conjugados (por ejemplo los de
tipo fenilenovinilideno) y la experiencia adquirida en el grupo en su síntesis. Típicamente la
síntesis de este tipo de materiales que requiere la formación del enlace triple carbono-carbono
(CC) se realiza a través de una reacción de acoplamiento. Históricamente Sonogashira48, Heck49
y Cassar estudiaron este tipo de reacción que involucra el acoplamiento de un bromuro o yoduro
de arilo con un alquino terminal bajo la acción de un complejo de paladio. En el año de 1975,
Sonogashira y colaboradores reportaron que un hidrógeno acetilénico terminal puede ser
fácilmente sustituido por un bromuro o yoduro de arilo por la acción catalítica del complejo dicloro
bis(trifenilfosfina) paladio(II) en presencia de dietilamina como solvente, bajo condiciones de
reacción media ~ 60ºC, que dependen del halogenuro de arilo empleado. Cassar y Heck reportaron
en el mismo año, la misma reacción de acoplamiento utilizando catalizadores similares. Sin
embargo, el mecanismo, descrito por primera vez por Heck concuerda con el de Sonogashira, sólo
difieren en que éste último emplea yoduro cuproso (CuI) como co-catalizador en presencia de
amina, el cual reduce el paladio de Pd(II) a Pd(0), además acelera la sustitución del halogenuro
por el acetileno al complejo de paladio en forma de acetiluros de Cobre(II), obteniendo resultados
más favorables para la síntesis. Cassar por su parte propuso que en el caso de utilizar
tetrakis(trifenilfosfina) paladio(0), el producto de adición oxidativa se forma primero y después es
seguida la ruta de la sustitución nucleofílica, la eliminación reductiva, etc.
Amatore, en 1993, publicó un trabajo referente a las sustituciones aromáticas nucleofílicas
catalizadas por complejos de paladio cero-valentes y propone un mecanismo simplificado que
consiste en cuatro etapas. La primera etapa (Figura 13) consta de la reducción de paladio(II) a
paladio(0), al hacer reaccionar cloruro de paladio y trifenilfosfina para obtener un complejo de
paladio(II) tetracoordinado (diclorobis(trifenilfosfina)). En esta primer etapa, la presencia de
cationes, que son producto del CuI actúan como reductores contribuyendo a la formación de pares
de iones a partir del [N+NH2Et2]Cl

35
Figura 13.-Especies producidas durante la primera etapa descrita por Amatore.
La segunda etapa (Figura 14) consiste en una adición oxidativa de los halogenuros aromáticos al
complejo paladio(0). La adición Ar-X es inmediata a la especie catalítica, esto provoca la
oxidación de Pd cero-valente a Pd di-valente, como producto se obtiene un intermediario
pentacoordinado estable de Pd(II), trans- Ar Pd2X(PPh3)2Cl, el cual es susceptible de reaccionar
con el nucleófilo formado a partir del acetileno.
Figura 14.- Adición oxidativa de la segunda etapa descrita por Amatore.
En la siguiente etapa es donde se lleva a cabo la sustitución nucleofílica del halógeno por el
acetiluro sobre el paladio(II), dando lugar a la formación de un nuevo complejo aniónico
pentacoordinado. La presencia de CuI permite la formación de acetiluros de cobre, los cuales son
buenos nucleófilos para atacar al trans-ArPd2X(PPh3)2Cl. De este ataque se libera un átomo de
halógeno, el cual junto con el protón acetilénico sufren la eliminación de HX que reacciona con la
trietilamina que se encuentra en el medio y precipitan en forma de sal insoluble como XH•NEt3.
La cuarta y última etapa consiste en una eliminación reductiva de los grupos arilo y acetiluro
generando el producto final del acoplamiento y regenerando el paladio(0). El nucleófilo (acetileno
E) queda unido al catalizador pentacoordinado; el cis-ArPd2E(PPh3)2Cl, el cual en un último
proceso de eliminación-reductiva del acoplamiento del Arilo con el nucléofilo Ar-E, quedando el

36
paladio en estado (0). La eliminación de Ar-E es causada por la fuerte atracción que hay entre estos
dos sustituyentes, y que se favorece por la posición cis en la que se encuentran dentro el complejo
catalítico. Con la regeneración del catalizador se inicia nuevamente el ciclo, dando origen a una
vía de acoplamiento muy eficaz que incluso se adaptó a la polimerización entre dihalogenuros de
arilo y diacetilenos para obtener poli(arilenoetinilenos). Todas estas etapas mencionadas, se
resumen en el ciclo catalítico que muestra en la Figura 15.
Existen varios factores o variables importantes que se deben considerar en esta reacción de
acoplamiento, tales como la elección del halogenuro de arilo, el efecto electrónico de los
sustituyentes sobre dicho halogenuro de arilo, el tipo de catalizador, así como el disolvente.
Figura 15.-Ciclo catalítico del Paladio propuesto por Amatore para el acoplamiento de halogenuros de arilo y
acetilenos.

37
Elección del halogenuro de arilo. La reactividad del halogenuro de arilo está directamente
relacionada con su electronegatividad, de forma que el más reactivo es el yodo y el menos es el
flúor de acuerdo al siguiente orden: I > Br > Cl > F. De hecho, los fluorarilos son prácticamente
inertes a esta reacción de acoplamiento. El acoplamiento de los cloroarilos con alquinos depende
de la naturaleza electrónica de los sustituyentes, por ejemplo aquellos arilos sustituidos con grupos
electroatractores, particularmente nitro, los rendimientos pueden ser hasta del 20%, mientras que
los cloruros de bencilo normalmente no reaccionan. Los compuestos más reactivos son los
bromoarilos y los yodoarilos, sin embargo existe una diferencia de reactividad entre los dos
halógenos. La reacción de los bromuros de arilo típicamente requieren de reacciones a altas
temperaturas (60-80ºC), mientras que las correspondientes con yoduro de arilo se sabe que
reaccionan a 0ºC, a temperatura ambiente o ligeramente arriba de esta. Se ha propuesto que en la
adición oxidativa del halogenuro de arilo al compuesto de paladio(0) es más rápida para los
yoduros de arilo que para los bromuros. La facilidad relativa de la adición oxidativa del yoduro de
arilo a la especie de Pd(0) es una función de la baja energía de ionización de este compuesto
comparada con la del bromuro de arilo.
Sustituyentes en el halogenuro de arilo. Comparando la reactividad de un haloareno rico y pobre
en electrones, es necesario examinar las especies activas a las que estos se adicionan. El complejo
de Pd(0) es rico en electrones y la adición oxidativa del halogenuro de arilo deberá ser influenciada
por las propiedades electrónicas de este compuesto. Es claramente evidente que entre más electrón
atractor es el haloareno más rápida es su adición al complejo de Pd(0) rico en electrones y por lo
tanto entre más electrón donador es el haloareno, más lenta es la adición oxidativa.
Tipo de catalizador. El complejo tetracoordinado se puede formar in situ partiendo del cloruro de
paladio PdCl2 y un exceso de trifenilfosfina PPh3. La presencia de una sal de cobre como CuI
permite la reducción a paladio(0). Sin embargo, existen comercialmente, complejos previamente
formados, del cual destaca el dicloro bis(trifenilfosfina) paladio(II), [PdCl2(PPh3)2], como el más
utilizado debido a su estabilidad al aire en relación a otros catalizadores como el Pd(PPh3)4, el
Pd(OAc)2 y el tris(dibenzilidenacetona)dipaladio (0) (Pd2(dba)3).
Disolvente. El disolvente debe cumplir con ciertos requisitos fundamentales para que se lleve a
cabo la reacción, éste debe estar compuesto principalmente de una base capaz de sustraer el protón
del acetileno terminal para realizar la adición oxidativa del alquino al Pd(0). En su mayoría, son
las aminas quienes tienen un buen desempeño, de las cuales destacan la trietilamina, la dietilamina

38
y la diisopropilamina. Además, se pueden emplear co-solventes como el tetrahidrofurano (THF),
el tolueno, la DMF, el metanol, etc., su función radica en difundir el compuesto organometálico
en el medio y mantener las especies monoméricas, oligoméricas y macromoleculares en solución.
Cabe mencionar que el THF es el más utilizado ya que tiene buenas propiedades de solvatación
para los complejos organometálicos y promueve la solubilidad de las cadenas conjugadas
manteniéndolas en solución51.
En esta reacción es muy importante tener en cuenta las siguientes condiciones: La ausencia de O2
a lo largo de la reacción, esto debido a que en el medio reactivo hay la formación de acetiluros de
cobre intermediarios, los cuales en presencia de O2 tienden a formar diacetilenos. También se debe
evitar toda traza de humedad, el agua actúa como nucleófilo y rompe los complejos formados a lo
largo de la polimerización.
1.2.3.- Síntesis de nanocomposites de puntos cuánticos con polímeros
De forma general para obtener un nanocomposite se pueden utilizar métodos in situ o ex situ. Los
procesos ex situ consisten en sintetizar previamente la nanoestructura e incorporarla a la matriz
por mezclado. Aunque el proceso de mezclado o blending ha sido muy utilizado en celdas solares
para aprovechar las propiedades ópticas y eléctricas de diferentes materiales, sin embargo es difícil
controlar la dispersión de un material con respecto a otro provocando problemas de
microseparación de fases y posibles aglomeraciones que pueden afectar el mecanismo de
separación y transporte de carga. Por otra parte, los métodos in situ incluyen intercambio iónico
y/o síntesis de puntos cuánticos funcionalizados con surfactantes que tengan grupos funcionales
específicos los cuales sucesivamente permitan la introducción de los segmentos conjugados. Un
ejemplo es el reporte por Skaff3 en el cual primeramente se funcionaliza el quantum dot con una
fosfina orgánica portadora de un bromobenzeno. Este queda al exterior de la superficie de la
partícula y puede acoplarse con un monómero vinilidenico dando lugar a CdSe pasivados con
fenilenovinilidenos (Figura 16)

39
Figura 16.- Esquema de la formación de nanocristales funcionalizados con PPV, adaptada de ref.3
Otra referencia muy interesante es el trabajo de Newkome y colaboradores donde se aprovecha la
funcionalización de dendrímeros conjugados para pasivar CdS. En este caso se añaden lentamente
las sales precursoras (nitrato de cadmio y sulfuro de sodio) a una solución del dendrímero a 0°C50.
Estas estrategias sintéticas permiten teóricamente obtener un mejor control en el tamaño y
distribución de los puntos cuánticos en la matriz poliméricas ya que la pasivación ocurre en sitios
específicos de la molécula conjugada. Para poderlos diferenciar de los materiales compuestos
obtenidos por mezcla física entre los puntos cuánticos y los polímeros, este tipo de composite se
llamará a continuación nanohíbrido.
1.3.- Estado del arte
Como se ha mencionado previamente, la tecnología de las celdas solares empezó con el empleo
de materiales semiconductores inorgánicos entre los cuales el más empleado es el silicio51. Los
primeros dispositivos estaban principalmente constituidos por Si y uniones simples p-n teniendo
una eficiencia de 7 a 8 %. En el desarrollo de dispositivos con mejor eficiencia se obtuvieron los
de Si monocristalinos los cuales lograron una eficiencia de 18% sin embargo el costo de
producción era alto.
En una siguiente etapa se empezaron a desarrollar dispositivos que pudieran competir con los altos
costo del Si monocristalino lográndose obtener estos a partir de Si amorfo pero con una eficiencias

40
de 10%52 en este mismo periodo se lograron obtener celdas a partir de semiconductores de los
grupos III y V alcanzando una eficiencia de 18% con el GaAs las cuales fueron elaboradas
mediante depositación epitaxial de fase liquida.53 Consiguientemente se desarrollaron celdas con
multiuniones o también llamadas tándem. Estos dispositivos consisten en celdas de doble uniones
GaInP2/GaAs y más tarde se desarrollaron de triple uniones GaInP2/GaAs/Ge, llegando hasta un
22 % de eficiencia en la de doble uniones y las de triple a 29.9%53 la principal características de
estos dispositivos es que la unión de la interfaz se controla para poder obtener celdas de doble o
triple uniones a fin de optimizar el uso de las longitudes de onda de distintas zonas del espectro y
por lo tanto lograr una mayor eficiencia54-56.
A partir del descubrimiento de las propiedades de semiconducción en materiales orgánicos en la
década de los ’80, se empezó el desarrollo de celdas solares orgánicas. La primera generación era
basada en una configuración sencilla o de monocapa57. En 1986, Tang reportó la primera celda de
bicapa utilizando derivado de la ftalocianina como semiconductor tipo p, y un perileno como
semiconductor n, teniendo un eficiencia de aproximadamente 1%58. Sin embargo, solamente
despúes del descubrimiento por parte de Heeger y col. de la transferencia de energía fotoinducida
de polímeros conjugados a la molécula de fulereno (C60), la tecnología de dispositivos orgánicos
despegó. Estudios sistemáticos en este aspecto se han realizado con fenilenovinilidenos
/fulerenos59 y politiofenos/fulerenos60 en configuración de heterounión o de cable molecular (ver
estructuras en ref.61). En este último caso es más factible el control a nivel nanoscópico de la
morfología por medio de técnicas de depositación de películas. En este sentido, un claro ejemplo
es el trabajo que desarrolla el grupo de Nierengarten, quien sintetiza fulerenos C60 portadores de
dendrones hidrofílicos y unidos a derivados -conjugados, dando lugar a nanopelículas multicapas
bien estructuradas por la técnica de Langmuir-Blodgett y con buenas propiedades fotovoltaicas.41
En años recientes se ha reportado el modelo de celdas solares tándem orgánicas en las cuales se
acoplan dos celdas de tipo heterounión en serie aprovechando materiales con diferente región
espectral con la finalidad de ampliar el rango de espectro solar absorbido. Bajo esta configuración,
Heeger obtuvo eficiencias de hasta 6.5%. (Figura 17)

41
Figura 17.- Celda solar tandem orgánica62
La estrategia de ampliar el espectro de absorción de la capa activa se utiliza también en los
dispositivos híbridos de puntos cuánticos. Los primeros estudios en estos materiales híbridos lo
realizo Grenham y colaboradores donde se utilizaron nanopartículas de CdS o CdSe embebidas en
MEH-PPV.63 Como una prevención de agregación en las nanopartículas se utilizó TOPO como
surfactante.63 Básicamente los procedimientos en los primeros trabajos en la estos materiales
híbridos fueron realizando mezclas de polímeros y nanocristales, el crecimiento de este último se
realizaba por separado.
Huynh y colaboradores fueron de los primeros en implementar el crecimientos de nanocristales
directamente en el polímeros donde utilizaron el CdSe en el P3HT64, en esta nueva ruta de síntesis
se pudo obtener un alto grado en el control y la minimización de defectos del cristal garantizando
la pureza química y la orientación optima en la conducción de las cadenas poliméricas
obteniéndose una eficiencia de 1.7%.
Pientka y colaboradores estudiaron la fotoindución de transferencia de carga para los nanocristales
del InP en MDMO-PPV65, en donde los autores demuestran que el uso de piridina favorece la
transferencia de cargas comparado con los ligantes orgánicos oltilfosfino normalmente utilizados.
Una estrategía muy interesante para formar nanocomposites es la reportada por Sebastian
Westenhoff y Nicholas A. Kotov quien obtienen el autoensamblaje de puntos cuánticos de CdTe
arriba de un poli(fenilenoetinileno) portador de grupos laterales con carga iónica por medio de la

42
técnica de Layer-by-layer (LBL, autoensamblaje) electroestático. Los autores demuestran que la
eficiencia en la transferencia de energía del polímero a las partículas depende de su distancia la
cual se manipula con las condiciones de LBL. Sin embargo, lamentablemente no reportan la
fabricación de celdas.66
Otros sistemas basados en diferentes nanopartículas han sido introducidos tal es el caso del CdTe67,
PdSe68, HgTe69, algunos de estos muestran una buena respuesta en la región infrarroja del espectro
solar. Dayal y col. 70 reportaron la fabricación de una celda solar con una eficiencia de 3.2% usando
un polímero conjugado poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-
b]dithiophene)-alt-4,7-(2,1,3-benzothia- diazole)] y nanopartículas de CdSe.
Jeltsch y col.71 obtuvieron un dispositivo fotovoltaico híbrido orgánico-inorgánico basado en
nanocristales de CdSe y el polímero semiconductor PCPDTBT obteniendo de 2.8 a 3.6 % de
eficiencia dependiendo la forma del nanocristal.

43
Capitulo 2: Enfoque de la tesis
2.1.- Justificación
El consumo de combustibles fósiles ha afectado a los cambios climáticos y el calentamiento global,
esto ha intensificado los esfuerzos por buscar nuevas fuentes de energía. Una de las energías
alternativas que continua siendo de las más interesantes es la energía solar. El estudio de mercado
demuestra que la producción mundial de módulos fotovoltaicos se ha incrementado
considerablemente en los últimos años con un aumento del 32 % solo del 2002 a 2003. Las celdas
fotovoltaicas constituyen actualmente una industria de $5.2 billones de dólares por año y su
capacidad ha aumentado 3 veces en los últimos 4 años y la tendencia es exponencial.72 Como se
ha mencionado previamente, la tecnología de celdas solares es muy viable en México debido a la
localización geográfica del país. Como parte del Plan Nacional de Desarrollo, el gobierno
mexicano ya ha dado impulso a la investigación en sistemas alternativos para energía sustentable
particularmente la energía solar. Adentro de este proyecto de largo alcance, el CIQA participa
realizando varios proyectos principalmente enfocados al desarrollo de nuevos materiales
orgánicos, en línea con su misión y la expertice de sus investigadores. El presente trabajo de tesis
se enmarca así en este proyecto de mayor alcance.
De acuerdo al análisis del estado del arte, la eficiencia de las celdas orgánicas e híbridas ha
mejorado exponencialmente en los últimos años. Un estudio exhaustivo realizado por la
Universidad de Stanford permite graficar la evolución de desempeño de estos dispositivos (Figura
18). Si bien las eficiencias de las celdas híbridas de puntos cuánticos son todavía menores a las de
las celdas orgánicas, su estudio es muy reciente por lo que quedan abiertas muchas estrategias para
mejorar su eficiencia.
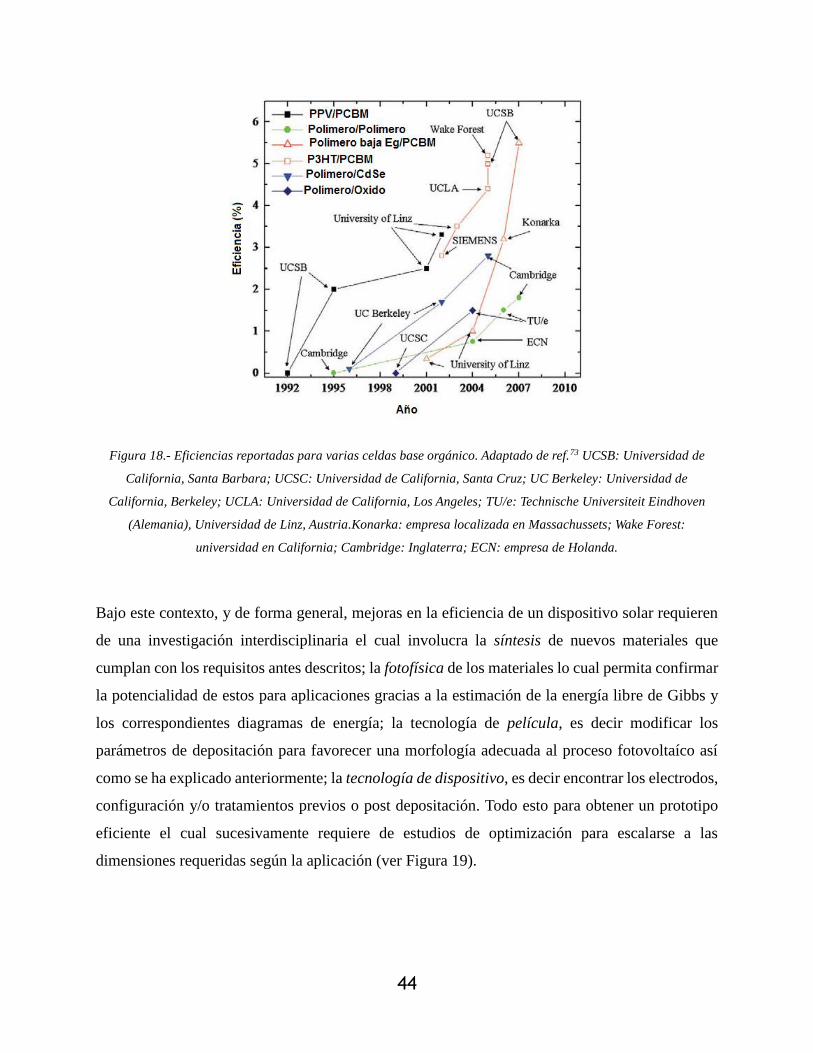
44
Figura 18.- Eficiencias reportadas para varias celdas base orgánico. Adaptado de ref.73 UCSB: Universidad de
California, Santa Barbara; UCSC: Universidad de California, Santa Cruz; UC Berkeley: Universidad de
California, Berkeley; UCLA: Universidad de California, Los Angeles; TU/e: Technische Universiteit Eindhoven
(Alemania), Universidad de Linz, Austria.Konarka: empresa localizada en Massachussets; Wake Forest:
universidad en California; Cambridge: Inglaterra; ECN: empresa de Holanda.
Bajo este contexto, y de forma general, mejoras en la eficiencia de un dispositivo solar requieren
de una investigación interdisciplinaria el cual involucra la síntesis de nuevos materiales que
cumplan con los requisitos antes descritos; la fotofísica de los materiales lo cual permita confirmar
la potencialidad de estos para aplicaciones gracias a la estimación de la energía libre de Gibbs y
los correspondientes diagramas de energía; la tecnología de película, es decir modificar los
parámetros de depositación para favorecer una morfología adecuada al proceso fotovoltaíco así
como se ha explicado anteriormente; la tecnología de dispositivo, es decir encontrar los electrodos,
configuración y/o tratamientos previos o post depositación. Todo esto para obtener un prototipo
eficiente el cual sucesivamente requiere de estudios de optimización para escalarse a las
dimensiones requeridas según la aplicación (ver Figura 19).
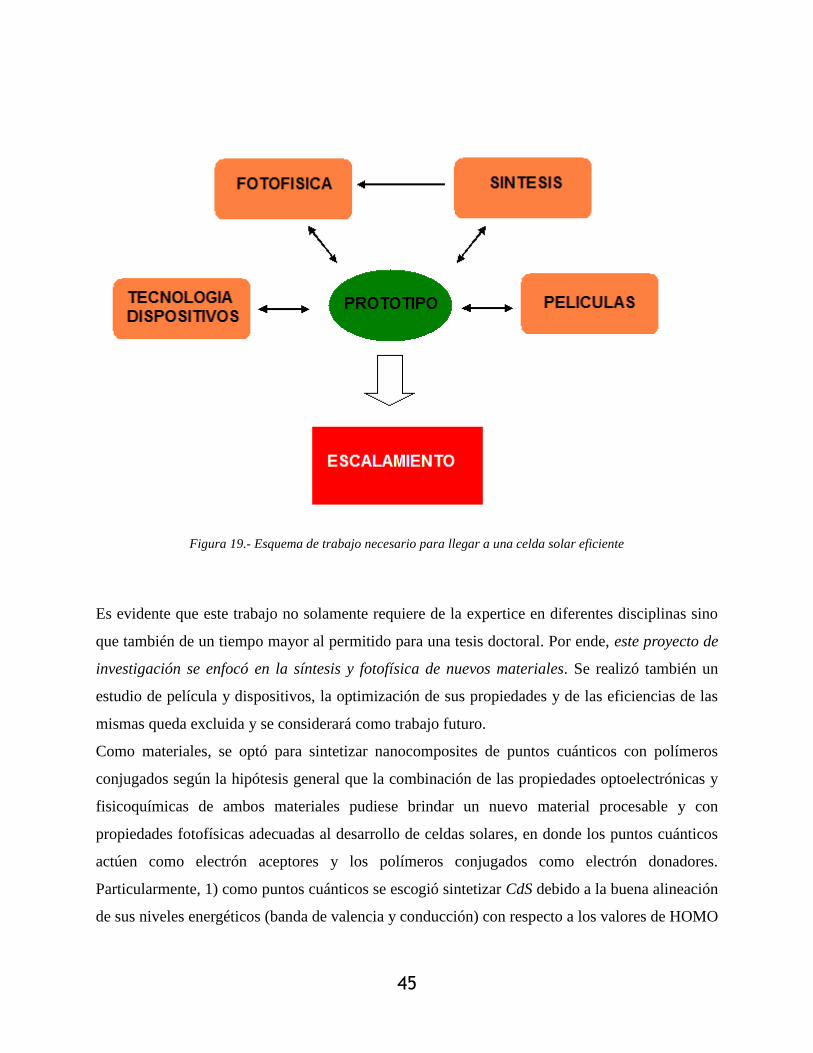
45
Figura 19.- Esquema de trabajo necesario para llegar a una celda solar eficiente
Es evidente que este trabajo no solamente requiere de la expertice en diferentes disciplinas sino
que también de un tiempo mayor al permitido para una tesis doctoral. Por ende, este proyecto de
investigación se enfocó en la síntesis y fotofísica de nuevos materiales. Se realizó también un
estudio de película y dispositivos, la optimización de sus propiedades y de las eficiencias de las
mismas queda excluida y se considerará como trabajo futuro.
Como materiales, se optó para sintetizar nanocomposites de puntos cuánticos con polímeros
conjugados según la hipótesis general que la combinación de las propiedades optoelectrónicas y
fisicoquímicas de ambos materiales pudiese brindar un nuevo material procesable y con
propiedades fotofísicas adecuadas al desarrollo de celdas solares, en donde los puntos cuánticos
actúen como electrón aceptores y los polímeros conjugados como electrón donadores.
Particularmente, 1) como puntos cuánticos se escogió sintetizar CdS debido a la buena alineación
de sus niveles energéticos (banda de valencia y conducción) con respecto a los valores de HOMO

46
y LUMO típicos de polímeros conjugados (Figura 11) y alto rendimiento cuántico lo cual puede
permitir un incremento en la formación de excitones.
Como ligante se escogió el 3-(mercaptopropil)-trimetoxisilano (MPS) el cual ha sido reportado
como un buen estabilizador de puntos cuánticos de CdS74-77 y que permitiría la funcionalización
de los puntos cuánticos con el polímero conjugado.
2) como polímero se optó por un poli(fenilenoetinileno). Los poli(fenilenoetinilenos) presentan las
siguientes ventajas con respecto a los polímeros conjugados más estudiados en literatura, los
poli(fenilenovinilidenos) PPV y los poli(tiofenos) PT. i) rendimientos cuánticos muy altos, en
algunos casos casi de 178 (mucho mayor a los poli(tiofenos)) con emisión estable en el tiempo a
diferencia de los PPV cuya fotoluminiscencia disminuye con el tiempo por photobleaching.79 ii)
son mucho más estables térmicamente y químicamente disminuyendo los problemas de
degradación por calor que seguido ocurren en los dispositivos optoelectrónicos y que afectan el
tiempo de vida de los mismos iii) debido a su estructura química, no pueden presentar isómeros
(cis-trans como en los PPV o regio isómeros como en los PT) de manera que hay menos
probabilidades de defectos estructurales.
La desventaja de estos polímeros para aplicaciones en celdas es que absorben normalmente en la
región de UV-azul (típicamente máximos en película alrededor de 420-460 nm), es decir un poco
desplazado hacía al azul con respecto al máximo de emisión solar.
La estructura del polímero (pPE(OC12-Co-BzC11OH) se diseñó de manera que la alternancia de
un monómero sustituido con alcoxy y uno tipo benzoato permitiera una buena solubilidad para la
reacción con el CdS(MPS) y también para la fabricación de las celdas. Además el monómero
benzoato lleva el sustituyente con grupo terminal OH para dar lugar a una reacción de
esterificación con los puntos cuánticos. Para la investigación del mecanismo de formación del
nanohíbrido, se sintetizó también el homopolímero pPE(BzC11OH) y su composite así como el
composite de un homopolímero 2,5-didodecanoxy(benzenoetinileno) pPE(OC12), previamente
sintetizado en nuestros laboratorios.
En la Figura 20 se muestran las estructuras de los materiales de estudio y las siglas utilizadas a lo
largo del documento para su identificación.
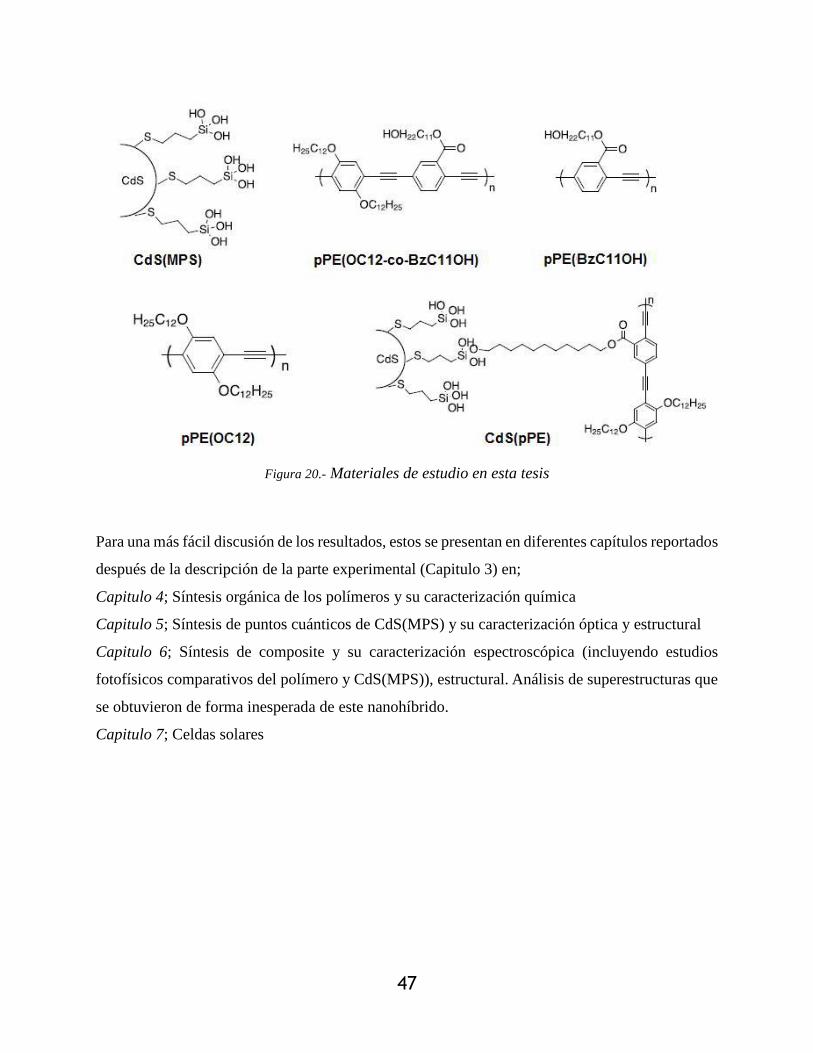
47
Figura 20.- Materiales de estudio en esta tesis
Para una más fácil discusión de los resultados, estos se presentan en diferentes capítulos reportados
después de la descripción de la parte experimental (Capitulo 3) en;
Capitulo 4; Síntesis orgánica de los polímeros y su caracterización química
Capitulo 5; Síntesis de puntos cuánticos de CdS(MPS) y su caracterización óptica y estructural
Capitulo 6; Síntesis de composite y su caracterización espectroscópica (incluyendo estudios
fotofísicos comparativos del polímero y CdS(MPS)), estructural. Análisis de superestructuras que
se obtuvieron de forma inesperada de este nanohíbrido.
Capitulo 7; Celdas solares

48
2.2.- Hipótesis
Es posible obtener nuevos nanohíbridos de interés en aplicaciones fotovoltaicas a partir de puntos
cuánticos de Sulfuro de Cadmio y polímeros conjugados del tipo fenilenoetinileno por medio de
la funcionalización a partir de una reacción de condensación entre los grupos hidroxilos de la
cadena lateral del polímero y los grupos metoxi terminales del agente estabilizante de los puntos
cuánticos, 3-(mercaptopropil)-trimetoxisilano.
2.3.- Objetivos
El objetivo general de este proyecto de tesis doctoral es obtener nanohíbridos de puntos cuánticos
con polímeros conjugados para aplicaciones en dispositivos fotovoltaicos.
2.3.1. Objetivo particulares
1.- Síntesis y caracterización de los monómeros 1,4-bis(dodecanoxy)-2-5diyodobenceno y 11-
hidroxiundecil2,5-dietinilbenzoato
2.- Síntesis y caracterización del polímero conjugado pPE(OC12-Co-BzC11OH)
3.- Síntesis y caracterización por de los puntos cuánticos de CdS(MPS)
4.- Síntesis y caracterización del nanohíbrido CdS(pPE)
5.- Fabricación de los dispositivos fotovoltaicos y su caracterización.
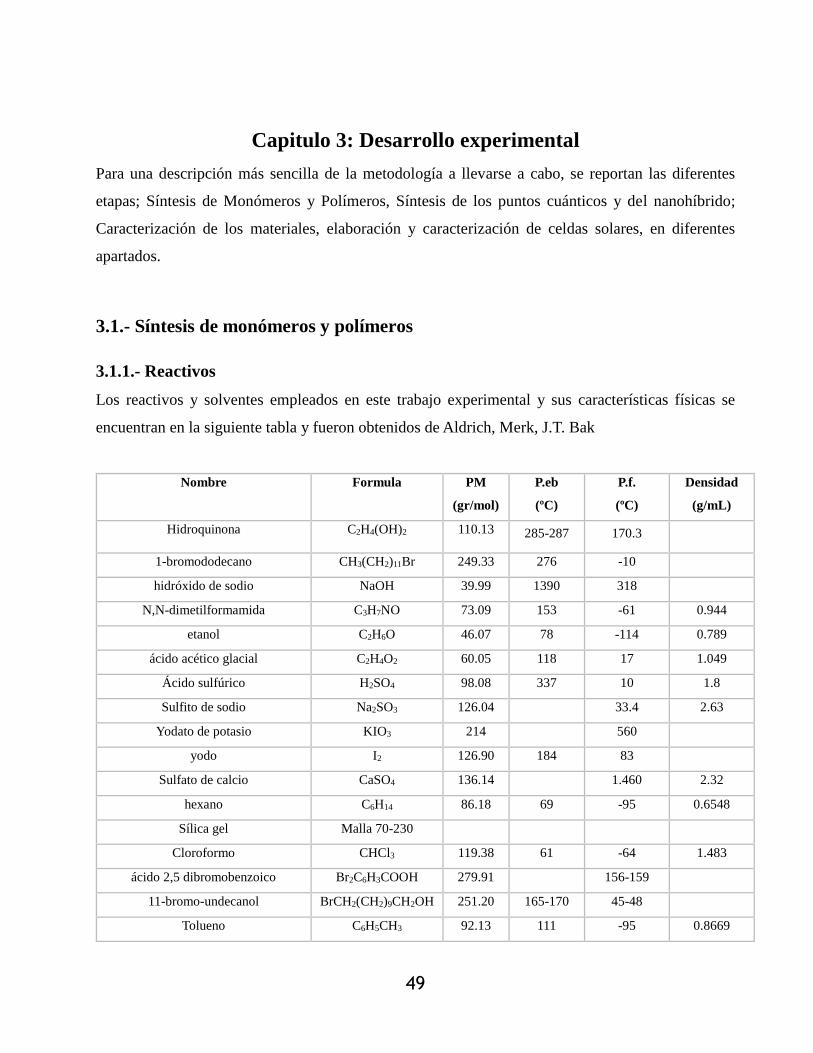
49
Capitulo 3: Desarrollo experimental
Para una descripción más sencilla de la metodología a llevarse a cabo, se reportan las diferentes
etapas; Síntesis de Monómeros y Polímeros, Síntesis de los puntos cuánticos y del nanohíbrido;
Caracterización de los materiales, elaboración y caracterización de celdas solares, en diferentes
apartados.
3.1.- Síntesis de monómeros y polímeros
3.1.1.- Reactivos
Los reactivos y solventes empleados en este trabajo experimental y sus características físicas se
encuentran en la siguiente tabla y fueron obtenidos de Aldrich, Merk, J.T. Bak
Nombre Formula PM
(gr/mol)
P.eb
(ºC)
P.f.
(ºC)
Densidad
(g/mL)
Hidroquinona C2H4(OH)2 110.13 285-287 170.3
1-bromododecano CH3(CH2)11Br 249.33 276 -10
hidróxido de sodio NaOH 39.99 1390 318
N,N-dimetilformamida C3H7NO 73.09 153 -61 0.944
etanol C2H6O 46.07 78 -114 0.789
ácido acético glacial C2H4O2 60.05 118 17 1.049
Ácido sulfúrico H2SO4 98.08 337 10 1.8
Sulfito de sodio Na2SO3 126.04 33.4 2.63
Yodato de potasio KIO3 214 560
yodo I2 126.90 184 83
Sulfato de calcio CaSO4 136.14 1.460 2.32
hexano C6H14 86.18 69 -95 0.6548
Sílica gel Malla 70-230
Cloroformo CHCl3 119.38 61 -64 1.483
ácido 2,5 dibromobenzoico Br2C6H3COOH 279.91 156-159
11-bromo-undecanol BrCH2(CH2)9CH2OH 251.20 165-170 45-48
Tolueno C6H5CH3 92.13 111 -95 0.8669
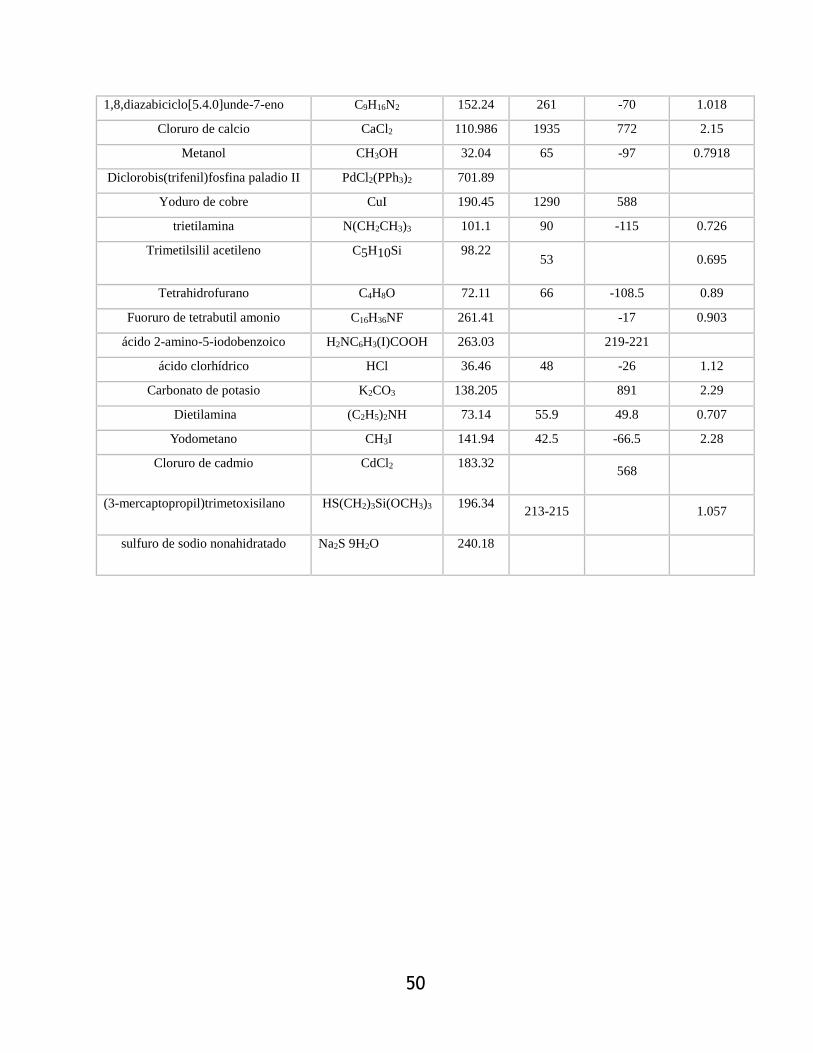
50
1,8,diazabiciclo[5.4.0]unde-7-eno C9H16N2 152.24 261 -70 1.018
Cloruro de calcio CaCl2 110.986 1935 772 2.15
Metanol CH3OH 32.04 65 -97 0.7918
Diclorobis(trifenil)fosfina paladio II PdCl2(PPh3)2 701.89
Yoduro de cobre CuI 190.45 1290 588
trietilamina N(CH2CH3)3 101.1 90 -115 0.726
Trimetilsilil acetileno C5H10Si 98.22 53 0.695
Tetrahidrofurano C4H8O 72.11 66 -108.5 0.89
Fuoruro de tetrabutil amonio C16H36NF 261.41 -17 0.903
ácido 2-amino-5-iodobenzoico H2NC6H3(I)COOH 263.03 219-221
ácido clorhídrico HCl 36.46 48 -26 1.12
Carbonato de potasio K2CO3 138.205 891 2.29
Dietilamina (C2H5)2NH 73.14 55.9 49.8 0.707
Yodometano CH3I 141.94 42.5 -66.5 2.28
Cloruro de cadmio CdCl2 183.32 568
(3-mercaptopropil)trimetoxisilano HS(CH2)3Si(OCH3)3 196.34 213-215 1.057
sulfuro de sodio nonahidratado Na2S 9H2O 240.18

51
3.1.2.- Síntesis de monómeros halogenados
3.1.2.1.- 1,4-bis(dodecanoxi)benceno(DOB)(2)
Figura 21.- Esquema de obtención de DOB
En un matraz bola de dos bocas provisto de agitación magnética, se colocaron 0.09082 mol (10 g) de
hidroquinona (1), 1-bromododecano 0.1816 mol (46.9895 g), hidróxido de sodio 0.1816 mol (7.26
g) y 80 ml de N,N-dimetilformamida. La mezcla se calentó aproximadamente a 120-130°C por 18 h.
Terminado este tiempo, la mezcla de reacción se dejó enfriar y posteriormente se le agregó 400 ml
de agua fría para hacer precipitar el producto. El precipitado se recolectó por filtración y
posteriormente se pusieron en un vaso conteniendo 200ml de etanol tibio (60°C) y se dejó en
agitación por un periodo de 30 minutos. La mezcla se enfrió a temperatura ambiente y luego se colocó
en un baño de hielo para hacer precipitar el producto. El producto se recolectó por filtración, se lavó
con etanol frío hasta que el líquido filtrado fuera transparente y posteriormente se secó a vacío para
obtener 23.52 g (58 %) del producto final (2) en forma de hojuelas de color blancas grisáceas. p.f.
74-75°C. 1H RMN (300 MHz, CDCl3) δ (ppm) 6.81 (s, 4H, H-Ar), 3.88 (t, 4H, J=6.60Hz, CH2-O-
Ar), 1.74 (q, 4H, CH2-β-O), 1.43 (q, 4H, CH2-γ-O), 1.25 (s, 32H, CH2), 0.87 (t, 6H, J=6.88Hz, CH3).
3.1.2.2.- 1,4-bis(dodecanoxi)-2-5diiodobenceno (DIDOB) (3)

52
Figura 22.- Esquema de obtención de DIDOB(3)
En un matraz bola provisto de agitación magnética y condensador se colocó el DOB (2) 0.0134 mol
(6 g), KIO3 0.0071 mol (1.52 g) y I2 0.0147 mol (3.756 g), posteriormente se agregó una mezcla de
92 mL de ácido acético glacial, 7 mL de H2O y 1 mL de H2SO4 concentrado y se dejó reaccionar
durante 24 horas a una temperatura de 120-130°C. Transcurrido este tiempo, la reacción se dejó
enfriar a temperatura ambiente y enseguida se le adicionó una disolución acuosa al 20% de Na2SO3
hasta que el color café del yodo desapareciera, indicando que el yodo residual había sido desactivado.
La mezcla se vertió en agua fría para precipitar el producto y se transfirió a un embudo de separación
en donde la fase orgánica se recolectó y la fase acuosa se extrajo con hexano (3x100 mL). Las fases
orgánicas se secaron con CaSO4 y se concentraron para obtener un sólido blanco esponjoso. El
producto se purificó por cromatografia en columna de silica empleando hexano:CHCl3 4:1 como fase
móvil para obtener (3) 8.23 g (88%). p.f. 66-68°C. 1H RMN (300 MHz, CDCl3) δ (ppm) 7.16 (s, 2H,
H-Ar), 3.91 (t, 4H, J=6.60 Hz, CH2-O-Ar), 1.78 (q, 4H, CH2-β-O), 1.48 (q, 4H, CH2-γ-O), 1.25(s,
32H, -CH2-), 0.88 (t, 6H, J=6.88 Hz, CH3).
3.1.2.3.- (11-undecanol) 2,5-dibromobenzoato(5)

53
Figura 23.- Esquema de obtención de (5)
En un matraz de dos bocas, se colocó el ácido 2,5 dibromobenzoico (3) 0.0107 mol (3 g), el 11-
bromo-undecanol 0.0107 mol (2.687g), 100 ml de tolueno y el 1,8,diazabiciclo[5.4.0]unde-7-ceno
0.117 mol (1.7811 g). La reacción se dejó a reflujo durante 15 horas. Terminado la reacción, se dejó
enfriar y se filtró, la solución filtrada se concentró y se purificó en una columna empacada de silica
con una mezcla eluente de 250:1 de CHCl3:MeOH (Rf) 0.2 para obtener (4) 3.83g (79.50% ). 1H
RMN (300 MHz, CDCl3) δ (ppm) 7.88(d, 1H, J=2.20Hz, H-Ar), 7.51(d, 1H, J=8.53Hz, H-Ar), 7.44,
7.41 (dd, 1H,J=2.48Hz, J=3.03Hz, Ar-H), 4.32(t, 2H, J=6.60Hz, CH2COO-Ar ), 3.63(t, 2H, J=6.0Hz,
CH2OH), 1.76(q, 2H, CH2-β-O), 1.55(q, 2H, CH2-γ-O), 1.29(s, 14H, CH2)
3.1.2.4.- (11-undecanol) 2,5-bis((trimetilsilil)etinilen)benzoato (6)
Figura 24.- Esquema de obtención de (6)
En un matraz de bola de dos bocas provisto de agitador magnético, llave para gas, septum y bajo
atmosfera de nitrógeno, se colocó (5) 2 g (0.0044 mol) y los catalizadores PdCl2(PPh3)2 0.1544 g
(0.0002 mol), CuI 0.00006 mol (0.0125 g). En otro matraz se desgasificó (ciclos de N2-vacío)
trietilamina (NEt3) recién destilada de KOH, la cual se agregó 100 mL vía cánula al matraz

54
conteniendo los reactivos hasta disolverlos. La mezcla reactiva bajo atmosfera de nitrógeno se calentó
a 60ºC por 15 min y después se agregó el trimetilsililacetileno 1.2965 g (0.0132 mol), teniendo
cuidado de burbujearlo en la solución lentamente. La reacción se dejó con agitación a 60°C durante
18 horas. Transcurrido este tiempo, la mezcla de reacción se filtró y se lavaron las sales con THF. El
solvente se eliminó por rota-evaporación. El producto en bruto fue purificado por cromatografía en
columna empacada con silica gel y empleando CHCl3:MeOH 250:1. 1 como eluente ( Rf 0.2) para
dar 2.73 g (6) (81.2%). RMN 1H (300 MHz, CDCl3) δ (ppm) 7.96(s, 1H, H-Ar), 7.49(s, 1H, H-Ar),
4.30(t, J=6.88Hz, 2H,CH2COO-Ar ), 3.62(t, 2H, J=6.88Hz, CH2OH), 1.77(q, 2H, CH2-β-O), 1.55(q,
2H, CH2-γ-O), 1.28(s, 14H, CH2)
3.1.2.5.- (11-undecanol) 2,5-bis(etinil)benzoato (7)
Figura 25.- Esquema de obtención de (7)
En un matraz bola de 100 mL se colocó el compuesto (6) 0.8972 mol (0.435 mg), luego se agregaron
20 ml de THF y dos gotas de agua des ionizada, una vez disuelto el compuesto se agregó NFBU4
392.805 mg (0.0107 mol) y se dejó reaccionar por 15 s. Posteriormente, la solución del matraz se
pasó por una columna de silica gel pequeña (seca) y luego se baja el producto con THF, el cual es
eliminado por rota evaporación. Se obtuvieron 296.5 mg de (7) (97.06 %). RMN 1H (300 MHz,
CDCl3) δ (ppm) 8.03(s, 1H, H-Ar), 7.55(s, 1H, H-Ar), 4.32(t, 2H, J=6.60Hz, CH2-α-COO-Ar ),
3.62(t, 2H, J=6.60Hz, CH2-α-OH), 3.46(s, 1H,C≡CH), 3.21(s, 1H,C≡CH ) 1.76(q, 2H, CH2-β-COO-
Ar), 1.55(q, 2H, CH2-γ-COO-Ar), 1.28(s, 14H, CH2)
3.1.2.6.- ácido 2-(3,3-dietil) triazenil)-5-iodobenzoico (9)
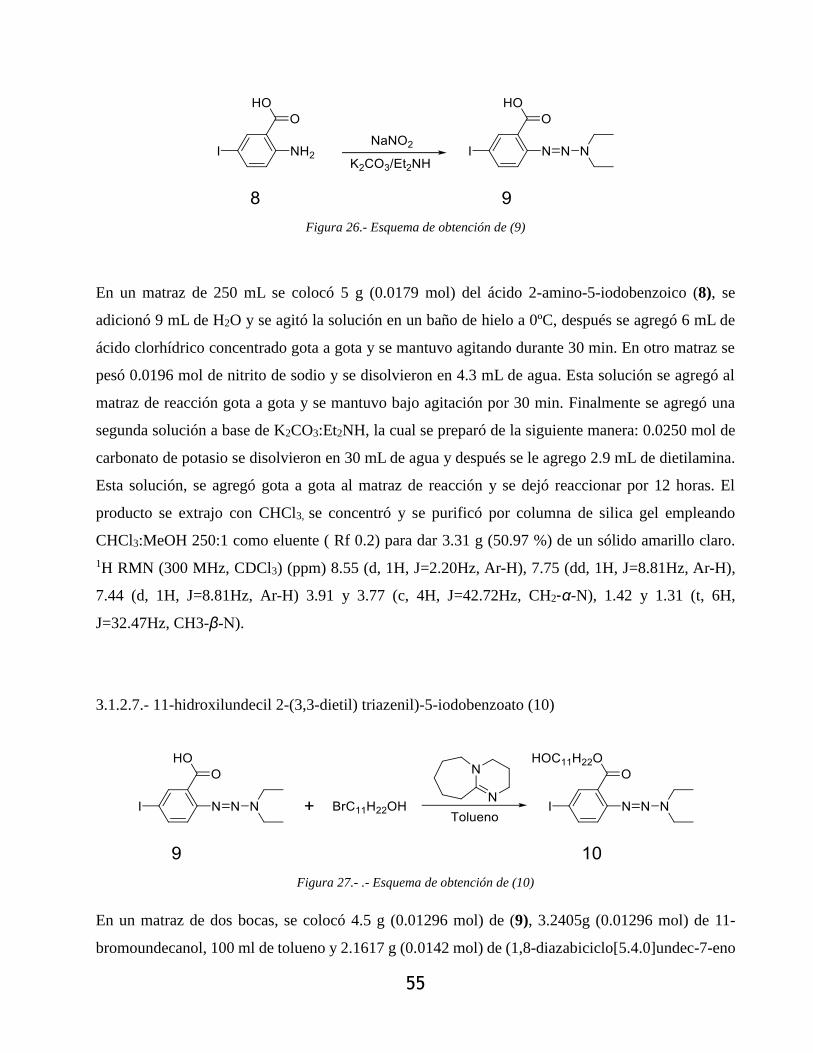
55
Figura 26.- Esquema de obtención de (9)
En un matraz de 250 mL se colocó 5 g (0.0179 mol) del ácido 2-amino-5-iodobenzoico (8), se
adicionó 9 mL de H2O y se agitó la solución en un baño de hielo a 0ºC, después se agregó 6 mL de
ácido clorhídrico concentrado gota a gota y se mantuvo agitando durante 30 min. En otro matraz se
pesó 0.0196 mol de nitrito de sodio y se disolvieron en 4.3 mL de agua. Esta solución se agregó al
matraz de reacción gota a gota y se mantuvo bajo agitación por 30 min. Finalmente se agregó una
segunda solución a base de K2CO3:Et2NH, la cual se preparó de la siguiente manera: 0.0250 mol de
carbonato de potasio se disolvieron en 30 mL de agua y después se le agrego 2.9 mL de dietilamina.
Esta solución, se agregó gota a gota al matraz de reacción y se dejó reaccionar por 12 horas. El
producto se extrajo con CHCl3, se concentró y se purificó por columna de silica gel empleando
CHCl3:MeOH 250:1 como eluente ( Rf 0.2) para dar 3.31 g (50.97 %) de un sólido amarillo claro.
1H RMN (300 MHz, CDCl3) (ppm) 8.55 (d, 1H, J=2.20Hz, Ar-H), 7.75 (dd, 1H, J=8.81Hz, Ar-H),
7.44 (d, 1H, J=8.81Hz, Ar-H) 3.91 y 3.77 (c, 4H, J=42.72Hz, CH2-α-N), 1.42 y 1.31 (t, 6H,
J=32.47Hz, CH3-β-N).
3.1.2.7.- 11-hidroxilundecil 2-(3,3-dietil) triazenil)-5-iodobenzoato (10)
Figura 27.- .- Esquema de obtención de (10)
En un matraz de dos bocas, se colocó 4.5 g (0.01296 mol) de (9), 3.2405g (0.01296 mol) de 11-
bromoundecanol, 100 ml de tolueno y 2.1617 g (0.0142 mol) de (1,8-diazabiciclo[5.4.0]undec-7-eno

56
(DBU). La reacción se dejó a reflujo con un condensador y trampa con CaCl2 durante 15 horas.
Terminado la reacción, se dejó enfriar y se filtró. La solución filtrada se concentró y se purificó por
columna de silica gel empleando CHCl3:MeOH 250:1 (Rf. 0.2). Se obtuvieron 5.74g (85.73%) de un
sólido amarillo claro. RMN 1H (300 MHz, CDCl3) (ppm) 7.86 (d, 1H, J=1.93Hz, Ar-H), 7.67 y
7.64(dd, 1H, J=8.53Hz, Ar-H), 7.16 (d, 1H, J=8.81Hz, Ar-H) 4.23(t, 2H, J=6.88, CH2-α-COO-Ar),
3.74 y 3.72 (c, 4H, J=13.76Hz, CH2-α-N) 3.62(t, 2H, J=6.60Hz, CH2-α-OH), 1.70(q, 2H, CH2-β-
COO-Ar), 1.55(q, 2H, CH2-γ-COO-Ar), 1.27(s, 14H, CH2), 1.24(t, 6H, CH3-β-N).
3.1.2.8.-(11-undecanol) 2-(3,3-dietil)triazenil)-5-((trimetilsilil)etinilen)benzoato (11)
Figura 28.- Esquema de obtención de (11)
En un matraz bola de 2 bocas, con agitador magnético, llave para gas, septum y bajo atmósfera de
nitrógeno, se colocaron (10) 2.3 g (0.0044 mol), PdCl2(PPh3)2 0.1544 g (0.00022 mol), CuI 0.0125 g
(0.00006 moles). En otro matraz se desgasificó (ciclos de N2-vacío) trietilamina (NEt3) recién
destilada de KOH, la cual se agregó 100 mL vía cánula al matraz conteniendo los reactivos hasta
disolverlos. La mezcla reactiva en atmosfera de nitrógeno se calentó a 60ºC por 15 min y después se
agregó el trimetilsililacetileno 0.47 g (0.0048 mol), teniendo cuidado de burbujearlo en la solución
lentamente. La reacción se dejó con agitación a 60°C durante 18 horas. Transcurrido este tiempo, la
mezcla de reacción se filtró y se lavaron las sales con THF. El solvente se eliminó por rota-
evaporación. El producto fue purificado por cromatografía en columna empacada con silica gel y
empleando CHCl3:MeOH 250:1 como eluente para obtener 1.6752g (78.05 %) de (10). 1H RMN (300
MHz, CDCl3) (ppm) 7.68 (d, 1H, J=1.93Hz, Ar-H), 7.44 (dd, 1H, J=8.53Hz, Ar-H), 7.35 (d, 1H,
J=8.53Hz, Ar-H) 4.23(t, 2H, J=6.88, CH2-α-COO-Ar), 3.76 y 3.72 (c, 4H, J=13.35Hz, CH2-α-N)
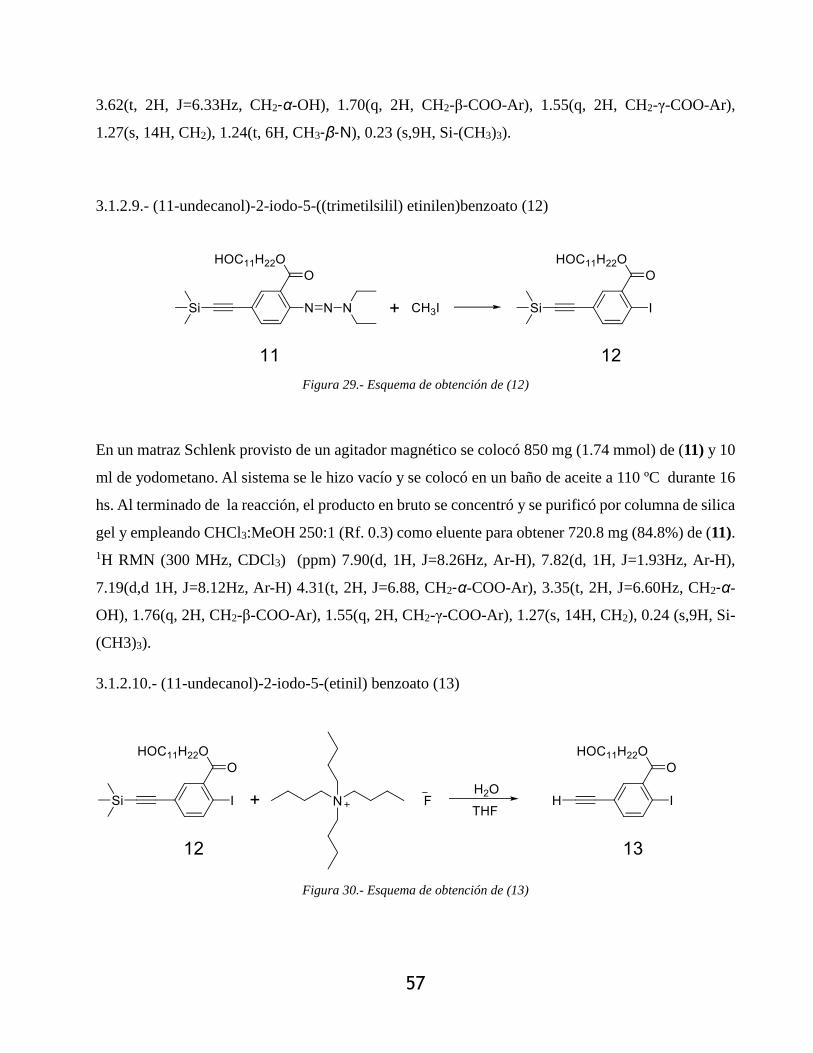
57
3.62(t, 2H, J=6.33Hz, CH2-α-OH), 1.70(q, 2H, CH2-β-COO-Ar), 1.55(q, 2H, CH2-γ-COO-Ar),
1.27(s, 14H, CH2), 1.24(t, 6H, CH3-β-N), 0.23 (s,9H, Si-(CH3)3).
3.1.2.9.- (11-undecanol)-2-iodo-5-((trimetilsilil) etinilen)benzoato (12)
Figura 29.- Esquema de obtención de (12)
En un matraz Schlenk provisto de un agitador magnético se colocó 850 mg (1.74 mmol) de (11) y 10
ml de yodometano. Al sistema se le hizo vacío y se colocó en un baño de aceite a 110 ºC durante 16
hs. Al terminado de la reacción, el producto en bruto se concentró y se purificó por columna de silica
gel y empleando CHCl3:MeOH 250:1 (Rf. 0.3) como eluente para obtener 720.8 mg (84.8%) de (11).
1H RMN (300 MHz, CDCl3) (ppm) 7.90(d, 1H, J=8.26Hz, Ar-H), 7.82(d, 1H, J=1.93Hz, Ar-H),
7.19(d,d 1H, J=8.12Hz, Ar-H) 4.31(t, 2H, J=6.88, CH2-α-COO-Ar), 3.35(t, 2H, J=6.60Hz, CH2-α-
OH), 1.76(q, 2H, CH2-β-COO-Ar), 1.55(q, 2H, CH2-γ-COO-Ar), 1.27(s, 14H, CH2), 0.24 (s,9H, Si-
(CH3)3).
3.1.2.10.- (11-undecanol)-2-iodo-5-(etinil) benzoato (13)
Figura 30.- Esquema de obtención de (13)

58
En un matraz bola de 100 ml provisto de un agitador magnético, se colocaron 300 mg (0.5830 mmol)
de (12), más 2 gotas de agua y 20 ml de THF. Posteriormente se agregó el NFBU4 0.5 mL (1 M en
THF) y se dejó reaccionar 15 seg. Posteriormente, la solución del matraz se pasó por una columna de
silica gel pequeña (seca) y luego se baja el producto con THF, el cual es eliminado por rota
evaporación. Se obtuvieron 235 mg (91.11 %) de (12). 1H RMN (300 MHz, CDCl3) (ppm) 7.94(d,
1H, J=7.98Hz, Ar-H), 7.87(d, 1H, J=2.20Hz, Ar-H), 7.22(d,d 1H, J=8.26Hz, Ar-H) 4.32(t, 2H,
J=6.60, CH2-α-COO-Ar), 3.36(t, 2H, J=6.60Hz, CH2-α-OH), 3.185(s, 1H, CC), 1.76(q, 2H, CH2-β-
COO-Ar), 1.55(q, 2H, CH2-γ-COO-Ar), 1.27(s, 14H, CH2), 0.24 (s,9H, Si-(CH3)3).
3.1.3. Síntesis de polímeros conjugados
3.1.3.1.- co-polímero pPE(OC12-Co-BzC11OH) (14)
Figura 31.- Esquema de obtención de (14)
En un matraz bola de 2 bocas, con agitador magnético, llave para gas, septum y bajo atmósfera de
nitrógeno, se colocaron (3) 340.46 mg (1.3511 mmol), (7) 698.54 mg (1.4862 mmol), PdCl2(PPh3)2
47.41 mg (0.0675 mmol), CuI 4 mg (0.0202 mmol). En otro matraz se desgasificó (ciclos de N2-
vacío) trietilamina (NEt3) recién destilada de KOH, la cual se agregó 100 mL vía cánula al matraz
conteniendo los reactivos hasta disolverlos. La mezcla reactiva se dejó en agitación a 60°C durante
18 horas. Transcurrido este tiempo, se filtró y se lavaron las sales con THF. El solvente se eliminó al
mínimo volumen por rota-evaporación y se precipitó-centrifugo en metanol por tres veces
consecutivas. Finalmente el producto se filtró y se concentró al mínimo en volumen, la solución
madre se guardó en cloroformo en refrigeración hasta su uso. 1H RMN (300 MHz, CDCl3) δ(ppm)
8.15( s, COOAr-H), 7.6 (s, C≡C-Ar- H), 7.3(t, C≡C-ArH-), 7.05 (d, OArH), 6.9 (d, OArH), 4.35 (t,

59
CH2COO-Ar-), 4.0 (t, CH2-OAr), 3.6 (t, CH2OH), 1.85 (q, CH2-β-O), 1.5 (q, CH2- γ-O), 1.25 (s,
CH2-), 0.85 (t, CH3). MWPS = 33729, Mn =25668, Mw/Mn= 1.31.
3.1.3.2.- Síntesis del homo-polímero conjugado pPE(BzC11OH)(15)
Figura 32.- Esquema de obtención de (15)
En un matraz bola de 2 bocas, con agitador magnético, llave para gas, septum y bajo atmosfera de
nitrógeno, se colocó (13) 185 mg (0.418 mol), PdCl2(PPh3)2 14.6697 mg (0.0209 mol), CuI 1.1807
mg (0.0062 mol). En otro matraz se desgasificó (ciclos de N2-vacío) trietilamina (NEt3) recién
destilada de KOH, la cual se agregó 40 mL vía cánula al matraz conteniendo los reactivos hasta
disolverlos. La mezcla reactiva se dejó en agitación a 60°C durante 18 horas. Transcurrido este
tiempo, se filtró y se lavaron las sales con THF. El solvente se eliminó al mínimo volumen por rota-
evaporación y se precipitó-centrifugo en metanol por tres veces consecutivas. Finalmente el producto
se filtró y se concentró al mínimo en volumen, la solución madre se guardó en cloroformo en
refrigeración hasta su uso. 1H RMN (300 MHz, CDCl3) δ(ppm) 8.18(s,Ar-H), 7.65(d, Ar- H), 4.36
(t, CH2-COO-Ar), 3.30(t, CH2OH), 1.85 (q, CH2-β-O), 1.5 (q, CH2- γ-O), 1.25 (s, CH2). MWPS =
7140, Mn = 4243, Mw/Mn = 1.683.

60
3.1.4.- Síntesis de puntos cuánticos
3.1.4.1.- Síntesis de puntos cuánticos de CdS(MPS) (17)
Figura 33.- Esquema de obtención de (17)
Los puntos cuánticos de CdS fueron sintetizados basados en la literatura75. La solución (A) precursora
de Cd2+ fue preparada disolviendo 0.5499 g de CdCl2 en 1 mL de agua desionizada bajo agitación
vigorosa durante 10 min. Por otro lado se preparó una solución (B) de 1.67 mL de MPS en 60mL de
DMF y se dejó en agitación. Una vez disuelto por completo el CdCl2, se agregó gota a gota, a la
solución (B) que contenía el MPS en DMF; ésta se dejó agitando vigorosamente por 10 min (soln.
C). Se preparó otra solución (D) precursora de S2− con 0.2161 g de Na2S 9H2O en 0.5 mL de agua
desionizada. Por último se añadió la solución (D) de sulfuro de sodio gota a gota, a la solución de
Cd2+:MPS en DMF (C) y se observó un cambio de incoloro a amarillo tenue; indicativo de la
formación de las nanopartículas de CdS. La reacción se llevó a cabo durante cuatro horas con
agitación a temperatura ambiente. Posteriormente se realizó una destilación azeotrópica con benceno
para extraer el agua desionizada, este procedimiento se llevó a cabo durante dos horas a 80ºC. Los
sólidos fueron eliminados por decantación.
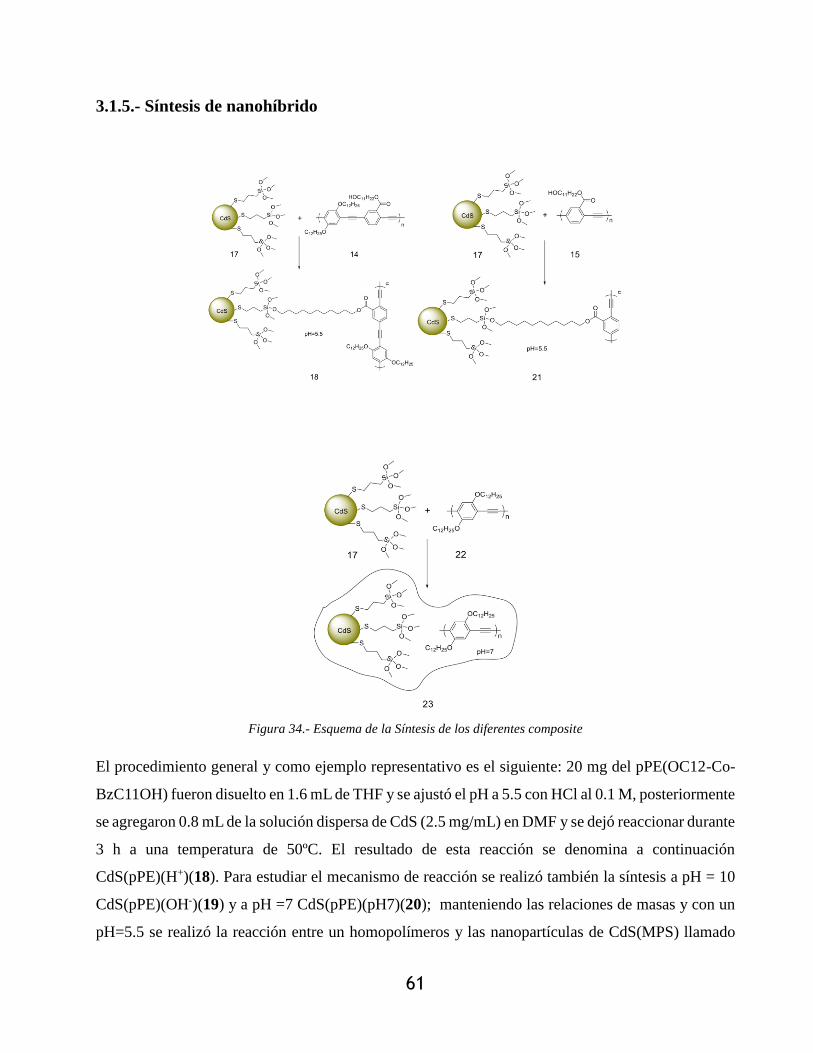
61
3.1.5.- Síntesis de nanohíbrido
Figura 34.- Esquema de la Síntesis de los diferentes composite
El procedimiento general y como ejemplo representativo es el siguiente: 20 mg del pPE(OC12-Co-
BzC11OH) fueron disuelto en 1.6 mL de THF y se ajustó el pH a 5.5 con HCl al 0.1 M, posteriormente
se agregaron 0.8 mL de la solución dispersa de CdS (2.5 mg/mL) en DMF y se dejó reaccionar durante
3 h a una temperatura de 50ºC. El resultado de esta reacción se denomina a continuación
CdS(pPE)(H+)(18). Para estudiar el mecanismo de reacción se realizó también la síntesis a pH = 10
CdS(pPE)(OH-)(19) y a pH =7 CdS(pPE)(pH7)(20); manteniendo las relaciones de masas y con un
pH=5.5 se realizó la reacción entre un homopolímeros y las nanopartículas de CdS(MPS) llamado

62
CdS(pPE(Co-BzC11OH))(21). Adicionalmente se realizó también el mismo procedimiento con el
polímero pPE(OC12)(22) bajo las mismas condiciones y relación en masa con pH=7 CdS(pPEOC12)
(23) en donde el polímero utilizado no tiene en su estructura grupos hidroxilos.
3.2.- Instrumentos y métodos de caracterización
La caracterización RMN 1H se llevó a cabo usando un espectrómetro Jeol Eclipse de 300 MHz,
empleando tetrametilsilano (TMS) como referencia y cloroformo deuterado (CDCl3) como
disolvente. La de estado sólido de 13C (100.5 MHz), 29Si (79.4 MHz) se realizó en un equipo Varian
INOVA 400 MHz (9.4T). Las muestras fueron empacadas en rotores de cerámica de 4 mm y todos
los espectros se obtuvieron a una velocidad de giro de 12 kHz. La referencia del desplazamiento
químico de 13C fue el hexametilbenceno (17.3 ppm, metil), para el 29Si fue el 2,2-dimetil-2-
silapentano sulfonado (1.46 ppm). i
La caracterización microscópica se realizó por STEM en un microscopio electrónico de barrido de
emisión de campo (FE-SEM) JEOL JSM-7401F a 30 kV con una distancia de trabajo de 6mm y por
TEM en un microscopio FEI-TITAN con 300 kV de emisión de campo, utilizándose área selectas de
difracción de electrones (SAED) y alta resolución (HRTEM) éste equipo tiene un lente simétrico tipo
S-TWIN (con una aberración esférica Cs=1.25 mm). Las imágenes fueron adquiridas con una cámara
CCD. Las muestras para STEM Y TEM fueron preparadas por casting sobre una rejilla de Lacey
Carbon.
El punto de fusión de los materiales se determinó en capilar con un fusiómetro
Barnstead/Electrothermal Mel-Temp bajo condiciones ambientales. El estudio termogravimétrico
(TGA) se llevó a cabo con un analizador DuPont Instruments 951 bajo atmósfera inerte en un rango
de 0 a 600 °C a una velocidad de 10 °C/min. De 600 a 800 °C el barrido se realizó con inyección de
aire. Las observaciones por microscopía óptica en luz polarizada (POM) se efectuaron con un
microscopio Olympus BH2- UMA equipado con una cámara Hitachi KP-D50 color y una platina de
calentamiento programado Mettler FP82HT. Los patrones de difracción de rayos X (XRD) se
i Estudio realizado en la universidad de Akron

63
obtuvieron en polvo en un difractrometro Bruker con fuente de radiación de CuKα de longitud de
onda de 0.1542 nm. El rango de barrido de 2θ fue de 0 a 40° con un paso de 0.02 °/s.
Los experimentos de XPS se realizaron en un espectrómetro SPECS Sage HR 100 con una fuente de
rayos X monocromática (línea Kα de Magnesio de 1253.6 eV de energía y potencia de 250 W,
calibrada usando la línea 3d5/2 de Ag con un ancho de media altura (FWHM) de 1.1 eV). Un flujo de
electrones se incidió en la superficie de la muestra para compensar la carga durante la adquisición de
los datos. Los espectros generales de XPS se tomaron con un paso de 15 eV y los espectros de alta
resolución fueron tomados con un paso de 0.15 eV. Todas las mediciones se llevaron a cabo en una
campana de ultra alto vacío a una presión de aprox. 5·10-8 mbar. Las muestras fueron bombardeadas
con un haz de iones Ar+ de energía de 3 keV entre 30 y 300 segundos para remover las impurezas del
medio ambiente. Las muestras se estudiaron en un portamuestra de polvos. En el fitting, se usaron
funciones tipo Gaussianas y Lorenzianas (después de corrección de la línea de base según el método
de Shirley).ii
La voltametría cíclica en solución se realizó agregando 3.6 mg de cada material: pPE(OC12-Co-
BzC11OH), CdS(MPS) y CdS(pPE) a una solución 0.1 M. de hexafluorofosfato de tetrabutil amonio
en CHCl3 (para el polímero y composite) y DMF para los puntos cuánticos. El equipo que se utilizó
es un potenciostato/galvanostato ACM Gill AC provisto de un sistema de tres electrodos, un electrodo
de referencia (Hg/Hg2Cl2), un electrodo auxiliar de platino y un electrodo de trabajo de carbono con
una velocidad de barrido de 50 mV/s para los tres materiales, las pruebas se realizaron bajo atmósfera
inerte con una rango electroquímico de 3000 a -2000 mV. La banda gap electroquímica (Eg) se
calculó con los potenciales de oxidación y reducción de acuerdo a la ecuación Eg=Eox-Ered, los
niveles de energía HOMO y LUMO se estimaron a partir de los potenciales de oxidación y reducción
del material de estudio usando como estándar interno el nivel de energía del ferroceno en el vacío
igual a 4.8 eV80 como se presenta en el conjunto de ecuaciones siguientes HOMO=-[Eox+4.8eV] y
LUMO=- [Ered+4.8eV].
ii Estudio realizado en CIC Biomagune, España

64
La caracterización espectroscópica del polímero, CdS y composite se realizó en DMF. Los espectros
UV-Vis se obtuvieron en un espectrofotómetro Shimadzu mientras que los de emisión en un
espectrofluorimetro Perkin Elmer LS50B. La banda gap óptica se calculó a partir del inicio de las
transiciones electrónicas en el espectro de absorción81 del material aplicando la ecuación de Planck
(3.1).
ecuación 3.1
Donde la banda gap (Eg) representa la brecha energética entre los niveles HOMO y LUMO, h es la
constante de Planck equivalente a 4.14 x 10-15
eV∙s, c es la velocidad de la luz en el vacío igual a 3
x 108
m/s y λ corresponde a la longitud de onda en metros que se identifica mediante el trazo de una
tangente al inicio del espectro de absorción.
El rendimiento cuántico se calculó aplicando la fórmula:
2)(est
m
estm
mest
n
n
FA
FA
Donde Aest y Am son las absorbancia a la longitud de onda de excitación para el estándar y la muestra,
respectivamente. Fest y Fm son las areas bajo la curva de emisión del estándar y de la muestra y nm,est
es el índice de refracción del solvente de la muestra (DMF) y del estándar (H2SO4 0.1 M).
Se analizaron tres soluciones con absorbancia a la longitud de onda de excitación <0.1. Para las
muestras la longitud de onda de excitación fue 384 nm mientras que para el sulfato de quinina fue
310 nm ya que a esta longitud esta reportado su rendimiento cuántico (0.54).82 Se utilizó un baño de
recirculación de agua a 25.0±0.3 °C para evitar efectos de temperatura en la determinación. El valor
de obtenido por cada solución fue promediado. Las curvas de decaimiento de fluorescencia se
obtuvieron con la técnica de recuento de fotones individuales (“Single Photon Counting) in un equipo
TemPro con láser NanoLED de excitación de 370 nm. Los tiempos de vida se determinaron con el
fit a la curva realizado por el software de manera que el cuadrado de la desviación cuadrática estándar
(2) fuese menor a 1.2. La respuesta del equipo fue medida usando una suspensión LUDOX (Aldrich)
al 0.01% en agua ultra pura.

65
Las películas se depositaron por spin coating en equipo WS-400B-6NPP-LITE marca Laurell
Technologies. Los espesores de las películas se determinaron mediante perfilometria utilizando un
perfilometro Dektak 5; con un bisturí se hace una hendidura sobre la película en forma de cruz y se
hacen cuatro barridos pasando por dicha hendidura. El espesor se determina calculando el promedio
entre los valores de alturas obtenidos para las diferentes mediciones.
Para la microscopia de fuerza atómica, se analizaron películas depositadas sobre ITO utilizando un
microscopio Digital Instruments 3100 en modalidad tapping a una velocidad de barrido de 0.2 Hz.
3.3- Celdas solares
3.3.1.-Materiales
El fenil C61 ácido butírico metil éster (PCBM) y la aleación de Woods, fueron suministrados por
Aldrich Chemical Company. El poli (3-4 etilen dioxitiofeno):poli(estirensulfonato) (PEDOT:PSS)
fue suministrado por Clevius, los sustratos de ITO (8-10 /cm2) fueron de Spi.Inc.
3.3.2- Elaboración y caracterización de las celdas
Las celdas se prepararon con la siguiente configuración; ITO/PEDOT:PSS/capa activa/Woods metal
(Figura 35).
Figura 35.- Configuración de las celdas solares
Los substratos de óxido de indio estaño (ITO) se limpiaron tres veces en ultrasonido con cloruro de
metileno y hexano en ciclos de 10 min por cada solvente y dos lavados en metanol uno de 20 y otro
de 40 min. El PEDOT:PSS fue filtrado y depositado sobre el ITO mediante spin coating a 5000 rpm

66
para obtener un espesor de ~20 nm, una vez depositado el PEDOT:PSS las películas se someten a un
recocido a una temperatura de 100 °C por 30 min, para después depositar la capa activa. Se estudiaron
tres tipos de capas activas; nanohíbrido, co-polímero con PCBM y nanohíbrido con PCBM. La
deposición de las películas se realizó por spin-coating se realizaron a partir de las soluciones
DMF/THF (2:1) para la capa activa a una concentración de 1.2 mg/mL utilizándose tres etapas 1)
1:00 min a 1000 rpm 2) 20 seg a 2600 rpm 3) 30 seg a 3000 rpm por spin coating. Como cátodo se
utilizó aleación de Woods que fue depositada a una temperatura de 85 °C y la caracterización de la
celda solar se llevó acabo utilizando una fuente de poder source meter Kiethley 2400 y un simulador
solar (Solar Light Co. Model XPS 400) con una irradiancia AM1.5 de 100 mW/cm2.

67
Capítulo 4: Resultados y discusiones- Síntesis orgánica de los
polímeros y su caracterización química
4.1-Síntesis de monómeros
En este capítulo reportamos la síntesis de los tres monómeros que dan lugar a la obtención, tanto de
un homopolímero como de un co-polímero portadores de cadenas con terminación –OH; grupo
funcional que emplearemos posteriormente en su condensación con grupos trimetilsililmetoxi para la
formación de superestructuras a base de puntos cuánticos de CdS. La estrategia consistió en formar
el (11-undecanol)2,5-dietinil benzoato (A), el 1,4-bis(dodecanoxi)-2,5-diiodobenceno (B), de los
cuales mediante la reacción de Sonogashira se obtiene el co-polímero correspondiente (A-B)n. En lo
que concierne al homopolímero (C)n, el cual a fin de tener una alta regio-regularidad estructural, se
tuvo que sintetizar el monómero (C) en una ruta que consistió de 5 pasos.
I
n
C12H25O
OC12H25
O
OC11H22HO
I I
I
n
C12H25O
OC12H25
O
OC11H22HO
H I
O
OC11H22HO
H
O
OC11H22HO
H H
A (A-B)nB
C (C)n
4.1.1- Ruta de síntesis
Partiendo de la hidroquinona (1) que es un producto comercial, se llevó a cabo su alquilación de
Williamson con el 12-bromododecano en medio básico para obtener (2), el cual posteriormente se
sometió a una reacción de yodación para obtener el monómero halogenado (3), Figura 36.
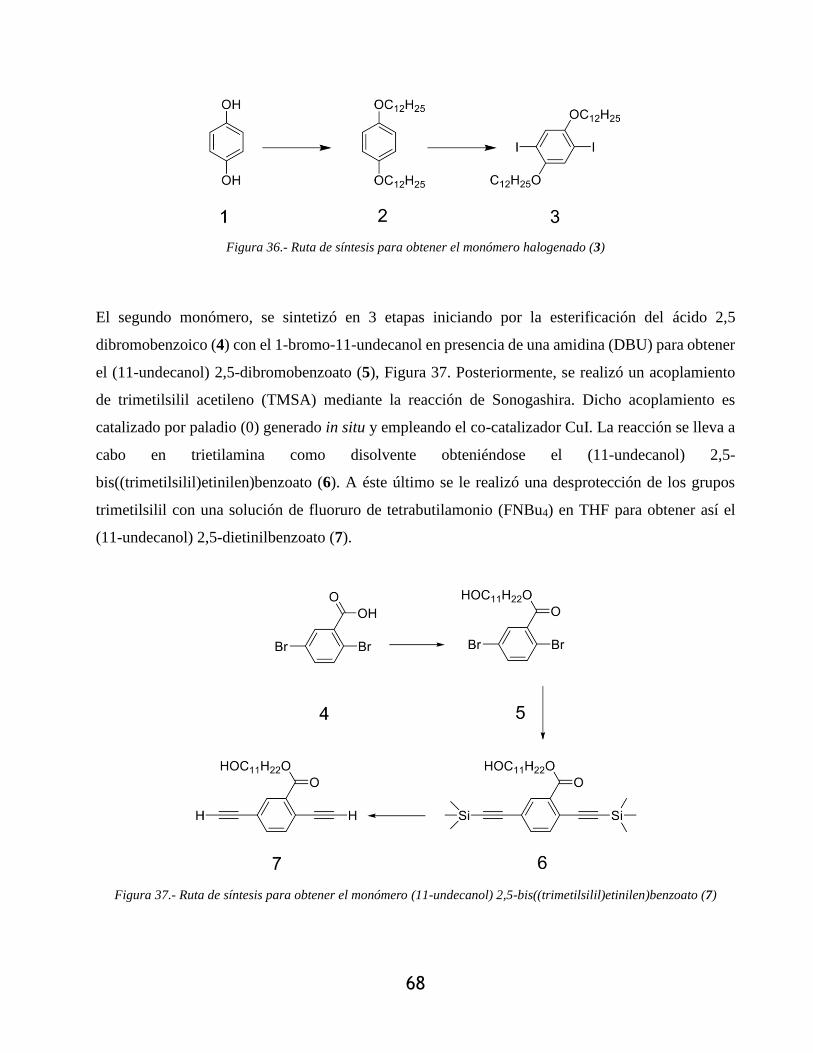
68
Figura 36.- Ruta de síntesis para obtener el monómero halogenado (3)
El segundo monómero, se sintetizó en 3 etapas iniciando por la esterificación del ácido 2,5
dibromobenzoico (4) con el 1-bromo-11-undecanol en presencia de una amidina (DBU) para obtener
el (11-undecanol) 2,5-dibromobenzoato (5), Figura 37. Posteriormente, se realizó un acoplamiento
de trimetilsilil acetileno (TMSA) mediante la reacción de Sonogashira. Dicho acoplamiento es
catalizado por paladio (0) generado in situ y empleando el co-catalizador CuI. La reacción se lleva a
cabo en trietilamina como disolvente obteniéndose el (11-undecanol) 2,5-
bis((trimetilsilil)etinilen)benzoato (6). A éste último se le realizó una desprotección de los grupos
trimetilsilil con una solución de fluoruro de tetrabutilamonio (FNBu4) en THF para obtener así el
(11-undecanol) 2,5-dietinilbenzoato (7).
Figura 37.- Ruta de síntesis para obtener el monómero (11-undecanol) 2,5-bis((trimetilsilil)etinilen)benzoato (7)

69
El tercer monómero da comienzo con la formación de una sal de diazonio a partir del ácido 2-amino-
5-yodo benzoico (8). Posteriormente, al adicionarle dietilamina se obtiene el triazeno (9). El ácido 2-
[(3,3-dietil) triazenil]-5-yodo benzoico (9) es sometido a un proceso de esterificación con el (11-
bromo-1-undecanol) para obtener el (11-undecanol) 2-[(3,3-dietil) triazenil]-5-yodo benzoato (10).
La etapa posterior consiste en acoplarle el trimetilsilil acetileno (TMSA) por la acción catalítica del
paladio (0) siguiendo el procedimiento establecido para la reacción de Sonogashira, la cual consiste
en el acoplamiento de un haluro de arilo con un acetileno terminal, usando como catalizador y co-
catalizador, dicloro bis(trifenil) fosfina paladio II y CuI, respectivamente, en presencia de trietilamina
como disolvente para generar el (11-undecanol) 2-[(3,3-dietil) triazenil]-5-[(trimetilsilil) etinilen]
benzoato (11). Subsecuentemente se le realiza una yodación para obtener el (11-undecanol)-2-yodo-
5-((trimetilsilil)etinilen)benzoato (12). Finalmente, el monómero bifuncional, el (11-undecanol) 2-
[(3,3-dietil) trazenil]-5-etinil benzoato (13), se obtiene a través de una reacción rápida y sencilla que
consiste en desproteger el grupo acetileno con una solución de fluoruro de tetrabutil amonio (FTBA)
en THF. Este monómero es de gran importancia ya que permite el crecimiento bidireccional de los
oligómeros,
Figura 38.
Figura 38.- Ruta de síntesis para obtener el monómero bifuncional (13).

70
4.1.2. Mecanismos de reacción.
Los siguientes mecanismos de reacción que se presentan son de gran ayuda para poder comprender
mejor las diferentes etapas que se llevaron a cabo en todo el proceso involucrado en la síntesis de los
diversos monómeros.
4.1.2.1. Mecanismos de reacción involucrados en la síntesis del 1,4-bis(dodecanoxi)-2,5-
diiodobenceno.
La reacción da comienzo cuando una base como el NaOH abstrae el protón ácido del hidroxilo para
formar el fenóxido de sodio, el cual actúa como nucleófilo que ataca al ion halogenuro, de acuerdo a
un mecanismo de reacción del tipo SN2. Figura 3983.
Figura 39.- Mecanismos de reacción en la alquilación de Williamson.
La yodación del compuesto (2) se lleva a cabo en presencia de KIO3 como agente oxidante, siendo
el objetivo de este último oxidar al yodo para generar el ion yodonio (I+), el cual es un electrófilo y
es atacado por el arilo, dándose la yodación. Los grupos dodecanoxi siendo electron-donadores
activan al anillo aromático orientando al primer sustituyente en posiciones orto-para, Figura 40, por
lo tanto el producto mayoritario es el compuesto monohalogenado o bien si se emplean 2 equivalentes
de Yodo bimolecular se obtiene el compuesto diiodado 3.

71
Figura 40.- Mecanismo de reacción para la obtención del (3)
4.1.2.2. Mecanismos de reacción involucrados en la síntesis del (11-undecanol) 2,5-dietinilbenzoato.
El principio de la esterificación del ácido carboxílico por el halogenuro de alquilo, en presencia de
1,8-diaza biciclo[5.4.0]undec-7-eno (DBU), se puede explicar de la siguiente manera: el DBU
reacciona con el protón del ácido carboxílico formado un puente de hidrógeno, aumentando el
carácter nucleofílico del ión carboxilato formado. El ión carboxilato ataca al carbón del
bromoundecano. El bromo junto con el protón del ácido carboxílico (HBr) son atrapados por la DBU
precipitando en forma de sal. En general, el mecanismo obedece al tipo de SN2, Figura 41.
Br
O
OC11H22HO
Br
BrBr
+
+
H
Br OH
N NO
O
N
NHBr
Figura 41.- Mecanismo de esterificación mediante una amidina (DBU)

72
En las reacciones de acoplamiento se emplean catalizadores de metales de transición, tales como
paladio, rutenio, níquel, entre otros. En particular en el acoplamiento de Sonogashira como ya se
había mencionado en los antecedentes se involucra el acoplamiento entre un acetileno terminal
aromático o alifático y un haloagenuro de arilo, en presencia de cantidades catalíticas de paladio y
yoduro de cobre. Se trata de un proceso cíclico en el que se presentan tres etapas, Figura 42:
Figura 42.- Ciclo catalítico de reacción de Sonogashira para acoplar un acetileno con un halogenuro de arilo.
La primer etapa (I) consiste en la formación de un complejo de Pd(0) (activación). En el siguiente
paso (II), la adición oxidativa del yoduro de arilo, que sucede inmediatamente al proceso de
formación de la especie catalítica del Pd(0), provoca la oxidación de Pd(0) a Pd(II) generando un
intermediario pentacoordinado de Pd (II), el trans-ArPd+2X(PPh3)2Cl. La transmetalación (III)
consiste en el intercambio de X por el fragmento acetilénico proveniente del acetiluro de cobre
formando entre el acetileno terminal y el CuI. Finalmente en la eliminación–reductora (IV) se libera

73
el producto acoplado oxidándose el paladio nuevamente a valencia cero lo que permite iniciar otra
vez el ciclo catalítico 84-86. La eliminación del Ar-R es debida a la fuerte atracción que hay entre estos
dos sustituyentes, la cual es favorecida por la posición cis en la que se encuentran dentro del complejo
catalítico. Este ciclo es válido tanto para la síntesis de monómeros como de polímeros.
El mecanismo de reacción involucrado en la desprotección del grupo acetileno se realiza bajo la
acción del fluoruro de tetrabutilamonio (Bu4NF) en THF como medio de reacción y una pequeña
cantidad de agua. Al momento en que el flúor entra en contacto con el compuesto sililado se forma
un intermediario fluorosiliconato pentavalente, posteriormente se rompe el enlace Csp-Si liberándose
fluoruro de trimetilsilil y el acetiluro, el cual se estabiliza en el medio con el contraión del
butilamonio. En una siguiente etapa el agua que está en el medio permite protonar y regenerar la
función acetilénica terminal que es estable a temperatura ambiente, Figura 43 .
Figura 43.- Mecanismo de reacción involucrado en la desprotección del grupo acetileno.
Al mismo tiempo, al entrar en contacto el fluoruro de trimetilsilil con el agua se forma ácido
fluorhídrico y trimetilsilanol. En una última etapa la unión de dos moléculas de éste último genera

74
disiloxanos, mientras que el HF es capaz de desproteger otra molécula sililada generándose así un
ciclo de desprotección n veces hasta el consumo total de la molécula silil-protegida 83,87.
4.1.2.3. Mecanismos de reacción involucrados en la síntesis del monómero (11-undecanol) 2-[(3,3-
dietil) trazenil]-5-etinil benzoato (13)
La formación del grupo triazeno se lleva a cabo en cuatro etapas, Figura 44: 1) formación del ión
nitrosilo, 2) ataque nucleofílico de la amina al ión nitrosilo, 3) formación de la sal de diazonio y 4)
formación del triazeno.
Figura 44.- Mecanismo de reacción para la síntesis del monómero triazeno (9).
La sustitución del grupo triazeno por un yoduro se lleva a cabo mediante una sustitución nucleofílica,
en donde el yoduro se genera in situ a partir del yodometano y por efecto de la temperatura. Así
mismo, el yodometano actúa también como solvente. Durante la reacción se observa la formación de

75
un precipitado de una sal negra que es el yoduro de dietildimetilamonio, además de que en el medio
se libera N2, Figura 45. Es una reacción explosiva si se emplean más de un gramo por reacción, ya
que en el medio se forman radicales libres, sin embargo es una reacción cuantitativa.
Figura 45.- Mecanismo de reacción involucrado en la sustitución del triazeno por el iodo
4.1.3.- Caracterización por resonancia magnética nuclear de 1H
La técnica de resonancia magnética nuclear de protón es de gran utilidad, ya que permite determinar
el entorno molecular que rodea a los átomos de hidrógeno y corroborar las estructuras químicas de
los compuestos obtenidos. A continuación se muestran cada uno de los espectros de resonancia
magnética nuclear de 1H correspondientes a los monómeros sintetizados, se describe también a detalle
la asignación y desplazamientos de señales.
En la Figura 46 se observa el monitoreo de la ruta de síntesis del monómero halogenado (3).
Empezando con el análisis del primer precursor (2), se pueden apreciar las siguientes señales; a δ
6.81 ppm, los protones del arilo (pico a), δ=3.88 ppm, un triplete el cual se debe a los protones
metileno en CH2-O-Ar (pico b), a δ=1.74 ppm, la señal correspondiente a los protones del grupo
(CH2-β-O) (pico c), a δ=1.43 ppm los que se asignan a los protones en (CH2-γ-O) (pico d), a δ=1.25
ppm se presenta la señal debida a los protones metileno CH2 (pico e), y por último la señal en δ=0.87
ppm se atribuye a protones metílico CH3 (pico f) de la cadena lateral. Siguiendo la ruta de síntesis,
las señales b-f debidas a las cadenas laterales quedan invariadas y la discusión se centra únicamente
en los desplazamientos de la señal de los protones aromáticos y aparición/desaparición de nuevos
picos según las diferentes reacciones. La yodación del compuesto (3) provoca un despantallamiento
de los protones del fenilo y por lo tanto las señales correspondientes se desplazan hacia campos más
bajos δ= 7.16 ppm (pico a).
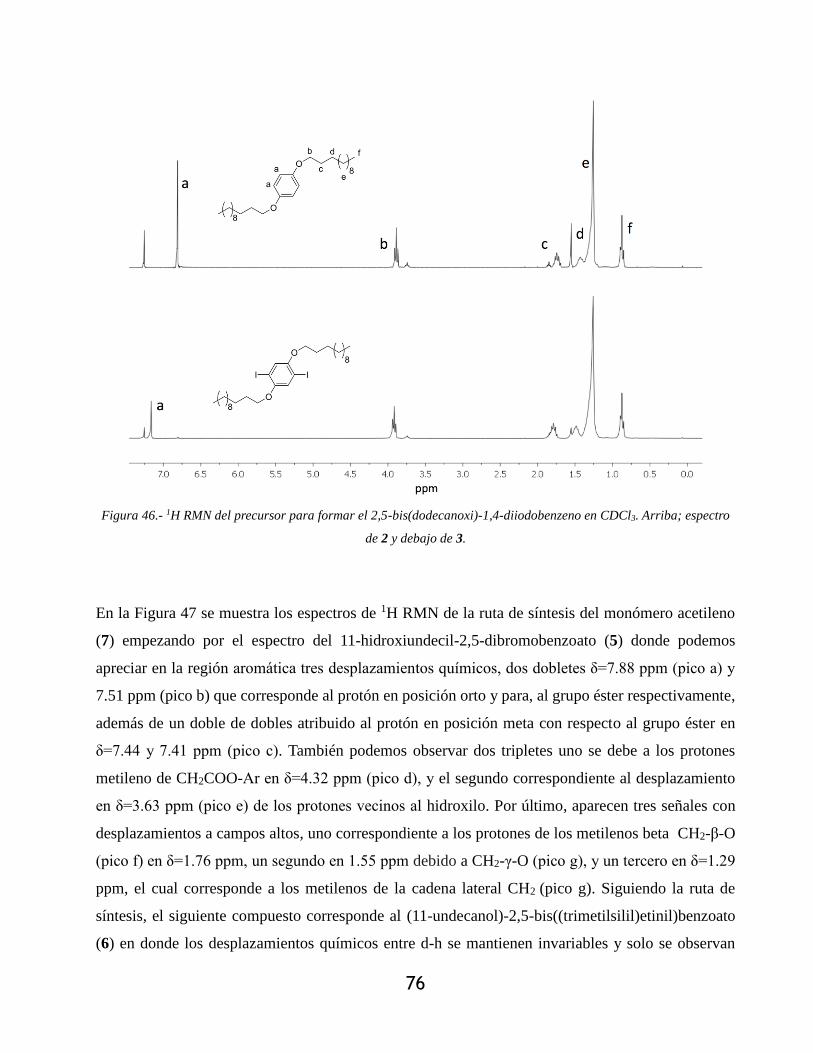
76
Figura 46.- 1H RMN del precursor para formar el 2,5-bis(dodecanoxi)-1,4-diiodobenzeno en CDCl3. Arriba; espectro
de 2 y debajo de 3.
En la Figura 47 se muestra los espectros de 1H RMN de la ruta de síntesis del monómero acetileno
(7) empezando por el espectro del 11-hidroxiundecil-2,5-dibromobenzoato (5) donde podemos
apreciar en la región aromática tres desplazamientos químicos, dos dobletes δ=7.88 ppm (pico a) y
7.51 ppm (pico b) que corresponde al protón en posición orto y para, al grupo éster respectivamente,
además de un doble de dobles atribuido al protón en posición meta con respecto al grupo éster en
δ=7.44 y 7.41 ppm (pico c). También podemos observar dos tripletes uno se debe a los protones
metileno de CH2COO-Ar en δ=4.32 ppm (pico d), y el segundo correspondiente al desplazamiento
en δ=3.63 ppm (pico e) de los protones vecinos al hidroxilo. Por último, aparecen tres señales con
desplazamientos a campos altos, uno correspondiente a los protones de los metilenos beta CH2-β-O
(pico f) en δ=1.76 ppm, un segundo en 1.55 ppm debido a CH2-γ-O (pico g), y un tercero en δ=1.29
ppm, el cual corresponde a los metilenos de la cadena lateral CH2 (pico g). Siguiendo la ruta de
síntesis, el siguiente compuesto corresponde al (11-undecanol)-2,5-bis((trimetilsilil)etinil)benzoato
(6) en donde los desplazamientos químicos entre d-h se mantienen invariables y solo se observan
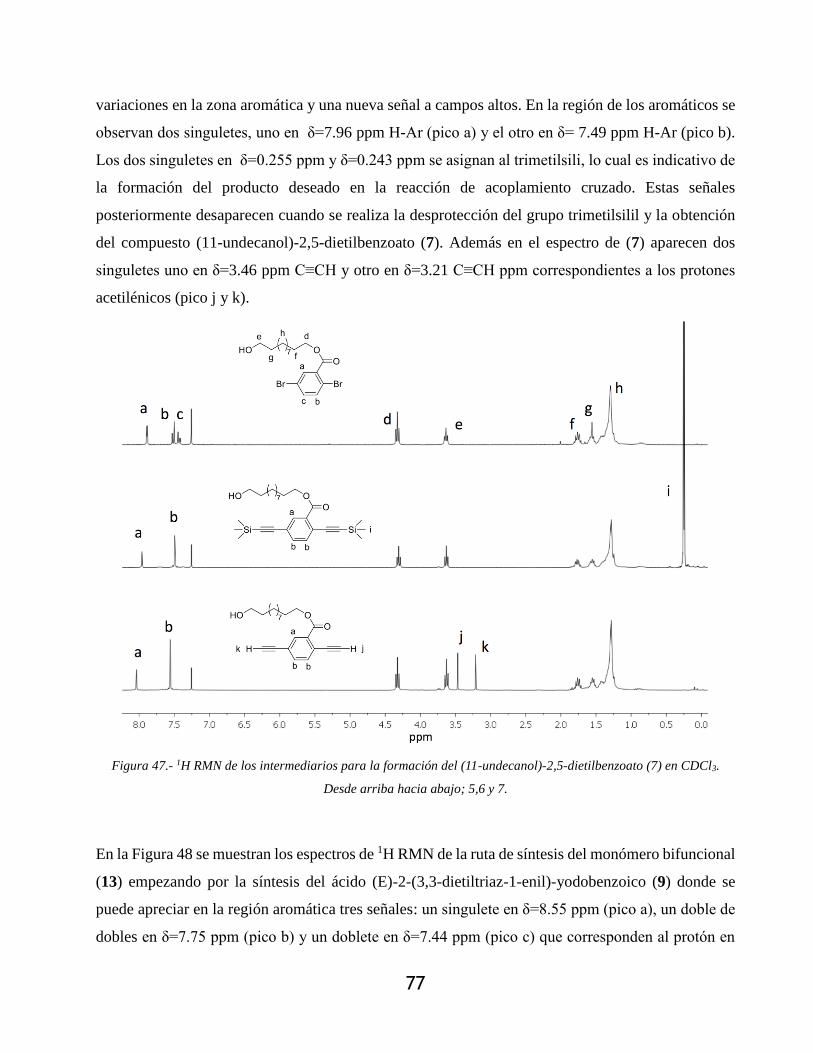
77
variaciones en la zona aromática y una nueva señal a campos altos. En la región de los aromáticos se
observan dos singuletes, uno en δ=7.96 ppm H-Ar (pico a) y el otro en δ= 7.49 ppm H-Ar (pico b).
Los dos singuletes en δ=0.255 ppm y δ=0.243 ppm se asignan al trimetilsili, lo cual es indicativo de
la formación del producto deseado en la reacción de acoplamiento cruzado. Estas señales
posteriormente desaparecen cuando se realiza la desprotección del grupo trimetilsilil y la obtención
del compuesto (11-undecanol)-2,5-dietilbenzoato (7). Además en el espectro de (7) aparecen dos
singuletes uno en δ=3.46 ppm C≡CH y otro en δ=3.21 C≡CH ppm correspondientes a los protones
acetilénicos (pico j y k).
Figura 47.- 1H RMN de los intermediarios para la formación del (11-undecanol)-2,5-dietilbenzoato (7) en CDCl3.
Desde arriba hacia abajo; 5,6 y 7.
En la Figura 48 se muestran los espectros de 1H RMN de la ruta de síntesis del monómero bifuncional
(13) empezando por la síntesis del ácido (E)-2-(3,3-dietiltriaz-1-enil)-yodobenzoico (9) donde se
puede apreciar en la región aromática tres señales: un singulete en δ=8.55 ppm (pico a), un doble de
dobles en δ=7.75 ppm (pico b) y un doblete en δ=7.44 ppm (pico c) que corresponden al protón en

78
posición orto, para y meta al grupo ácido. En δ=3.91 y 3.77 ppm se observa cuadruplete (pico d)
correspondiente a CH2-α-N y un doble de triplete en δ=1.42 y 1.31 debido a los protones CH3-β-N.
Ahora , de acuerdo a la ruta, el siguiente compuesto que se sintetizó fue el (11-undecanol)-2-(2,3-
dietiltriaz-1-enil)-5-yodobenzoato (10). Como producto de la esterificación en la región aromática se
observan desplazamientos significativos, el protón en posición orto al grupo éster aparece como un
doblete a δ=7.86 ppm (pico a) y en para un doble de dobles a δ=7.67 y 7.64 ppm (pico b), mientras
que a δ=7.16 ppm se observa un doblete que corresponde al protón ubicado en posición meta con
respecto al grupo éster. Además, es evidente la aparición de dos nuevas señales (pico f y g), dos
tripletes uno en δ=4.23 ppm debido a CH2-α-COO-Ar y otro a δ=3.62ppm correspondiente a los CH2-
α-OH. También es evidente la aparición de las señales de los metilenos de la cadena lateral en δ=1.70
ppm CH2-β-COO-Ar, en δ=1.55 ppm debido a CH2-γ-COO-Ar y en δ=1.27 ppm CH2. Por último las
señales provenientes de los metilenos CH2-α-N del sustituyente triazeno (pico d) sufren un ligero
desplazamiento hacia δ=3.74 y 3.72 ppm quedando semitraslapadas, mientras que los metilos CH3-
β-N no alcanzan a diferenciarse pues quedan sobrelapados con la señal en δ=1.27ppm CH2.
Posteriormente, se realizó una reacción de acoplamiento de Sonogashira para obtener el compuesto
(11-undecanol)-2-(3,3-dietiltriaz-1-enil)-5-((trimetilsilil) etinil) benzoato (11). Las señales d-j
debidas a las cadenas laterales y los protones correspondiente al grupo triazeno quedan invariadas,
no así en la zona de los protones aromáticos donde se observan cambios y un nuevo desplazamiento
en campos altos, un doblete en δ=7.68 ppm, un doble de dobles en δ=7.44ppm , y un doblete en
δ=7.35 ppm. La señal que da la pauta para decir que la reacción se llevó acabo es la que corresponde
al trimetilsilil, la cual se observa como un singulete en 0.23 ppm. Posteriormente se realizó una
yodación para sintetizar el compuesto (11-undecanol)-2-yodo-5-((trimetilsilil)etinil)benzoato (12),
las señales f-j debidas a la resonancia de las señales de los metilenos de la cadena lateral quedan
invariable, además se aprecia la desaparición de los desplazamientos del multiplete correspondiente
a los metilenos CH2-α-N y metilos CH3-β-N que corresponden al grupo triazeno, el cual fue
sustituido por yodo. También suceden cambios en la región aromática, como es la aparición de un
doblete en δ=7.90 ppm, el cual corresponde al protón orto al grupo éster, un doblete en δ=7.82 ppm
correspondiente al protón en posición orto con respecto al yodo y un doble de dobles en δ=7.19 ppm
debido al protón en posición para con respecto al grupo éster. Una vez obtenido este producto, en la
ruta de síntesis el último compuesto es generado mediante la desprotección del grupo trimetilsil. En
el espectro de protón solo se pueden apreciar dos diferencias con respecto al del precursor, un triplete

79
en δ=3.36 ppm que corresponde al CH2-α-OH, el cual se observa un pequeño desplazamiento a campo
alto esto debido a la influencia por el efecto de apantallamiento y por la proximidad al grupo
acetileno, de hecho esta es la señal que da la pauta para corroborar la obtención de la nueva molécula,
es decir hay la desaparición del desplazamiento químico de los protones del grupo trimetilsililo y la
aparición del protón que aparece en δ=3.18 ppm debido al grupo C≡CH.
Figura 48.- 1H RMN de los intermediarios para la formación del (11-undecanol)-2-yodo-5) etinil) benzoato (13) en
CDCl3. Desde arriba hacia abajo; 9-13.

80
4.2.- Síntesis de polímeros
4.2.1- Síntesis del co-polímero
4.2.1.1- Ruta de síntesis
Una vez sintetizados los intermediarios (11-undecanol)-2,5-dietilbenzoato (7) y el 1,4-
bis(dodecanoxi)-2,5-diyodobenceno (3), fue posible sintetizar el co-polímero (14) por medio del
acoplamiento de Sonogashira, Figura 49.
Figura 49.- Ruta de síntesis de Sonogashira para la preparación del co-polímero (14)
4.2.1.2- Mecanismo de reacción
El mecanismo de reacción para la polimerización se describió en la sección 4.1.2.2., el cual es el
mecanismo general de acoplamiento teniendo como catalizador una especie de paladio (0) y CuI
como co-catalizador.
4.2.2.- Caracterización por resonancia magnética nuclear
4.2.2.1.- Resonancia magnética nuclear de protón 1H
En la Figura 50 se muestra el espectro de 1H RMN correspondientes al co-polímero (14). En dicho
espectro se puede apreciar en la región aromática tres señales, δ=8.13 ppm (pico a) correspondiente
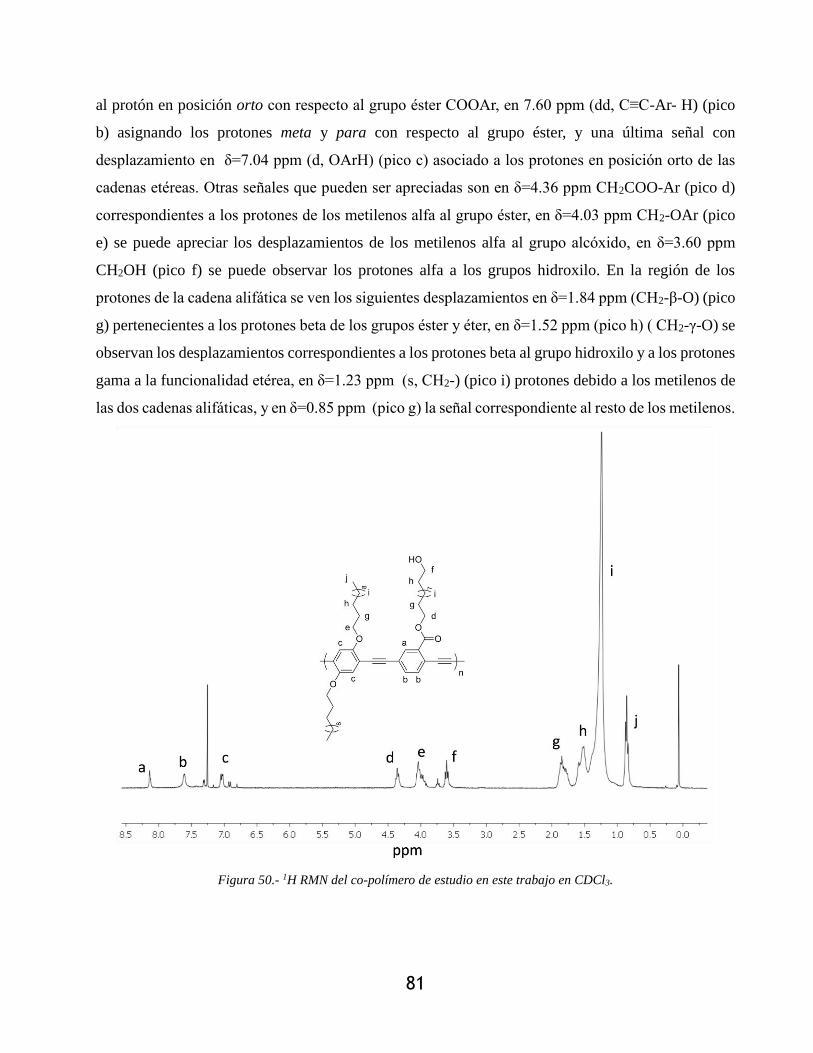
81
al protón en posición orto con respecto al grupo éster COOAr, en 7.60 ppm (dd, C≡C-Ar- H) (pico
b) asignando los protones meta y para con respecto al grupo éster, y una última señal con
desplazamiento en δ=7.04 ppm (d, OArH) (pico c) asociado a los protones en posición orto de las
cadenas etéreas. Otras señales que pueden ser apreciadas son en δ=4.36 ppm CH2COO-Ar (pico d)
correspondientes a los protones de los metilenos alfa al grupo éster, en δ=4.03 ppm CH2-OAr (pico
e) se puede apreciar los desplazamientos de los metilenos alfa al grupo alcóxido, en δ=3.60 ppm
CH2OH (pico f) se puede observar los protones alfa a los grupos hidroxilo. En la región de los
protones de la cadena alifática se ven los siguientes desplazamientos en δ=1.84 ppm (CH2-β-O) (pico
g) pertenecientes a los protones beta de los grupos éster y éter, en δ=1.52 ppm (pico h) ( CH2-γ-O) se
observan los desplazamientos correspondientes a los protones beta al grupo hidroxilo y a los protones
gama a la funcionalidad etérea, en δ=1.23 ppm (s, CH2-) (pico i) protones debido a los metilenos de
las dos cadenas alifáticas, y en δ=0.85 ppm (pico g) la señal correspondiente al resto de los metilenos.
Figura 50.- 1H RMN del co-polímero de estudio en este trabajo en CDCl3.

82
4.2.2.2.- Resonancia magnética nuclear de carbono 13C
El espectro de 13C RMN del co-polímero (14) se presenta en la Figura 51 es posible apreciar en la
región aromática los siguientes desplazamientos químicos, δ=165.65 ppm éste debido al carbonilo
(C=O), el desplazamiento en δ=153.98ppm corresponde a los carbonos C2, C5 de la unidad repetitiva
que tiene la cadena lateral alcoxida; se observa una sola señal debido a que estos carbones son
simétricos, otro desplazamientos que se puede apreciar es el de los carbonos C8, C9 en δ=134.17ppm
también observándose una sola señal, ya que su entorno químico es el mismo, δ=133.61ppm(C11).
El siguiente desplazamiento que se observa en δ=132.11ppm es debido a los carbonos C1, C4 de la
unidad repetitiva que tiene la cadena lateral alcoxida, otro desplazamiento que también se ve es en
δ=123.31ppm correspondiente al carbono C12, el cual se encuentra en la unidad repetitiva que
contiene la cadena lateral del éster, en δ=117.17ppm se observa los carbonos C7, C10 carbonos que
se encuentran cerca los grupos acetilenos, y los últimos dos desplazamientos de la región aromática
es en δ=114.03ppm debido al los carbones C1, C4, así como δ=94.04ppm C3, C6, desplazamiento
importante que marca la pauta, de haberse obtenido el compuesto deseado son las señales de los
carbonos acetilenos que se aprecian en δ=93.16ppm (C≡C) y δ=88.53ppm (C≡C). Como referencia
se ve la multiplicidad y desplazamiento del solvente CDCl3 a δ=77.54ppm (Cl3), δ=77.12ppm(Cl3),
δ=76.70ppm(Cl3). En la región alifática, se pueden ver los desplazamiento alfa a los oxígenos, que
dentro de ellos están en δ=69.80ppm correspondiente al carbono alfa al oxigeno del alcóxido
(COAr)(pico a), en δ=65.72ppm este desplazamiento corresponde al carbono alfa al grupo éster (C-
COO-Ar)(pico b), y en δ=63.13ppm el desplazamiento alfa al grupo hidroxilo(C-OH)(pico c).
También se pueden ver los siguientes desplazamientos correspondientes a la cadena alifática, en
δ=32.90ppm (C-β-OH), δ=32.02ppm (C-β-CH3), δ=29.76ppm(CH2), δ=29.46ppm(C-β-COO-Ar),
δ=28.83ppm(C-β-O-Ar), δ=26.19ppm (C-γ-O), δ=25.84ppm (C-γ-CH3),δ= 22.79ppm(C-CH3),
δ=14.22 ppm (CH3).
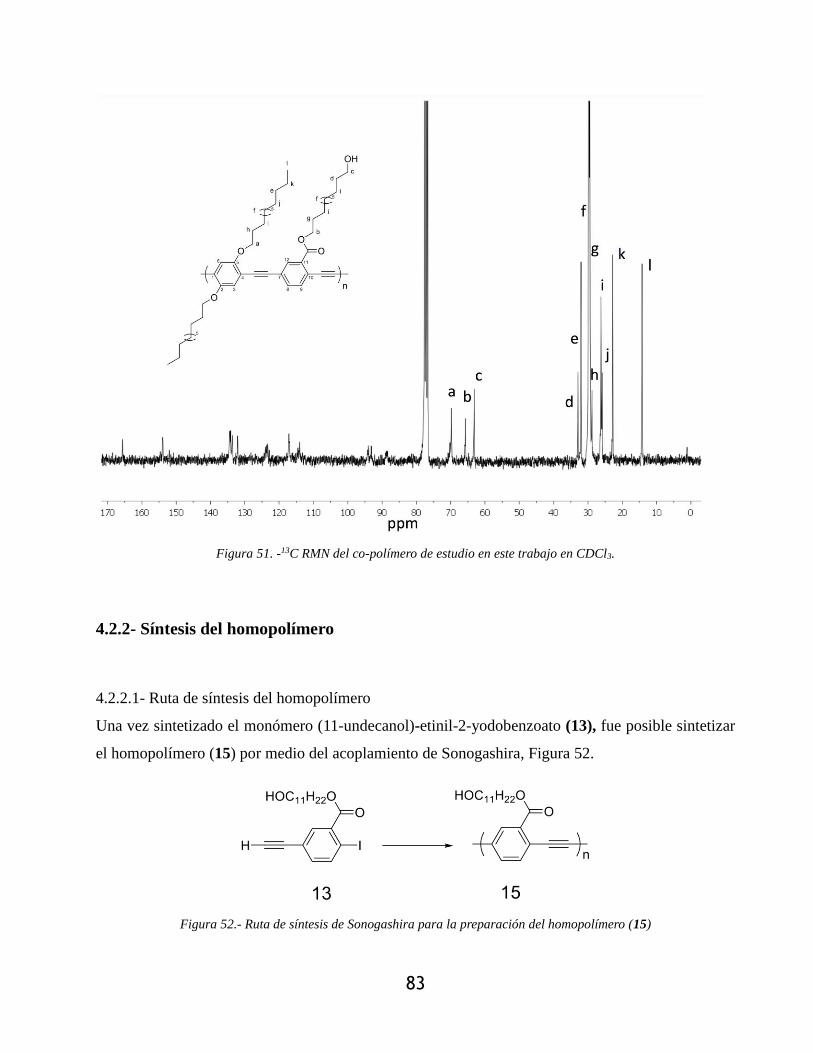
83
Figura 51. -13C RMN del co-polímero de estudio en este trabajo en CDCl3.
4.2.2- Síntesis del homopolímero
4.2.2.1- Ruta de síntesis del homopolímero
Una vez sintetizado el monómero (11-undecanol)-etinil-2-yodobenzoato (13), fue posible sintetizar
el homopolímero (15) por medio del acoplamiento de Sonogashira, Figura 52.
Figura 52.- Ruta de síntesis de Sonogashira para la preparación del homopolímero (15)
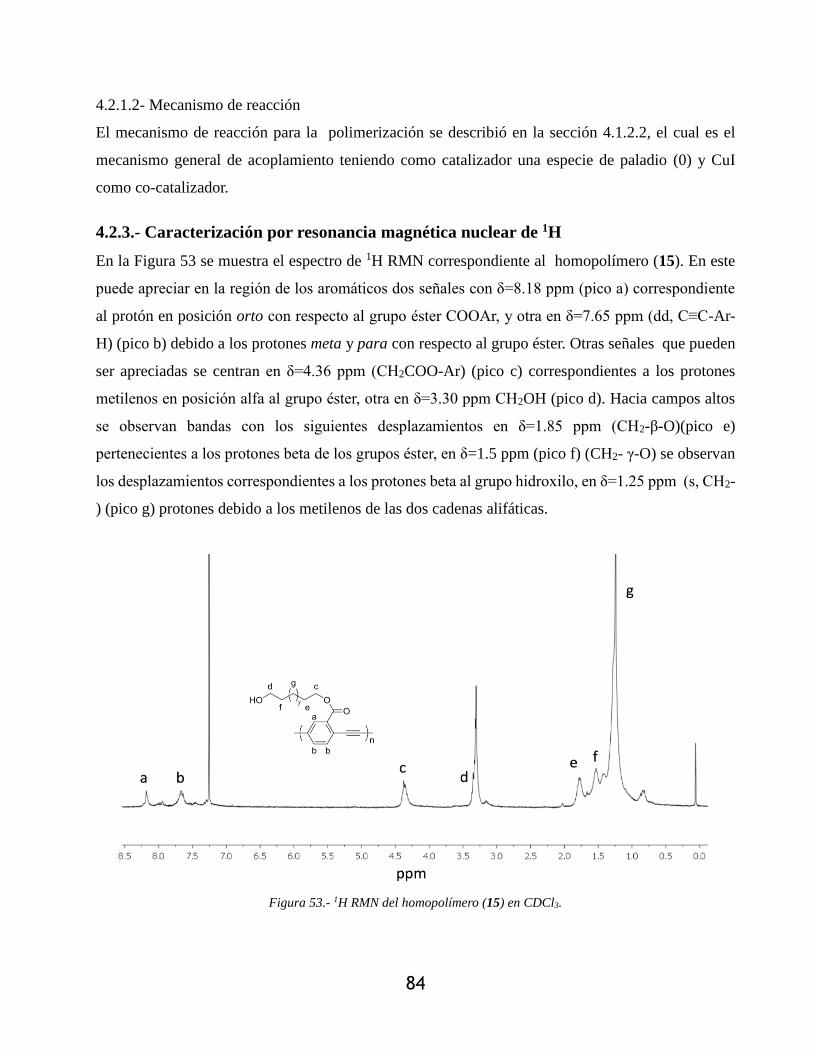
84
4.2.1.2- Mecanismo de reacción
El mecanismo de reacción para la polimerización se describió en la sección 4.1.2.2, el cual es el
mecanismo general de acoplamiento teniendo como catalizador una especie de paladio (0) y CuI
como co-catalizador.
4.2.3.- Caracterización por resonancia magnética nuclear de 1H
En la Figura 53 se muestra el espectro de 1H RMN correspondiente al homopolímero (15). En este
puede apreciar en la región de los aromáticos dos señales con δ=8.18 ppm (pico a) correspondiente
al protón en posición orto con respecto al grupo éster COOAr, y otra en δ=7.65 ppm (dd, C≡C-Ar-
H) (pico b) debido a los protones meta y para con respecto al grupo éster. Otras señales que pueden
ser apreciadas se centran en δ=4.36 ppm (CH2COO-Ar) (pico c) correspondientes a los protones
metilenos en posición alfa al grupo éster, otra en δ=3.30 ppm CH2OH (pico d). Hacia campos altos
se observan bandas con los siguientes desplazamientos en δ=1.85 ppm (CH2-β-O)(pico e)
pertenecientes a los protones beta de los grupos éster, en δ=1.5 ppm (pico f) (CH2- γ-O) se observan
los desplazamientos correspondientes a los protones beta al grupo hidroxilo, en δ=1.25 ppm (s, CH2-
) (pico g) protones debido a los metilenos de las dos cadenas alifáticas.
Figura 53.- 1H RMN del homopolímero (15) en CDCl3.

85
Capitulo 5: Resultados y discusiones-síntesis y caracterización de
puntos cuánticos CdS(MPS)
5.1.- Síntesis
La síntesis de los puntos cuánticos de sulfuro de cadmio con agente ligante (3-mercaptopropil)
trimetoxisilano (MPS), se llevó a cabo en N,N-dimetilformamida (DMF) modificando la metodología
reportada por Fang.75 Esta modificación involucra una destilación azeotrópica con benceno para
extraer el agua desionizada del medio de reacción, así como se eliminan los sólidos generados de los
subproductos por decantación.
El CdCl2 y Na2S en agua se disocian en iones, los cuales en ausencia de ligantes darían lugar a la
reacción de intercambio iónico con precipitación de CdS como producto. Sin embargo, el MPS actúa
como ligante restringiendo el crecimiento de las partículas, de hecho se ha reportado en otros
artículos76,88-90, que incluso puede formar enlaces Cd-S-metoxi (Figura 54, estructura 1, R=OCH3).
Los grupos metoxisilano se pueden hidrolizar en presencia de agua dando lugar a estructuras como
la del tipo 1 de la Figura 54, R=H y sucesivamente formar grupos Si-O-Si (Figura 54, estructura 2).
También es posible la poli(condensación) con otras moléculas de MPS dando estructuras como la del
tipo 3. Todas estas reacciones pueden ocurrir en todos los grupos metoxi o incluso en solo algunos,
por lo que las partículas de CdS(MPS) forman un sistema complejo no estequiométrico. Según
Kozhevnikova y colaboradores, también puede ocurrir la formación de enlaces Si-O-Si (estructura 4,
Figura 54) o de puentes disulfuro (estructura 5, Figura 54) entre partículas de CdS(MPS). Es
importante considerar que todos los grupos Si-OR de las estructuras 2-5 pueden seguir polimerizando
en redes de tipo poli(siloxano) tal y como se esquematiza para el caso de 2 en la estructura 6.
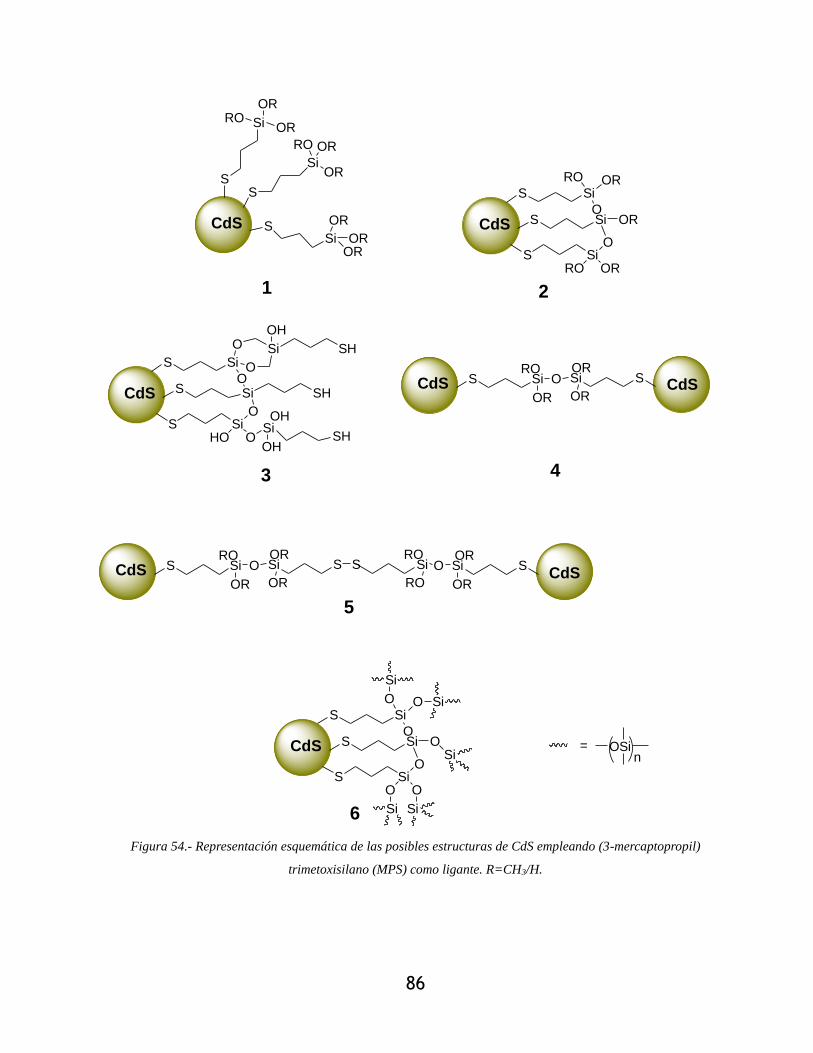
86
S
S
S
Si
Si
Si
RO OR
OR
OR
ORRO
OR
OROR
CdS CdS
S Si
S
S Si
RO OR
RO OR
O
O
1 2
CdS
S Si
S
S SiHO O
O
O
3
Si
OH
OH
SH
O
O Si
OH
SH
Si OR
Si SHCdS S
RO
4
Si CdSSORSiO
OR OR
CdS SRO
5
Si SORSiO
OR ORCdS
SORSi
ROSi O
ORRO
S
CdS
S Si
S
S Si
O O
O O
O
O
6
Si
Si
Si
SiSi
OSi
= OSin
Figura 54.- Representación esquemática de las posibles estructuras de CdS empleando (3-mercaptopropil)
trimetoxisilano (MPS) como ligante. R=CH3/H.

87
5.2.- Caracterización
5.2.1.- Espectroscopia de resonancia magnética nuclear de estado sólido (SS-NMR de
las siglas en ingles)iii
En la Figura 55 se muestra el espectro 13C (izquierda) y 29Si (derecha) de CdS (MPS). En el espectro
de carbono, se observan picos a 13, 24, 28 y 42 ppm asignados a los enlaces de Si-CH2, -CH2-, -CH2-
S(CdS)- y O-CH3, respectivamente. Los picos a 32, 38 y 165 ppm son de la DMF (b.p. 153°C)
atrapado en el sólido. En cuanto al espectro de 29Si, se observan dos picos intensos a -65 y -57 ppm
asignados a los enlaces -Si(-OSi)3 y -Si(-OSi)2O(R/H) y una señal débil a -50 ppm indicativa de la
presencia de una pequeña cantidad de -Si(-OSi)O(R/H)2. La presencia de estos picos comprueba
también las estructuras propuestas de la Figura 54. El hecho que las dos señales más intensas
presenten casi la misma intensidad sugiere que las estructuras con los grupos metoxi total o
parcialmente hidrolizados (es decir que presentan grupos OR o OH) se encuentran en la misma
proporción. No se observan sin embargo, los picos atribuidos al MPS libre (que no haya reaccionado),
los cuales se esperarían que resonaran a aprox. -40 y -45 ppm. Cabe mencionar, que en el caso del
CdS(MPS) no hay ninguna interacción que evite la conversión de grupos -Si(-OCH3)3 a -Si(OSi-
)2O(R/H) y -Si (-OSi)3 ya que en el sistema se siguen formando estas especies al estar expuestas a la
humedad del medio ambiente y en función del tiempo.
iii La muestra de CdS(MPS) preparada en CIQA fue enviada a la Universidad de Akron para su análisis por SS-NMR, utilizando como
probe 13C y 29Si.
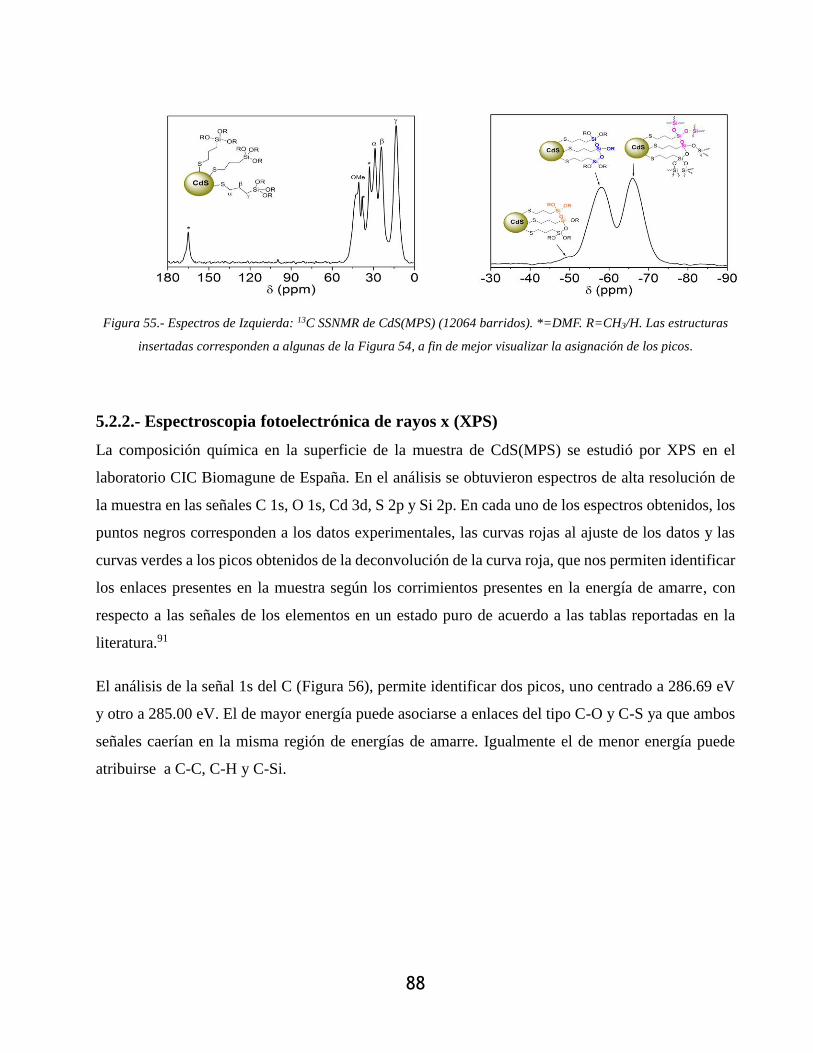
88
Figura 55.- Espectros de Izquierda: 13C SSNMR de CdS(MPS) (12064 barridos). *=DMF. R=CH3/H. Las estructuras
insertadas corresponden a algunas de la Figura 54, a fin de mejor visualizar la asignación de los picos.
5.2.2.- Espectroscopia fotoelectrónica de rayos x (XPS)
La composición química en la superficie de la muestra de CdS(MPS) se estudió por XPS en el
laboratorio CIC Biomagune de España. En el análisis se obtuvieron espectros de alta resolución de
la muestra en las señales C 1s, O 1s, Cd 3d, S 2p y Si 2p. En cada uno de los espectros obtenidos, los
puntos negros corresponden a los datos experimentales, las curvas rojas al ajuste de los datos y las
curvas verdes a los picos obtenidos de la deconvolución de la curva roja, que nos permiten identificar
los enlaces presentes en la muestra según los corrimientos presentes en la energía de amarre, con
respecto a las señales de los elementos en un estado puro de acuerdo a las tablas reportadas en la
literatura.91
El análisis de la señal 1s del C (Figura 56), permite identificar dos picos, uno centrado a 286.69 eV
y otro a 285.00 eV. El de mayor energía puede asociarse a enlaces del tipo C-O y C-S ya que ambos
señales caerían en la misma región de energías de amarre. Igualmente el de menor energía puede
atribuirse a C-C, C-H y C-Si.
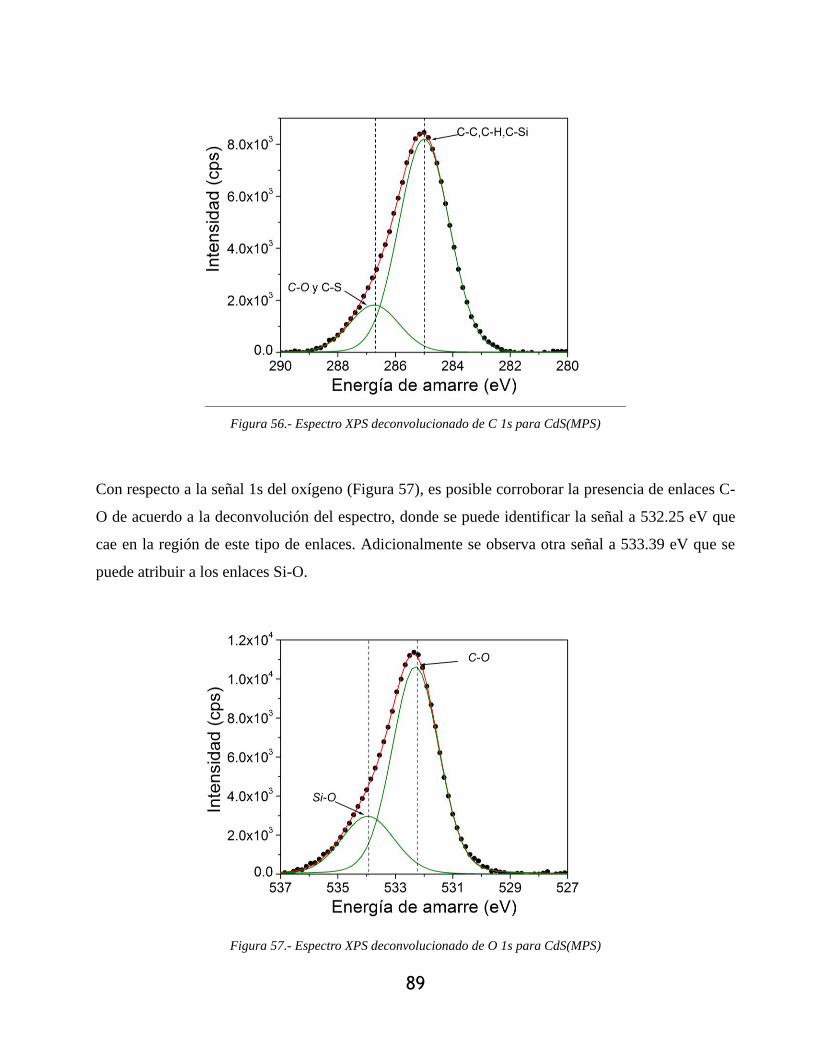
89
Figura 56.- Espectro XPS deconvolucionado de C 1s para CdS(MPS)
Con respecto a la señal 1s del oxígeno (Figura 57), es posible corroborar la presencia de enlaces C-
O de acuerdo a la deconvolución del espectro, donde se puede identificar la señal a 532.25 eV que
cae en la región de este tipo de enlaces. Adicionalmente se observa otra señal a 533.39 eV que se
puede atribuir a los enlaces Si-O.
Figura 57.- Espectro XPS deconvolucionado de O 1s para CdS(MPS)
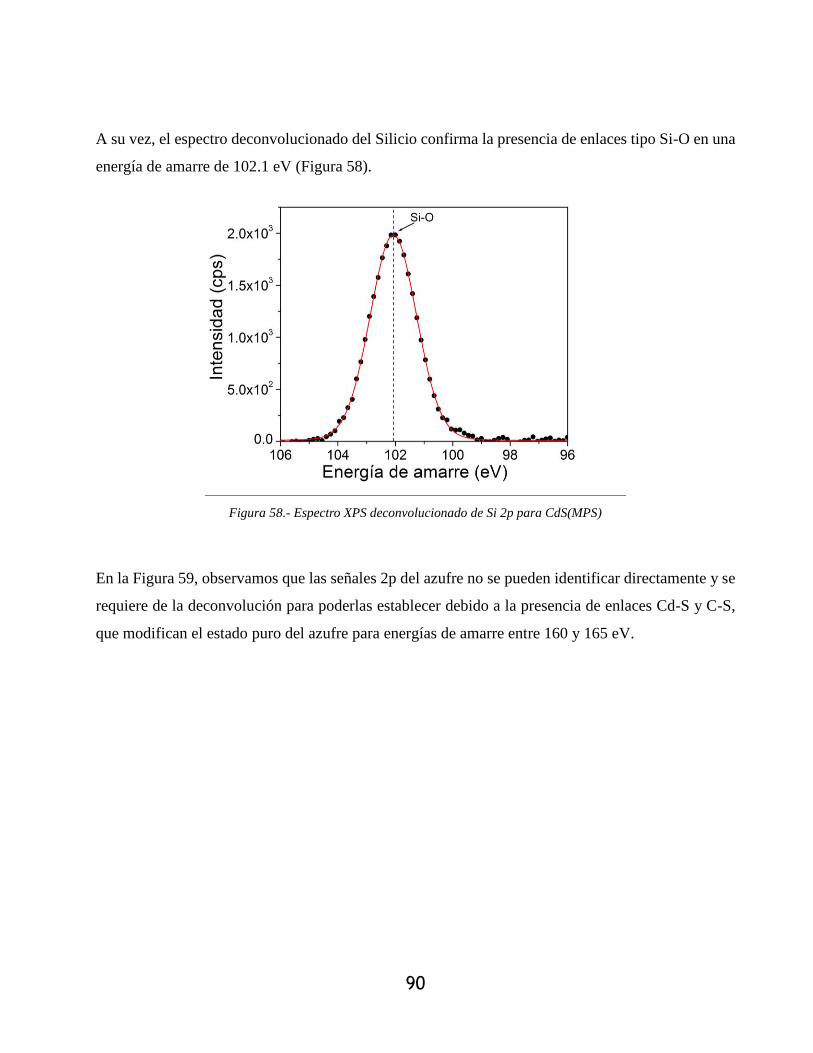
90
A su vez, el espectro deconvolucionado del Silicio confirma la presencia de enlaces tipo Si-O en una
energía de amarre de 102.1 eV (Figura 58).
Figura 58.- Espectro XPS deconvolucionado de Si 2p para CdS(MPS)
En la Figura 59, observamos que las señales 2p del azufre no se pueden identificar directamente y se
requiere de la deconvolución para poderlas establecer debido a la presencia de enlaces Cd-S y C-S,
que modifican el estado puro del azufre para energías de amarre entre 160 y 165 eV.
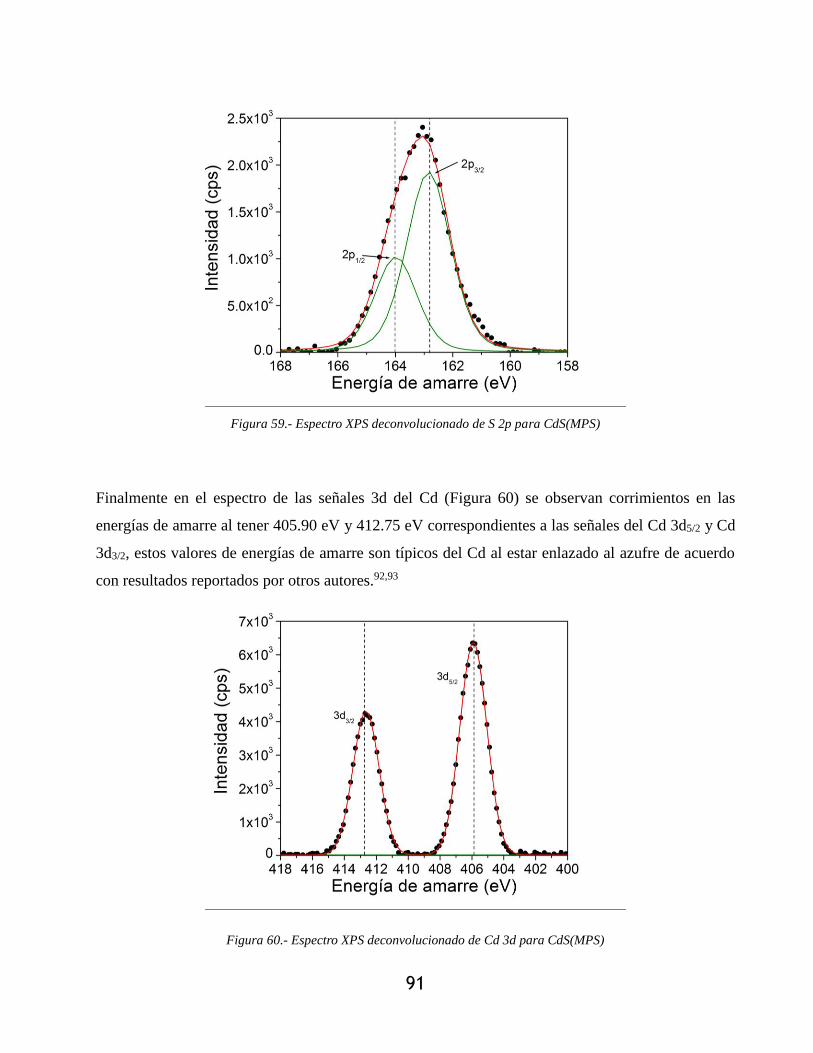
91
Figura 59.- Espectro XPS deconvolucionado de S 2p para CdS(MPS)
Finalmente en el espectro de las señales 3d del Cd (Figura 60) se observan corrimientos en las
energías de amarre al tener 405.90 eV y 412.75 eV correspondientes a las señales del Cd 3d5/2 y Cd
3d3/2, estos valores de energías de amarre son típicos del Cd al estar enlazado al azufre de acuerdo
con resultados reportados por otros autores.92,93
Figura 60.- Espectro XPS deconvolucionado de Cd 3d para CdS(MPS)

92
A partir de la integración de los espectros de carbono C 1s, oxigeno O 1s, cadmio Cd 3d, azufre S 2p
y silicio Si 2p se obtuvo la composición en porcentaje de átomo (Tabla 1).
Tabla 1.- Porcentaje en átomo de C,S,Si,O y C obtenidos a partir de la integración de los picos XPS correspondientes
Cd S Si O C
2.7 11.1 7.7 26.3 52.1
A partir de estos valores se pueden estimar las relaciones en átomo entre Cd y Si. dando ~ 2.8 Si por
átomo de Cd. En el MPS, HS(CH2)3Si(OCH3)3, hay 6 átomos de carbono y 3 de oxigeno por cada
átomo de silicio. Por ende, si consideremos como caso extremo que todo el 7.7 % de átomos de Si se
deba a las moléculas de MPS como ligante, entonces deberíamos tener aprox. 46% de átomos de C,
23% de oxigeno y 10.4% (2.7 de la relación Cd:S 1:1 de las nanopartículas +7.7% de MPS) . Para
todos los elementos se tiene un exceso; de aprox. 12% para C y O y 6% de S. Estos excesos pueden
explicarse con base a las estructuras parcialmente o totalmente hidrolizadas propuestas en la Figura
54 y en acuerdo con los resultados de SS-NMR. Para el azufre, es posible también un exceso debido
a la formación de nanopartículas de CdS ricas en S.94,95 Por ende podemos concluir que el número de
moléculas de MPS por nanopartícula no puede deducirse debido a que como se ha discutido
anteriormente, el sistema CdS(MPS) no es estequiométrico a causa de las diferentes posibles
estructuras derivadas de las reacciones de hidrólisis/condensación. Además no se conoce
exactamente la profundidad de penetración, aunque es notorio que el XPS es una técnica superficial.
Es importante mencionar que en el estudio XPS, no se detectó N atribuible a DMF (C3H7NO)
atrapado en CdS(MPS), a diferencia de lo observado por resonancia.
5.2.3.- Análisis termogravimétrico (TGA)
En la Figura 61 se muestra el termograma de la muestra de CdS(MPS) cuyo peso inicial fue de
27.7330 mg. La línea negra corresponde a la perdida en peso en porcentaje y la azul a su primera
derivada (línea azul). La primer pérdida en peso comienza en 42ºC con una temperatura máxima de
perdida (Tmax) de 100.9ºC, el cual representa el 9.220% y podría atribuirse la humedad de la muestra
o solvente (DMF) usado durante la síntesis, siendo el punto de ebullición del DMF relativamente alto
(153ºC) este puede quedar atrapado cuando se precipitan las nanopartículas, así como se comprobó
por SS-NMR.
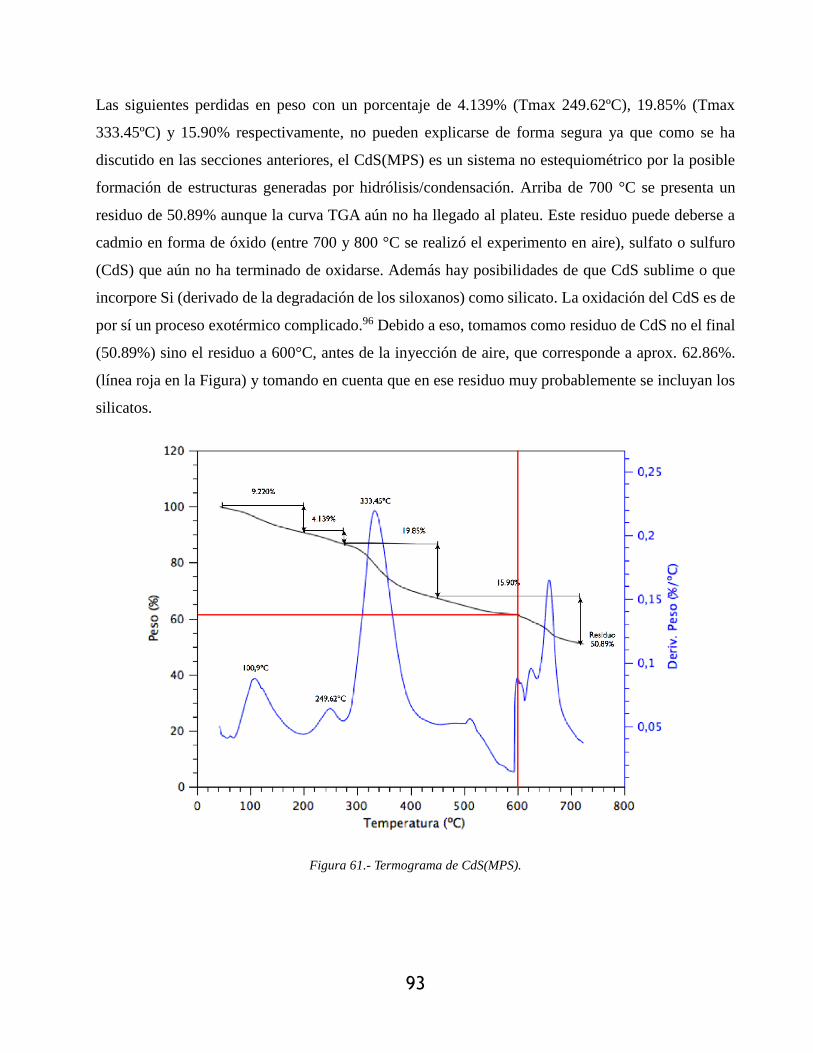
93
Las siguientes perdidas en peso con un porcentaje de 4.139% (Tmax 249.62ºC), 19.85% (Tmax
333.45ºC) y 15.90% respectivamente, no pueden explicarse de forma segura ya que como se ha
discutido en las secciones anteriores, el CdS(MPS) es un sistema no estequiométrico por la posible
formación de estructuras generadas por hidrólisis/condensación. Arriba de 700 °C se presenta un
residuo de 50.89% aunque la curva TGA aún no ha llegado al plateu. Este residuo puede deberse a
cadmio en forma de óxido (entre 700 y 800 °C se realizó el experimento en aire), sulfato o sulfuro
(CdS) que aún no ha terminado de oxidarse. Además hay posibilidades de que CdS sublime o que
incorpore Si (derivado de la degradación de los siloxanos) como silicato. La oxidación del CdS es de
por sí un proceso exotérmico complicado.96 Debido a eso, tomamos como residuo de CdS no el final
(50.89%) sino el residuo a 600°C, antes de la inyección de aire, que corresponde a aprox. 62.86%.
(línea roja en la Figura) y tomando en cuenta que en ese residuo muy probablemente se incluyan los
silicatos.
Figura 61.- Termograma de CdS(MPS).
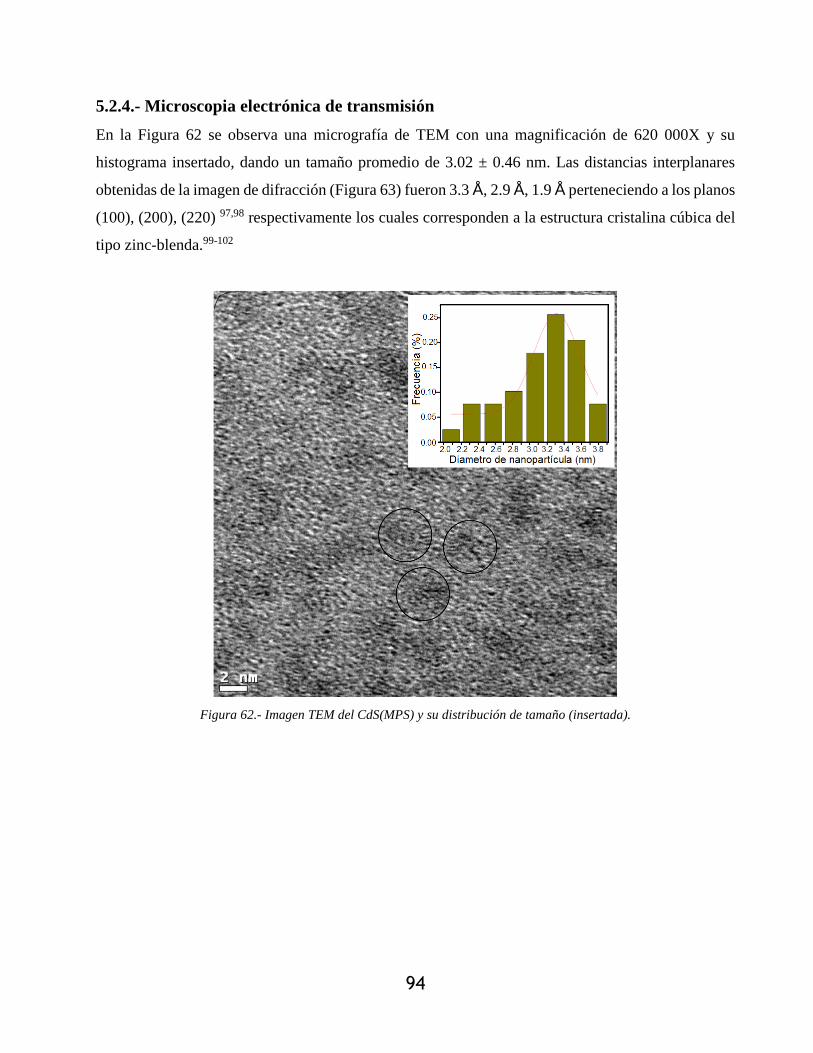
94
5.2.4.- Microscopia electrónica de transmisión
En la Figura 62 se observa una micrografía de TEM con una magnificación de 620 000X y su
histograma insertado, dando un tamaño promedio de 3.02 ± 0.46 nm. Las distancias interplanares
obtenidas de la imagen de difracción (Figura 63) fueron 3.3 Å, 2.9 Å, 1.9 Å perteneciendo a los planos
(100), (200), (220) 97,98 respectivamente los cuales corresponden a la estructura cristalina cúbica del
tipo zinc-blenda.99-102
Figura 62.- Imagen TEM del CdS(MPS) y su distribución de tamaño (insertada).

95
Figura 63.- Imagen de difracción de electrones del CdS(MPS) anillo 1, 2, 3 para los planos (100), (200), (220)
respectivamente.
5.4.- Propiedades ópticas
El espectro de absorción de UV-Vis para el CdS se puede apreciar en la Figura 64 (línea negra), la
banda de absorción es identificable como un hombro alrededor de 360 nm (gráfica insertada).
Utilizando el inicio o “onset” de absorción espectral (λe=416.05 nm), se obtiene un valor de 2.75 nm
para el diámetro de las nanopartículas de CdS por medio de la ecuación 2RCdS=0.1/(0.1338-
0.000234λe)nm103, valor cercano al obtenido por TEM. La banda prohibida óptica se calculó con la
ecuación de Planck Eg=hc/λe dando 2.99 eV. En el espectro UV se puede apreciar también un pico a
267 nm, misma longitud de onda del MPS. Ya que la resonancia SS-NMR no evidenció presencia de
MPS libre, este pico puede asociarse con transiciones electrónicas de las estructuras con grupos
metoxisilano no hidrolizados. La emisión (Figura 64, línea azul) presenta una banda amplia de 400-
600 nm centrado en 506.5 nm. El ancho de ésta banda podría deberse a la amplia distribución de
tamaños74 como se puede corroborar por la desviación estándar en el tamaño obtenida por TEM, que
es de 0.46 nm sobre 3.02 nm es decir un 15% del valor promedio. Sin embargo, Sander F. Wuister y
Andries Meijerink reportaron espectros de emisión muy anchos y longitud de onda máxima similar
para puntos cuánticos CdS(MPS) de tamaño similar. Los autores atribuyen el ancho de banda amplio
a emisión debida a electrones y/o huecos atrapados (“trapped emission”).74 El rendimiento quántico
de CdS(MPS) fue determinado en CIQA y en Universidad de Akron donde se realizaron estudios
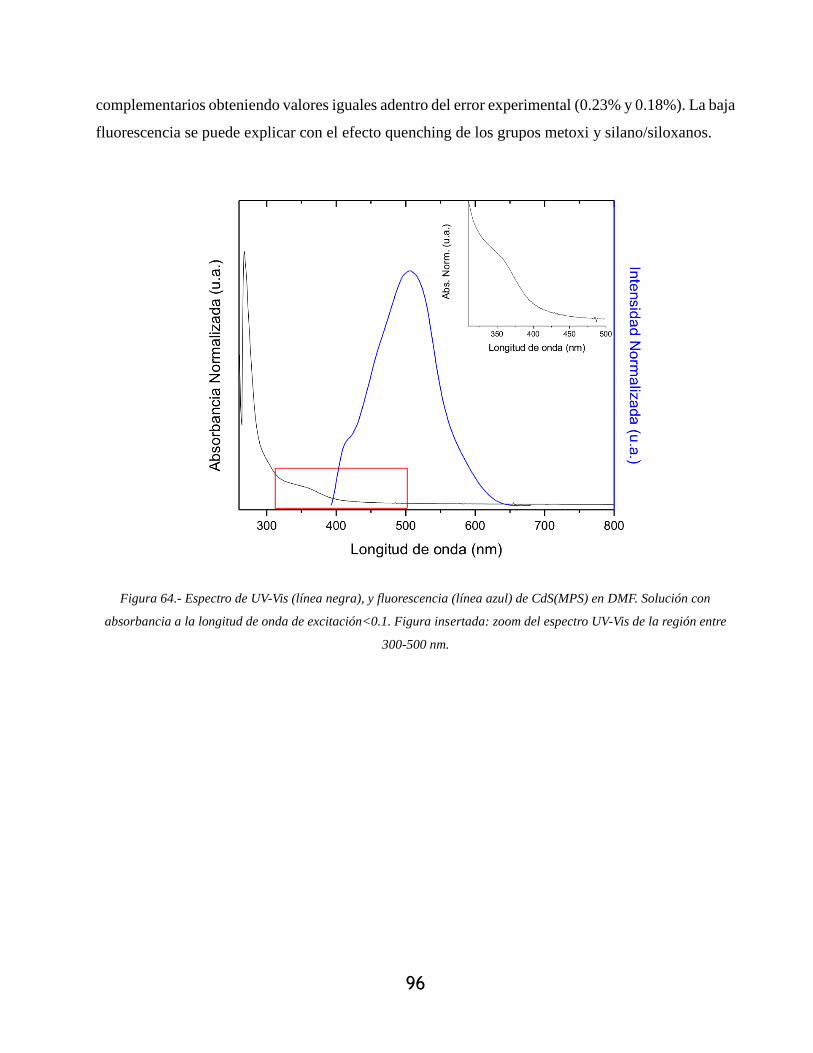
96
complementarios obteniendo valores iguales adentro del error experimental (0.23% y 0.18%). La baja
fluorescencia se puede explicar con el efecto quenching de los grupos metoxi y silano/siloxanos.
Figura 64.- Espectro de UV-Vis (línea negra), y fluorescencia (línea azul) de CdS(MPS) en DMF. Solución con
absorbancia a la longitud de onda de excitación<0.1. Figura insertada: zoom del espectro UV-Vis de la región entre
300-500 nm.

97
Capitulo 6: Resultados y discusiones-síntesis y caracterización de
nanohíbrido de puntos cuánticos con el co-polímero
6.1.- Síntesis
El nanohíbrido se obtuvo mediante una reacción de condensación entre los grupos metoxy del ligante
MPS y los hidroxilos del co-polímero. La reacción se realizó a diferente pH . Debido a la
identificación de superestructuras, también se puso a reaccionar bajo las mismas condiciones, los
puntos cuánticos con el homopolímero benzoato así como con el homopolímero dodecanoxy.
6.2.- Caracterización
6.2.1.- Resonancia magnética nuclear de estado sólido (SS-NMR)iv
En la Figura 65 se reporta el espectro 13C SS-NMR del composite obtenido en medio ácido
CdS(pPE)(H+) y en medio básico (CdS(pPE)(OH-) en DMF. En la misma figura se incluye para fines
comparativos el espectro de las nanopartículas CdS(MPS) y el del co-polímero pPE(OC12-Co-
BzC11OH) cuyas señales se discutieron en los capítulos anteriores. El espectro de 13C RMN del
CdS(pPE)(H+) presenta los mismos desplazamientos observados para el co-polímero en (ppm) 164
ppm (carbono del éster), en 154 ppm (carbono del fenoxi), en 114 ppm a 133 ppm (otros carbonos
aromáticos) y los dos carbonos del grupo acetileno se observan a 89 ppm y 95 ppm. Se pueden
apreciar tres tipos de carbono CH2, unido a los grupos O-Ar, COO- y -OH, a 70 ppm, 67 ppm y 63
ppm, respectivamente. Los desplazamientos en 33 ppm y 30 ppm son debidos a los grupos -CH2- de
las cadenas de alquilo, con el grupo terminal CH2 y CH3 a 23 ppm y 15 ppm, respectivamente.
Adicionalmente, sin embargo se puede apreciar un ensanchamiento de los picos a 15 ppm y 30 ppm
debido al sobrelapamiento con las señales del MPS. También se aprecia un pequeño hombro a 45
ppm debido a la presencia del carbono O-CH3 para el MPS.
iv Las muestras de composites preparadas en CIQA fueron enviadas a la Universidad de Akron para su análisis por SS-NMR, utilizando
como probe 13C y 29Si.
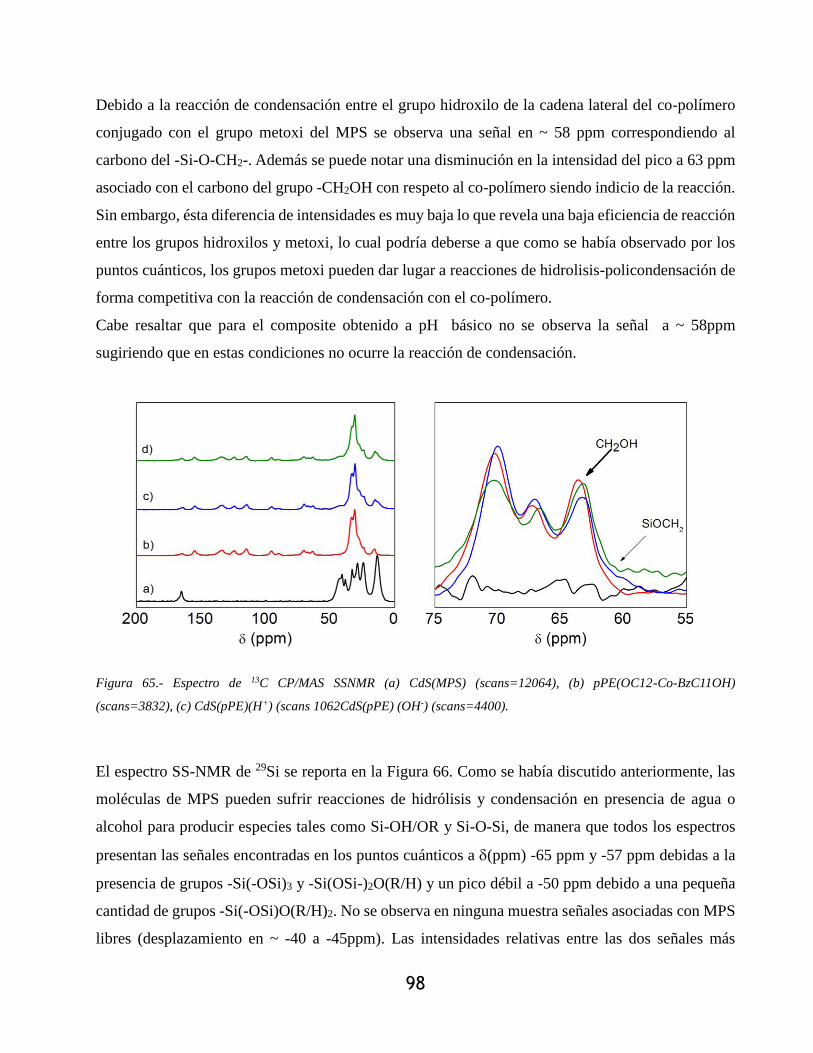
98
Debido a la reacción de condensación entre el grupo hidroxilo de la cadena lateral del co-polímero
conjugado con el grupo metoxi del MPS se observa una señal en ~ 58 ppm correspondiendo al
carbono del -Si-O-CH2-. Además se puede notar una disminución en la intensidad del pico a 63 ppm
asociado con el carbono del grupo -CH2OH con respeto al co-polímero siendo indicio de la reacción.
Sin embargo, ésta diferencia de intensidades es muy baja lo que revela una baja eficiencia de reacción
entre los grupos hidroxilos y metoxi, lo cual podría deberse a que como se había observado por los
puntos cuánticos, los grupos metoxi pueden dar lugar a reacciones de hidrolisis-policondensación de
forma competitiva con la reacción de condensación con el co-polímero.
Cabe resaltar que para el composite obtenido a pH básico no se observa la señal a ~ 58ppm
sugiriendo que en estas condiciones no ocurre la reacción de condensación.
Figura 65.- Espectro de 13C CP/MAS SSNMR (a) CdS(MPS) (scans=12064), (b) pPE(OC12-Co-BzC11OH)
(scans=3832), (c) CdS(pPE)(H+) (scans 1062CdS(pPE) (OH-) (scans=4400).
El espectro SS-NMR de 29Si se reporta en la Figura 66. Como se había discutido anteriormente, las
moléculas de MPS pueden sufrir reacciones de hidrólisis y condensación en presencia de agua o
alcohol para producir especies tales como Si-OH/OR y Si-O-Si, de manera que todos los espectros
presentan las señales encontradas en los puntos cuánticos a (ppm) -65 ppm y -57 ppm debidas a la
presencia de grupos -Si(-OSi)3 y -Si(OSi-)2O(R/H) y un pico débil a -50 ppm debido a una pequeña
cantidad de grupos -Si(-OSi)O(R/H)2. No se observa en ninguna muestra señales asociadas con MPS
libres (desplazamiento en ~ -40 a -45ppm). Las intensidades relativas entre las dos señales más
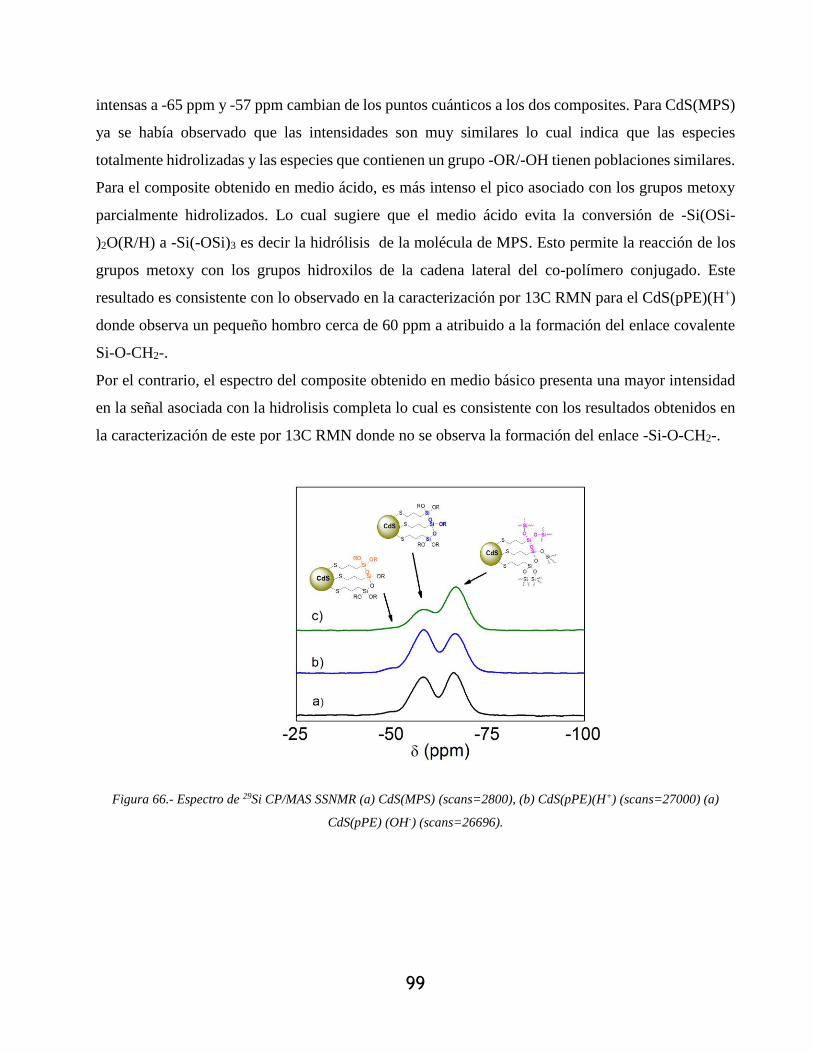
99
intensas a -65 ppm y -57 ppm cambian de los puntos cuánticos a los dos composites. Para CdS(MPS)
ya se había observado que las intensidades son muy similares lo cual indica que las especies
totalmente hidrolizadas y las especies que contienen un grupo -OR/-OH tienen poblaciones similares.
Para el composite obtenido en medio ácido, es más intenso el pico asociado con los grupos metoxy
parcialmente hidrolizados. Lo cual sugiere que el medio ácido evita la conversión de -Si(OSi-
)2O(R/H) a -Si(-OSi)3 es decir la hidrólisis de la molécula de MPS. Esto permite la reacción de los
grupos metoxy con los grupos hidroxilos de la cadena lateral del co-polímero conjugado. Este
resultado es consistente con lo observado en la caracterización por 13C RMN para el CdS(pPE)(H+)
donde observa un pequeño hombro cerca de 60 ppm a atribuido a la formación del enlace covalente
Si-O-CH2-.
Por el contrario, el espectro del composite obtenido en medio básico presenta una mayor intensidad
en la señal asociada con la hidrolisis completa lo cual es consistente con los resultados obtenidos en
la caracterización de este por 13C RMN donde no se observa la formación del enlace -Si-O-CH2-.
Figura 66.- Espectro de 29Si CP/MAS SSNMR (a) CdS(MPS) (scans=2800), (b) CdS(pPE)(H+) (scans=27000) (a)
CdS(pPE) (OH-) (scans=26696).

100
6.2.2.- Microscopia electrónica de transmisión y de barrido (STEM)
Una vez confirmada la reacción de condensación entre el polímero y los puntos cuánticos a pH acido
por medio de la espectroscopia SS-NMR, se realizó el estudio microscópico del composite con la
finalidad de observar la distribución de las nanopartículas en el mismo. De forma inesperada, se
observó (Figura 67) la formación de estructuras circulares con tamaño promedio de 242.21nm ±
79nm..
Figura 67.- Imagen STEM CdS(pPE)(H+)
Para investigar más a fondo el mecanismo de formación de estas estructuras, se analizó también la
muestra obtenida del co-polímero y CdS(MPS) en medio básico (CdS(pPE)(OH-) así como una
obtenida a pH neutro (Figura 68). Todas las muestras se prepararon en las mismas condiciones de
concentración y mezcla de solvente así como se reportó en el capítulo de la parte experimental.
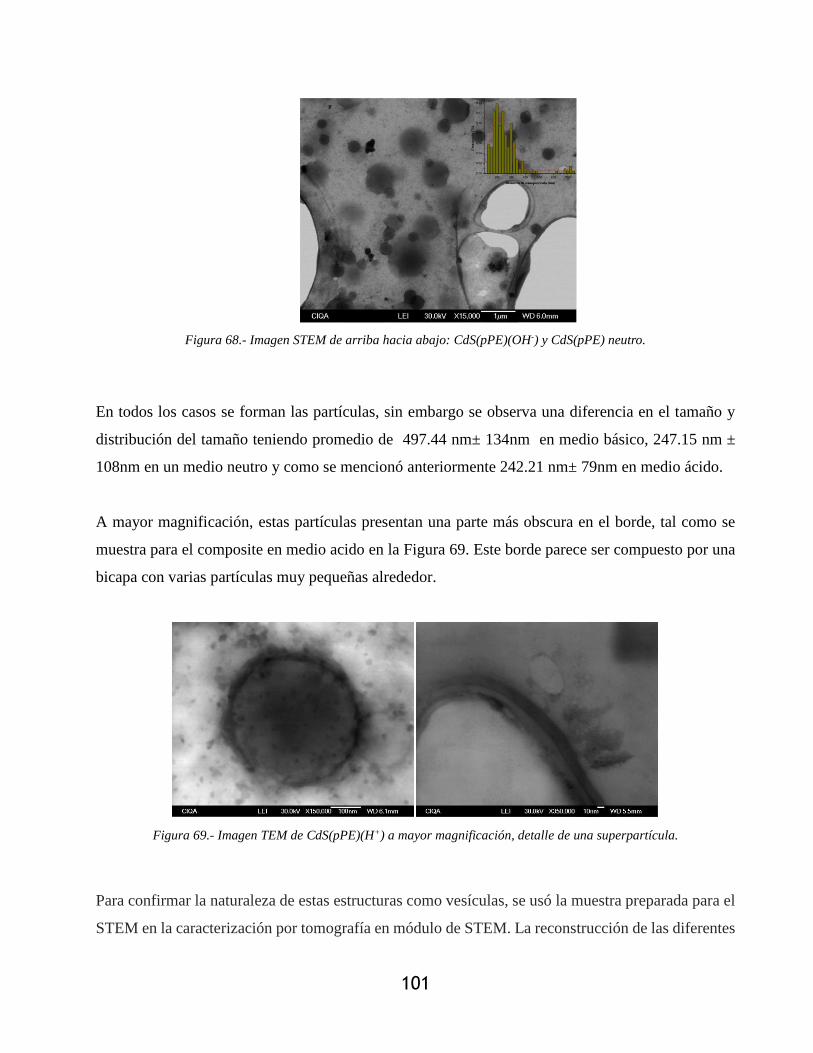
101
Figura 68.- Imagen STEM de arriba hacia abajo: CdS(pPE)(OH-) y CdS(pPE) neutro.
En todos los casos se forman las partículas, sin embargo se observa una diferencia en el tamaño y
distribución del tamaño teniendo promedio de 497.44 nm± 134nm en medio básico, 247.15 nm ±
108nm en un medio neutro y como se mencionó anteriormente 242.21 nm± 79nm en medio ácido.
A mayor magnificación, estas partículas presentan una parte más obscura en el borde, tal como se
muestra para el composite en medio acido en la Figura 69. Este borde parece ser compuesto por una
bicapa con varias partículas muy pequeñas alrededor.
Figura 69.- Imagen TEM de CdS(pPE)(H+) a mayor magnificación, detalle de una superpartícula.
Para confirmar la naturaleza de estas estructuras como vesículas, se usó la muestra preparada para el
STEM en la caracterización por tomografía en módulo de STEM. La reconstrucción de las diferentes

102
secciones se presenta en el video 1 anexo que muestra la adquisición secuencial de las imágenes del
análisis tomografico y en video 2 la reconstrucción de la imagen tridimensional a partir de estas. Se
puede observar claramente que las superestructuras corresponden a vesículas tridimensionales
“huecas” y transparentes como se podría esperar por un ensamblaje de tipo vesicular bicapa.
Es notorio que moléculas amfifilicas pueden ensamblarse en diferentes tipos de estructura como son
micelas globulares, elipsoides, discos, cilindros, vesículas y lamelas, dependiendo de la arquitectura
molecular, concentración y solvente. Particularmente la formación de estructuras tipo vesícula con
diámetro de alrededor de 500 nm, a partir del autoensamblaje de un macrociclo amfifilico tipo
fenilenoetinileno fue demostrada por Seo104. Cabe mencionar que estas superpartículas no se
presentan en el CdS(MPS) ni en el co-polímero ni en una muestra preparada mezclando los puntos
cuánticos y el polímero 2,5-didodecanoxyfenilenoetinileno (Ver Figura 70), lo cual sugieren que se
forman por la presencia del lígante MPS de los puntos cuánticos y los grupos OH del co-polímero,
bajo estas condiciones de preparación. A pesar de haber confirmado por SS-NMR que la interacción
entre el co-polímero y las partículas de CdS(MPS) es debida al enlace covalente, es imposible al
momento saber exactamente donde se encuentran los puntos cuánticos de CdS(MPS) en el
nanohíbrido polimérico particularmente debido a la complejidad de las estructuras asociadas con el
MPS y su hidrólisis. Finalmente es importante mencionar que las vesículas son estables en las
condiciones de alto vacío del análisis.
Debido a que de acuerdo a los estudios de SS-NMR únicamente a pH acido se forma el enlace
covalente entre el co-polímero y puntos cuánticos, en la continuación del trabajo de investigación se
consideró solamente este nanohíbrido utilizando la nomenclatura de CdS(pPE).
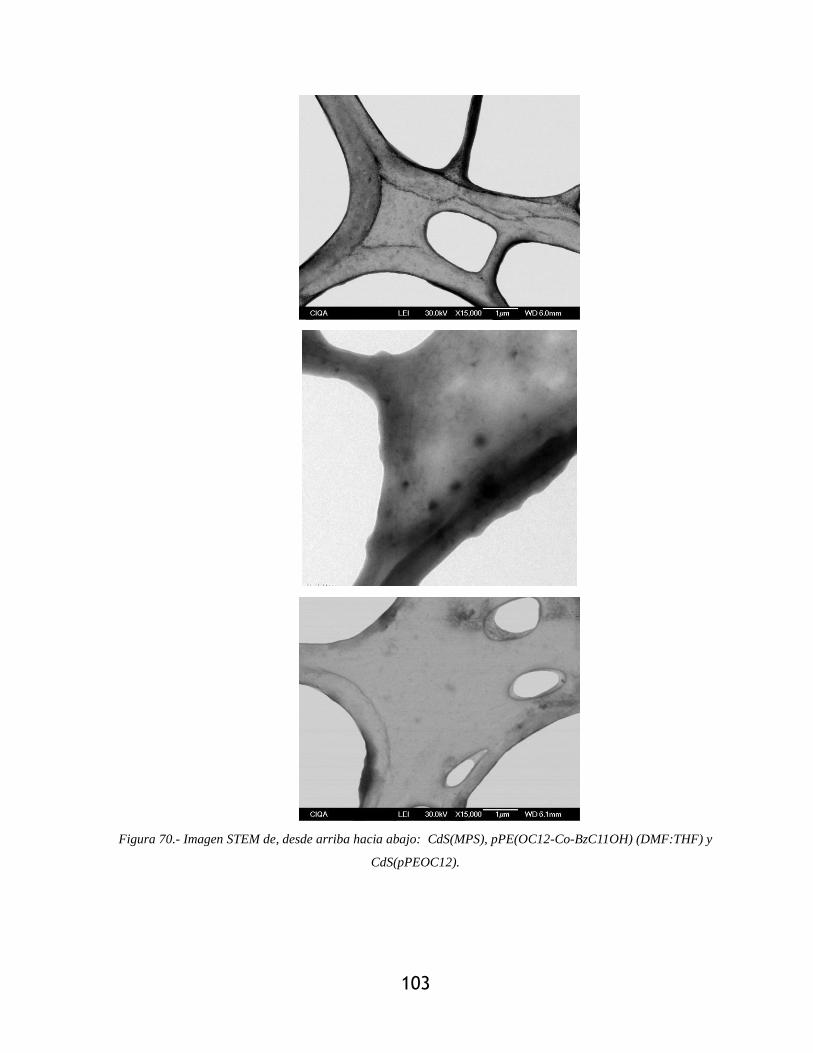
103
Figura 70.- Imagen STEM de, desde arriba hacia abajo: CdS(MPS), pPE(OC12-Co-BzC11OH) (DMF:THF) y
CdS(pPEOC12).
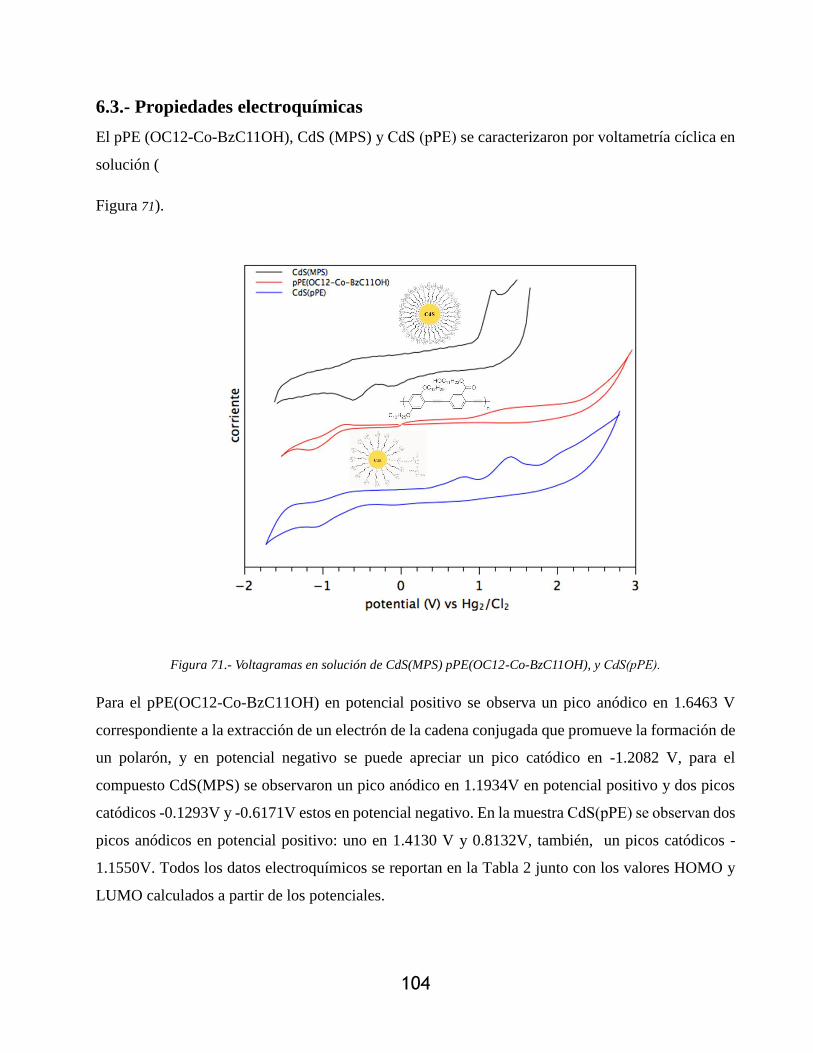
104
6.3.- Propiedades electroquímicas
El pPE (OC12-Co-BzC11OH), CdS (MPS) y CdS (pPE) se caracterizaron por voltametría cíclica en
solución (
Figura 71).
Figura 71.- Voltagramas en solución de CdS(MPS) pPE(OC12-Co-BzC11OH), y CdS(pPE).
Para el pPE(OC12-Co-BzC11OH) en potencial positivo se observa un pico anódico en 1.6463 V
correspondiente a la extracción de un electrón de la cadena conjugada que promueve la formación de
un polarón, y en potencial negativo se puede apreciar un pico catódico en -1.2082 V, para el
compuesto CdS(MPS) se observaron un pico anódico en 1.1934V en potencial positivo y dos picos
catódicos -0.1293V y -0.6171V estos en potencial negativo. En la muestra CdS(pPE) se observan dos
picos anódicos en potencial positivo: uno en 1.4130 V y 0.8132V, también, un picos catódicos -
1.1550V. Todos los datos electroquímicos se reportan en la Tabla 2 junto con los valores HOMO y
LUMO calculados a partir de los potenciales.
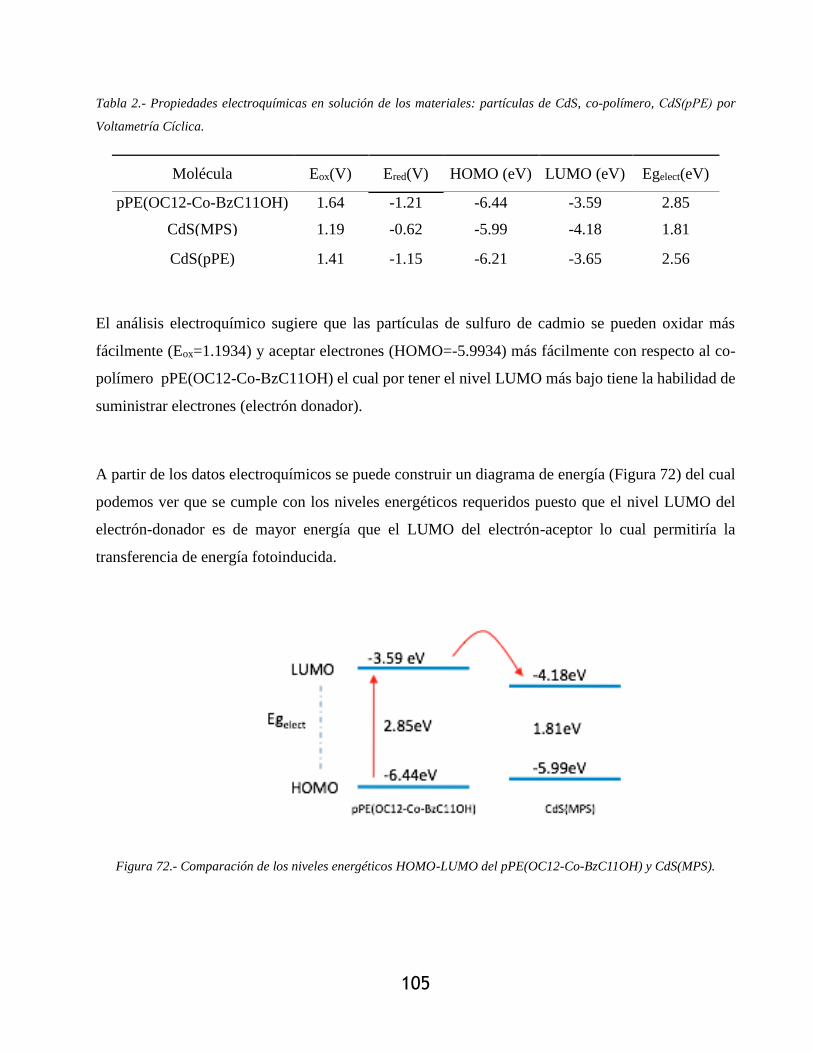
105
Tabla 2.- Propiedades electroquímicas en solución de los materiales: partículas de CdS, co-polímero, CdS(pPE) por
Voltametría Cíclica.
Molécula Eox(V) Ered(V) HOMO (eV) LUMO (eV) Egelect(eV)
pPE(OC12-Co-BzC11OH) 1.64 -1.21 -6.44 -3.59 2.85
CdS(MPS) 1.19 -0.62 -5.99 -4.18 1.81
-0.1293 CdS(pPE) 1.41 -1.15 -6.21 -3.65 2.56
El análisis electroquímico sugiere que las partículas de sulfuro de cadmio se pueden oxidar más
fácilmente (Eox=1.1934) y aceptar electrones (HOMO=-5.9934) más fácilmente con respecto al co-
polímero pPE(OC12-Co-BzC11OH) el cual por tener el nivel LUMO más bajo tiene la habilidad de
suministrar electrones (electrón donador).
A partir de los datos electroquímicos se puede construir un diagrama de energía (Figura 72) del cual
podemos ver que se cumple con los niveles energéticos requeridos puesto que el nivel LUMO del
electrón-donador es de mayor energía que el LUMO del electrón-aceptor lo cual permitiría la
transferencia de energía fotoinducida.
Figura 72.- Comparación de los niveles energéticos HOMO-LUMO del pPE(OC12-Co-BzC11OH) y CdS(MPS).
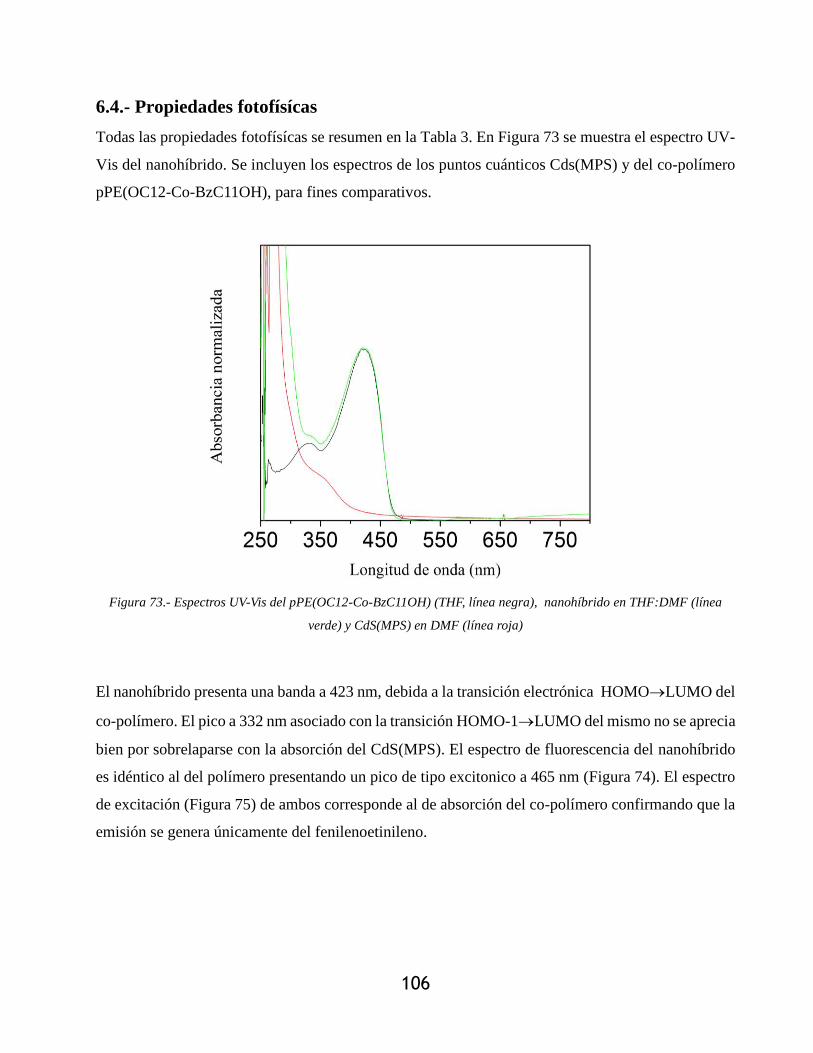
106
6.4.- Propiedades fotofísícas
Todas las propiedades fotofísícas se resumen en la Tabla 3. En Figura 73 se muestra el espectro UV-
Vis del nanohíbrido. Se incluyen los espectros de los puntos cuánticos Cds(MPS) y del co-polímero
pPE(OC12-Co-BzC11OH), para fines comparativos.
Figura 73.- Espectros UV-Vis del pPE(OC12-Co-BzC11OH) (THF, línea negra), nanohíbrido en THF:DMF (línea
verde) y CdS(MPS) en DMF (línea roja)
El nanohíbrido presenta una banda a 423 nm, debida a la transición electrónica HOMOLUMO del
co-polímero. El pico a 332 nm asociado con la transición HOMO-1LUMO del mismo no se aprecia
bien por sobrelaparse con la absorción del CdS(MPS). El espectro de fluorescencia del nanohíbrido
es idéntico al del polímero presentando un pico de tipo excitonico a 465 nm (Figura 74). El espectro
de excitación (Figura 75) de ambos corresponde al de absorción del co-polímero confirmando que la
emisión se genera únicamente del fenilenoetinileno.
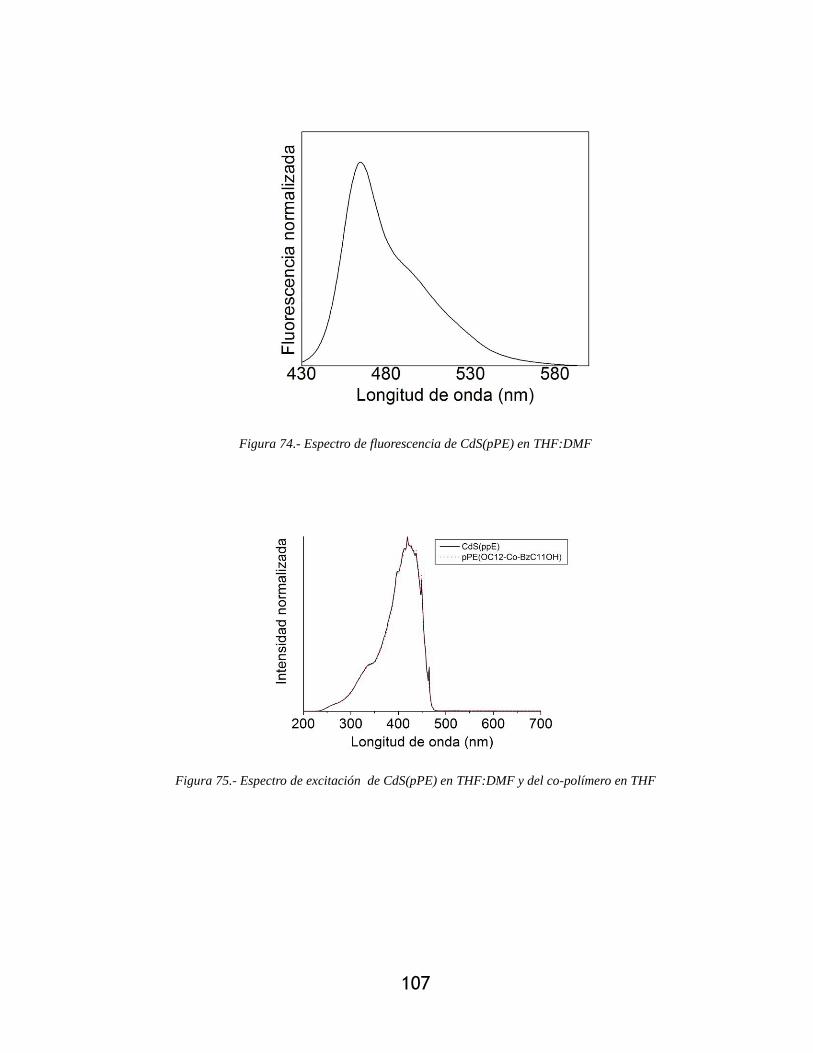
107
Figura 74.- Espectro de fluorescencia de CdS(pPE) en THF:DMF
Figura 75.- Espectro de excitación de CdS(pPE) en THF:DMF y del co-polímero en THF
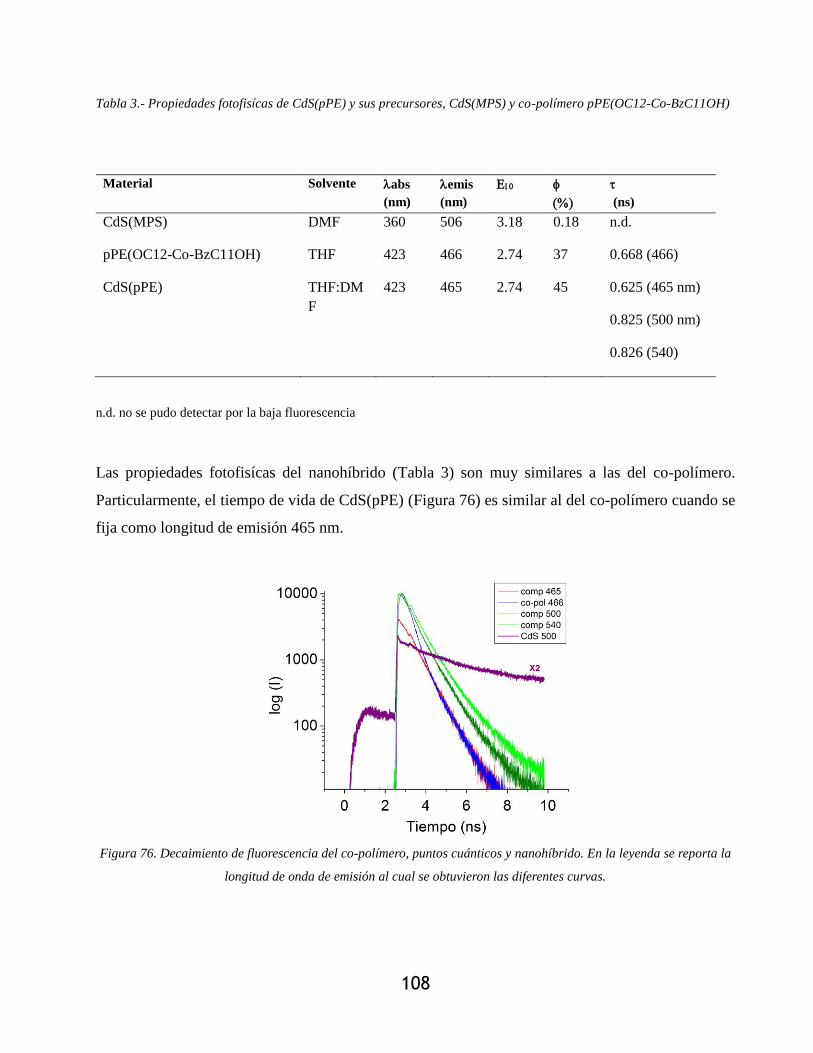
108
Tabla 3.- Propiedades fotofisícas de CdS(pPE) y sus precursores, CdS(MPS) y co-polímero pPE(OC12-Co-BzC11OH)
n.d. no se pudo detectar por la baja fluorescencia
Las propiedades fotofisícas del nanohíbrido (Tabla 3) son muy similares a las del co-polímero.
Particularmente, el tiempo de vida de CdS(pPE) (Figura 76) es similar al del co-polímero cuando se
fija como longitud de emisión 465 nm.
Figura 76. Decaimiento de fluorescencia del co-polímero, puntos cuánticos y nanohíbrido. En la leyenda se reporta la
longitud de onda de emisión al cual se obtuvieron las diferentes curvas.
Material Solvente abs
(nm)
emis
(nm)
(ns)
CdS(MPS) DMF 360 506 3.18 0.18 n.d.
pPE(OC12-Co-BzC11OH) THF 423 466 2.74 37 0.668 (466)
CdS(pPE) THF:DM
F
423 465 2.74 45 0.625 (465 nm)
0.825 (500 nm)
0.826 (540)

109
Si se mide a la longitud de emisión de los puntos cuánticosv se obtiene un tiempo de vida más largo
lo cual podría indicar un proceso de transferencia de energía del polímero a los CdS y sucesiva
emisión de estos. A pesar de que las propiedades electroquímicas son consistentes con la posibilidad
de transferencia de energía del polímero (electrón donador) a los puntos cuánticos (electrón aceptor),
las propiedades fotofisícas no confirman este proceso ya que el rendimiento cuántico del co-polímero
no disminuye y en general no se observa un efecto de la incorporación de CdS(MPS) en las
propiedades del co-polímero en el nanohíbrido. Lamentablemente no se puede realizar el cálculo de
la energía de Gibbs G necesaria para la transferencia de carga ya que las propiedades
electroquímicas se obtuvieron en diferentes solventes. Sin embargo si consideramos los primeros
términos de la ecuación, que no dependen de la constante dieléctrica del medio de voltametría (ref),
es decir e[Eox(D)-Ered(A)]-E00, e introduciendo los valores correspondientes (Eox(D)=1.64 eV,
Ered(A)=-0.62 eV y E10=2.74 eVvi, el valor de G sería -0.48 eV es decir debería ser posible la
transferencia. Es evidente que la distancia entre co-polímero y puntos cuánticos RCC es posiblemente
grande ya sea por la extensión de la cadenas alifáticas del co-polímero y el MPS de los puntos
cuánticos ya sea por la formación de las superestructuras provocando que el proceso de transferencia
fotoinducida ocurra probablemente en la región invertida de Marcus es decir sí hay separación de
cargas pero estas se recombinan antes de poderse transportar a los electrodos.
v debido al ancho amplio de fluorescencia de CdS(MPS) se hizo mediciones a 500 y 540 nm
vi Obtenido de cruce entre el espectro normalizado de absorción y el de fluorescencia
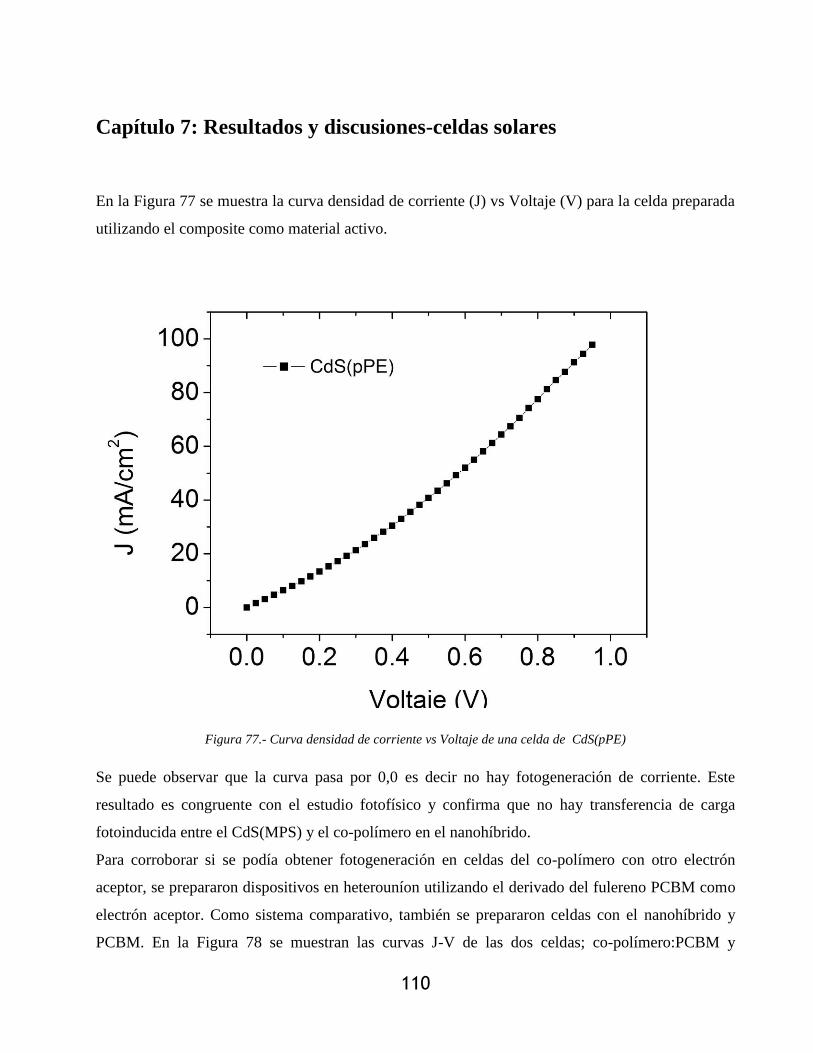
110
Capítulo 7: Resultados y discusiones-celdas solares
En la Figura 77 se muestra la curva densidad de corriente (J) vs Voltaje (V) para la celda preparada
utilizando el composite como material activo.
Figura 77.- Curva densidad de corriente vs Voltaje de una celda de CdS(pPE)
Se puede observar que la curva pasa por 0,0 es decir no hay fotogeneración de corriente. Este
resultado es congruente con el estudio fotofísico y confirma que no hay transferencia de carga
fotoinducida entre el CdS(MPS) y el co-polímero en el nanohíbrido.
Para corroborar si se podía obtener fotogeneración en celdas del co-polímero con otro electrón
aceptor, se prepararon dispositivos en heterouníon utilizando el derivado del fulereno PCBM como
electrón aceptor. Como sistema comparativo, también se prepararon celdas con el nanohíbrido y
PCBM. En la Figura 78 se muestran las curvas J-V de las dos celdas; co-polímero:PCBM y
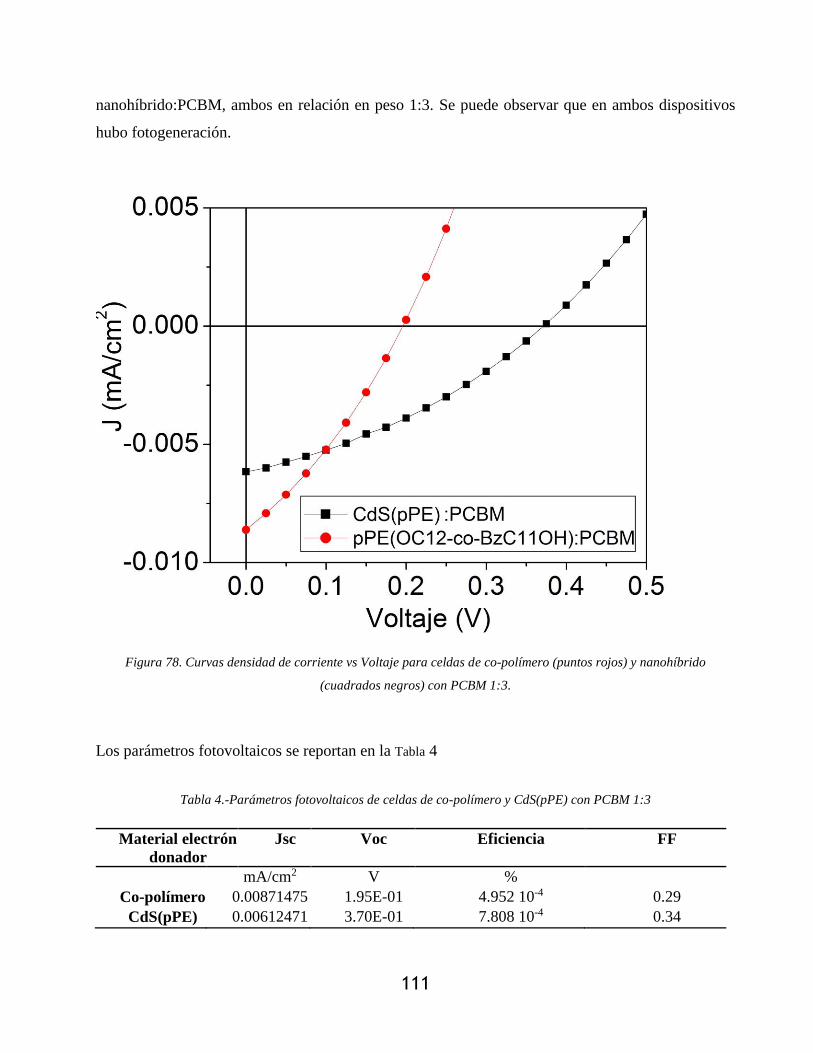
111
nanohíbrido:PCBM, ambos en relación en peso 1:3. Se puede observar que en ambos dispositivos
hubo fotogeneración.
Figura 78. Curvas densidad de corriente vs Voltaje para celdas de co-polímero (puntos rojos) y nanohíbrido
(cuadrados negros) con PCBM 1:3.
Los parámetros fotovoltaicos se reportan en la Tabla 4
Tabla 4.-Parámetros fotovoltaicos de celdas de co-polímero y CdS(pPE) con PCBM 1:3
Material electrón
donador
Jsc Voc Eficiencia FF
mA/cm2 V %
Co-polímero 0.00871475 1.95E-01 4.952 10-4 0.29
CdS(pPE) 0.00612471 3.70E-01 7.808 10-4 0.34
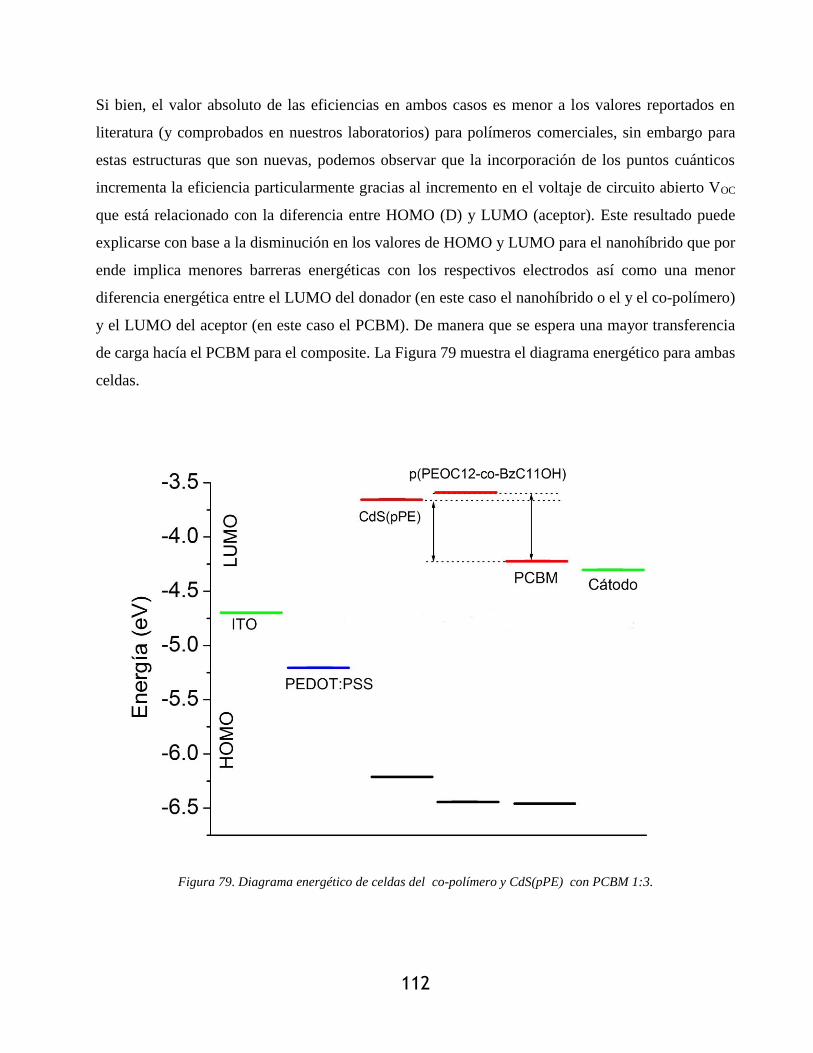
112
Si bien, el valor absoluto de las eficiencias en ambos casos es menor a los valores reportados en
literatura (y comprobados en nuestros laboratorios) para polímeros comerciales, sin embargo para
estas estructuras que son nuevas, podemos observar que la incorporación de los puntos cuánticos
incrementa la eficiencia particularmente gracias al incremento en el voltaje de circuito abierto VOC
que está relacionado con la diferencia entre HOMO (D) y LUMO (aceptor). Este resultado puede
explicarse con base a la disminución en los valores de HOMO y LUMO para el nanohíbrido que por
ende implica menores barreras energéticas con los respectivos electrodos así como una menor
diferencia energética entre el LUMO del donador (en este caso el nanohíbrido o el y el co-polímero)
y el LUMO del aceptor (en este caso el PCBM). De manera que se espera una mayor transferencia
de carga hacía el PCBM para el composite. La Figura 79 muestra el diagrama energético para ambas
celdas.
Figura 79. Diagrama energético de celdas del co-polímero y CdS(pPE) con PCBM 1:3.

113
Sin embargo se requiere profundizar en este aspecto, en cuanto a morfología de la capa activa. Este
estudio está afuera del alcance de esta tesis y se plantea como trabajo a futuro.
7.1.- Consideraciones finales
Como se ha mencionado en los antecedentes, la eficiencia de una celda solar orgánica depende de
varios factores. Unos son intrínsecos de las moléculas que se utilizan en la capa activa, entre los
cuales recordamos que los materiales activos deben presentar un alto coeficiente de extinción molar
cerca del máximo de emisión solar, buen rendimiento cuántico (el electrón donador) que se refleja en
formación de excitones, buena procesabilidad, alta movilidad de carga. Otros factores son extrínsecos
es decir dependen de los procesos de preparación del dispositivo y pueden variar según la
configuración de las celdas, electrodos, tratamientos post depositación (como el recocido),
encapsulado etc. El factor más importante sin embargo es la posibilidad de tener transferencia de
carga fotoinducida entre el material electrón donador y el electrón aceptor. Bajo este contexto, el co-
polímero presenta buena solubilidad y un valor de rendimiento cuántico bastante bueno pero absorbe
en una región que no corresponde al máximo de emisión solar. Es así que las celdas del co-polímero
con el PCBM fotogeneran pero su eficiencia es mucho menor a la reportada para celdas de
heterounión con este electrón aceptor para otros tipos de polímero conjugado. Por ejemplo para el
poli(3-hexyltiofeno) hemos reproducido en nuestros laboratorios eficiencias de hasta 2% aun
trabajando en condiciones de atmosfera no controlada, y sin encapsulado. Cabe mencionar que en la
literatura se reportan celdas de fenilenoetinilenos funcionalizados con fulereno (celdas tipo cable
molecular) o con nanotubos de carbono y las eficiencias varían entre 10-4 y 10-5%. Previamente
nosotros encontramos eficiencias de 10-5%-10-6% en celdas ya sea de cable molecular que de
heterounión105-108. El presente resultado entonces revela que se ha mejorado las eficiencias para este
tipo de material incluso con respecto a la literatura pero nos obliga evidentemente a direccionar la
parte de síntesis hacía estructuras más conjugadas, del tipo tiofeno o ditiofeno. La incorporación de
los puntos cuánticos se esperaba pudiese generar celdas de tipo cable molecular al tener el grupo
electrón donador y electrón aceptor en un solo material. Y en donde la eficiencia se pudiese
incrementar por la sinergia en las propiedades de absorción de los dos materiales. Efectivamente el
espectro UV del composite muestra sobrelapamiento de la absorción de los dos materiales aunque la
mejora se obtiene en la región UV. En este aspecto, puntos cuánticos de PbS o ZnS sería mejores

114
candidatos y estaban considerados al principio de este proyecto de investigación pero no se logró
realizar la síntesis. Los valores de HOMO y LUMO obtenidos por voltametría sugerían la posibilidad
de transferencia de carga al ser el LUMO del electrón donador menor al del electrón aceptor. Sin
embargo, los estudios fotofísicos sugieren lo contrario. El nanohíbrido presenta las mismas
propiedades de emisión que el co-polímero, es decir no se observa quenching de la fluorescencia.
Este resultado no necesariamente está en contradicción con los resultados de voltametría ya que por
sí no indica que no pueda haber transferencia de carga sino que esta no es favorecida. Se ha
mencionado que no pudimos calcular exactamente la energía libre de Gibbs ya que la complejidad
del sistema CdS(PE) no permite hacer un modelado y una estimación de la distancia entre electrón
donador y electrón aceptor. Sin embargo recordando la formula
2 2
10
0 0
1 1 1 1( )
4 8
D A
CS ox red
s cc r s
e eG e E E E
R r r
y tomando en cuenta el primer término, los valores de Eox/Ered de la voltametría y E10 de fluorescencia
se obtiene G=-0.48 eV. Por lo que teóricamente la transferencia es posible. Sin embargo la falta de
fotogeneración sugiere que una vez separadas las cargas, estas se recombinan generando finalmente
la misma fluorescencia que el co-polímero. Es decir el excitón se separa pero las cargas se recombinan
por la distancia entre el electrón donador y el electrón aceptor, debida a nivel molecular por la larga
distancia determinada por la extensión de la cadena alifática del co-polímero así como por la cadena
del MPS. Esta distancia es posiblemente incrementada a nivel supramolecular por la formación de
la superestructura aunque no se alcanzó realizar el estudio morfológico de la capa activa para
corroborar si esta superestructura persiste cuando se prepara la película para la celda, es decir a partir
de solución más concentrada que la utilizada para el TEM. Finalmente, en configuración de
heterounión, el composite presentó mejores eficiencias con respecto al co-polímero posiblemente
porqué la disminución de los valores de HOMO y LUMO (ver resultados de voltametría cíclica)
implican menores barrieras energéticas con los electrodos así como una menor diferencia energética
con el LUMO del PCBM. Es importante mencionar que no hay reportes de híbridos de CdS con
polímeros del tipo fenilenoetinileno ni su aplicación en celdas, por lo que a pesar de los resultados
negativos obtenidos en las celdas de cable molecular del nanohíbrido, la síntesis de este nanohíbrido,

115
la formación de la superestructura y finalmente la aplicación del nanohíbrido en las celdas de
heterounión con PCBM representan los puntos originales de esta tesis.

116
8. - Conclusiones
En este trabajo doctoral, se obtuvieron puntos cuánticos de 3.02 nm de CdS con estructura
cristalográfica cúbica de tipo Zinc blenda utilizando como ligante el (3-mercaptopropil)
trimetoxisilano (MPS). De acuerdo al estudio por Resonancia Magnética Nuclear de Estado Sólido
(SS-NMR), la muestra CdS(MPS) es en realidad un sistema no estequiométrico complejo por la
formación de estructuras originadas por reacciones de hidrólisis y condensación de los grupos metoxi.
Como consecuencia no se puede calcular el número de moléculas de MPS por nanopartìcula ni por
XPS ni por TGA. Sin embargo, la SS-NMR excluye la presencia de MPS libre y confirma la
estructuras con algún grupo metoxi (es decir no totalmente hidrolizadas) lo cual permite la
funcionalización de los puntos cuánticos de CdS(MPS) con un co-polímero poli(parafenilenetinileno)
a través de la condensación entre grupos -OH terminales de las cadenas laterales del pPE y los grupos
metoxi del ligante 3-(mercaptopropil)-trimetoxisilano de los puntos cuánticos. Esta reacción solo
ocurre en medio acido. La funcionalización se corrobora por la caracterización de SS-RMN al
observar la formación de un nuevo pico en -58ppm correspondiente al enlace Si-O-CH2- y la
disminución del pico del carbono del grupo -OH en 63 ppm con respecto al co-polímero. Por STEM
se observan partículas grandes para las muestras obtenidas mezclando los puntos cuánticos y el co-
polímero independientemente del pH, sin embargo cambia el tamaño promedio y la distribución de
las mismas adentro de la matriz polimérica. Estudios por tomografía STEM revelan que para el
nanohíbrido obtenido en medio ácido, estas partículas son vacías es decir son de tipo vesícula. El
estudio por voltametría cíclica en solución indica que el electrón-donador pPE(OC12-Co-BzC11OH)
y el electrón-aceptor CdS(MPS) cumplen con los requerimientos energéticos para poder funcionar en
un dispositivo fotovoltaico. Sin embargo, las propiedades fotofisicas no confirman la posibilidad de
transferencia electrónica fotoinducida ya que no hay quenching de la fluorescencia, lo que sería
indicativo de este proceso. Por lo tanto, su aplicación en celdas solares no se puede visualizar como
capa activa, sin embargo la incorporación de los puntos cuánticos permite disminuir la diferencia
energética entre LUMO del donador y LUMO del aceptor en celdas de heterounión con PCBM y por
ende da un incremento en el voltaje de circuito abierto y eficiencia en celdas obtenidas con el
nanohíbrido en esta configuración.

117
Trabajo a futuro
Esta tesis es la primera del grupo en investigar la posibilidad de utilizar nanohíbridos de puntos
cuánticos directamente funcionalizados con polímeros conjugados, por lo que se abren varías líneas
futuras a desarrollar;
Investigar más a detalle el mecanismo de formación de las vesículas, cambiando por ejemplo
parámetros de síntesis del nanohíbrido como son relación en peso co-polímero:puntos
cuánticos, temperatura, solvente
Sintetizar puntos cuántico de CdS en presencia de otros ligantes que permitan la
funcionalización covalente con el co-polímero pero sin dar reacciones secundarias como la
hidrólisis presentada por el MPS
Sintetizar puntos cuánticos de PbS o ZnS para ampliar la ventana de absorción del
nanohíbrido
Sintetizar un co-polímero alternando unidad fenilenoetinileno con una unidad monomerica
del tipo tiofeno para tener absorción más desplazada hacía al rojo y por ende mejorar la
eficiencia de las celdas
Probar el composite en celdas solares de heterounión de PCBM con poli(hexyltiofeno), en
donde el composite pueda funcionar como capa buffer para mejorar la inyección de huecos.

118
Referencias
(1) Arias-Marin, E.; Le Moigne, J.; Maillou, T.; Guillon, D.; Moggio, I.; Geffroy, B. Macromolecules 2003, 36,
3570.
(2) Skaff, H.; Ilker, M. F.; Coughlin, E. B.; Emrick, T. Journal of the American Chemical Society 2002, 124, 5729.
(3) Skaff, H.; Sill, K.; Emrick, T. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 2004, 45, 615.
(4) Heeger, A. J. Reviews of Modern Physics 2001, 73, 681.
(5) Bunz, U. H. F. Chemical Reviews 2000, 100, 1605.
(6) Guo, Y.; Li, Y.; Xu, J.; Liu, X.; Xu, J.; Lv, J.; Huang, C.; Zhu, M.; Cui, S.; Jiang, L.; Liu, H.; Wang, S. The
Journal of Physical Chemistry C 2008, 112, 8223.
(7) Jie, J. S.; Zhang, W. J.; Jiang, Y.; Meng, X. M.; Li, Y. Q.; Lee, S. T. Nano Letters 2006, 6, 1887.
(8) Nasr, C.; Hotchandani, S.; Kim, W. Y.; Schmehl, R. H.; Kamat, P. V. The Journal of Physical Chemistry B
1997, 101, 7480.
(9) Yong, K.-T.; Sahoo, Y.; Swihart, M. T.; Prasad, P. N. The Journal of Physical Chemistry C 2007, 111, 2447.
(10) Watt, A.; Thomsen, E.; Meredith, P.; Rubinsztein-Dunlop, H. Chemical Communications 2004, 2334.
(11) Yin, Z.; Tang, X. Solid-State Electronics 2007, 51, 6.
(12) Di Luccio, T.; Laera, A. M.; Tapfer, L.; Kempter, S.; Kraus, R.; Nickel, B. The Journal of Physical Chemistry B
2006, 110, 12603.
(13) Warner, J. H.; Watt, A. A. R. Materials Letters 2006, 60, 2375.
(14) Zhuang, H.; Liu, J.-b.; Zhang, C.-l.; Kong, X.-g. Chemical Research in Chinese Universities 2007, 23, 5.
(15) Sooklal, K.; Cullum, B. S.; Angel, S. M.; Murphy, C. J. The Journal of Physical Chemistry 1996, 100, 4551.
(16) Lucey, D. W.; MacRae, D. J.; Furis, M.; Sahoo, Y.; Cartwright, A. N.; Prasad, P. N. Chemistry of Materials
2005, 17, 3754.
(17) Wang, C.; Jiang, Y.; Li, G.; Zhang, Z.; Shi, J.; Li, N. Journal of Crystal Growth 2008, 310, 2890.
(18) Noone, K. M.; Strein, E.; Anderson, N. C.; Wu, P.-T.; Jenekhe, S. A.; Ginger, D. S. Nano Letters 2010, 10,
2635.
(19) Xu, J.; Wang, J.; Mitchell, M.; Mukherjee, P.; Jeffries-El, M.; Petrich, J. W.; Lin, Z. Journal of the American
Chemical Society 2007, 129, 12828.
(20) Carl E. Behrens, C. G. Energy-consumption "consumption by fuel, 1965-2008", CRS Report for Congress, 2006.
(21) The Outlook for Energy: A View to 2040, ExxonMobil, 2014.
(22) Case, M. A.; Owusu, Y. A.; Chapman, H.; Dargan, T.; Ruscher, P. Renewable Energy 2008, 33, 2645.
(23) Sun, s.-S.; Bonner, C. E. In Optimization of Organic Solar Cells in Both Space and Energy–Time Domains, Chp
8 Organic Photovoltaics: Mechanisms, Materials, and Devices; Sun, S.-S., Sariciftci, N. S., Eds.; CRC Press:
2005.
(24) Halls, J. J. M.; Walsh, C. A.; Greenham, N. C.; Marseglia, E. A.; Friend, R. H.; Moratti, S. C.; Holmes, A. B.
Nature 1995, 376, 498.
(25) Maligaspe, E.; Tkachenko, N. V.; Subbaiyan, N. K.; Chitta, R.; Zandler, M. E.; Lemmetyinen, H.; D’Souza, F.
The Journal of Physical Chemistry A 2009, 113, 8478.
(26) Hankache, J.; Niemi, M.; Lemmetyinen, H.; Wenger, O. S. Inorganic Chemistry 2012, 51, 6333.
(27) Salinas, J.-F.; Maldonado, J.-L.; Ramos-Ortíz, G.; Rodríguez, M.; Meneses-Nava, M.-A.; Barbosa-García, O.;
Santillan, R.; Farfán, N. Solar Energy Materials and Solar Cells 2011, 95, 595.
(28) Salto, C.; Salinas, J.-F.; Maldonado, J.-L.; Ramos-Ortíz, G.; Rodríguez, M.; Meneses-Nava, M.-A.; Barbosa-
García, O.; Del Oso, J.-A.; Ortíz-Gutiérrez, M. Synthetic Met 2011, 161, 2412.
(29) Maldonado, J. L.; Ramos-Ortíz, G.; Miranda, M. L.; Vázquez-Córdova, S.; Meneses-Nava, M. A.; Barbosa-
García, O.; Ortíz-Gutiérrez, M. American Journal of Physics 2008, 76, 1130.
(30) Antoniadis, H.; Hsieh, B. R.; Abkowitz, M. A.; Jenekhe, S. A.; Stolka, M. Synthetic Met 1994, 62, 265.
(31) Landi, B. J.; Raffaelle, R. P.; Castro, S. L.; Bailey, S. G. Progress in Photovoltaics: Research and Applications
2005, 13, 165.
(32) Kymakis, E.; Koudoumas, E.; Franghiadakis, I.; Amaratunga, G. A. J. Journal of Physics D: Applied Physics
2006, 39, 1058.

119
(33) Kymakis, E.; Alexandrou, I.; Amaratunga, G. A. J. J Appl Phys 2003, 93, 1764.
(34) Geng, J.; Zeng, T. J Am Chem Soc 2006, 128, 16827.
(35) Kymakis, E.; Amaratunga, G. A. J. Applied Physics Letters 2002, 80, 112.
(36) Liu, Z.; Liu, Q.; Huang, Y.; Ma, Y.; Yin, S.; Zhang, X.; Sun, W.; Chen, Y. Advanced Materials 2008, 20, 3924.
(37) Liu, Q.; Liu, Z.; Zhang, X.; Yang, L.; Zhang, N.; Pan, G.; Yin, S.; Chen, Y.; Wei, J. Advanced Functional
Materials 2009, 19, 894.
(38) Li, C.; Wonneberger, H. Advanced Materials 2012, 24, 613.
(39) Hoppe, H.; Sariciftci, N. S. Journal of Materials Research 2004, 19, 1924.
(40) Halls, J. J. M.; Pichler, K.; Friend, R. H.; Moratti, S. C.; Holmes, A. B. Applied Physics Letters 1996, 68, 3120.
(41) Nierengarten, J.-F. New Journal of Chemistry 2004, 28, 1177.
(42) Reiss, P.; Couderc, E.; De Girolamo, J.; Pron, A. Nanoscale 2011, 3, 446.
(43) Alysha, G.; Ibrahim, K.; John, W.; Elias, T. In Solar Energy: New Materials and Nanostructured Devices for
High Efficiency; Optical Society of America: 2008, p SWA4.
(44) LaMer, V. K.; Dinegar, R. H. Journal of the American Chemical Society 1950, 72, 4847.
(45) Murray, C. B.; Kagan, C. R.; Bawendi, M. G. Annual Review of Materials Science 2000, 30, 545.
(46) Rozenberg, B. A.; Tenne, R. Progress in Polymer Science 2008, 33, 40.
(47) Murray, C. B.; Norris, D. J.; Bawendi, M. G. Journal of the American Chemical Society 1993, 115, 8706.
(48) Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Letters 1975, 16, 4467.
(49) Dieck, H. A.; Heck, F. R. Journal of Organometallic Chemistry 1975, 93, 259.
(50) Hwang, S.-H.; Moorefield, C. N.; Wang, P.; Jeong, K.-U.; Cheng, S. Z. D.; Kotta, K. K.; Newkome, G. R.
Chemical Communications 2006, 0, 3495.
(51) Bogado, M. G. M., Universidad Nacional de General San Martín Comisión Nacional de Energía Atómica
Instituto de Tecnología "Prof. Jorge A. Sabato", 2004.
(52) Tamasi, M. J. L., Universidad Nacional de General San Martín Comisión Nacional de Energía Atómica Instituto
de Tecnología "Prof. Jorge A. Sabato", 2003.
(53) http://www.spectrolab.com/prd/space/cell-main.asp 2009.
(54) Park, M.; Chin, B. D.; Yu, J.-W.; Chun, M.-S.; Han, S.-H. Journal of Industrial and Engineering Chemistry
2008, 14, 382.
(55) Tong, S. W.; Zhang, C. F.; Jiang, C. Y.; Liu, G.; Ling, Q. D.; Kang, E. T.; Chan, D. S. H.; Zhu, C. Chemical
Physics Letters 2008, 453, 73.
(56) Lee, J.-Y.; Connor, S. T.; Cui, Y.; Peumans, P. Nano Letters 2008, 8, 689.
(57) Dieter Wöhrle, D. M. Advanced Materials 1991, 3, 129.
(58) Tang, C. W. Applied Physics Letters 1986, 48, 183.
(59) Martens, T.; D'Haen, J.; Munters, T.; Beelen, Z.; Goris, L.; Manca, J.; D'Olieslaeger, M.; Vanderzande, D.; De
Schepper, L.; Andriessen, R. Synthetic Metals 2003, 138, 243.
(60) Al-Ibrahim, M.; Roth, H. K.; Schroedner, M.; Konkin, A.; Zhokhavets, U.; Gobsch, G.; Scharff, P.; Sensfuss, S.
Organic Electronics 2005, 6, 65.
(61) Nierengarten, J. F.; Gu, T.; Aernouts, T.; Geens, W.; Poortmans, J.; Hadziioannou, G.; Tsamouras, D. Appl Phys
A 2004, 79, 47.
(62) Kim, J. Y.; Lee, K.; Coates, N. E.; Moses, D.; Nguyen, T.-Q.; Dante, M.; Heeger, A. J. Science 2007, 317, 222.
(63) Greenham, N. C.; Peng, X.; Alivisatos, A. P. Physical Review B 1996, 54, 17628.
(64) Liu, J.; Tanaka, T.; Sivula, K.; Alivisatos, A. P.; Frechet, J. M. J. Journal of the American Chemical Society
2004, 126, 6550.
(65) Pientka, M.; Dyakonov, V.; Meissner, D.; Rogach, A.; Talapin, D.; Weller, H.; Lutsen, L.; Vanderzande, D.
Nanotechnology 2004, 163.
(66) Westenhoff, S.; Kotov, N. A. J Am Chem Soc 2002, 124, 2448.
(67) Nguyen, B. T.; Gautrot, J. E.; Ji, C.; Brunner, P.-L.; Nguyen, M. T.; Zhu, X. X. Langmuir 2006, 22, 4799.
(68) Dehu, C.; Jian, X.; Ting, Z.; Gary, P.; Ashok, S.; Michael, G. Applied Physics Letters 2006, 88, 183111.
(69) Günes, S.; Heiss, H. N. N. S. S. J. R. M. K. G. P. W. Advanced Functional Materials 2006, 16, 1095.
(70) Dayal, S.; Kopidakis, N.; Olson, D. C.; Ginley, D. S.; Rumbles, G. Nano Letters 2009, 10, 239.
(71) Jeltsch, K. F.; Schädel, M.; Bonekamp, J.-B.; Niyamakom, P.; Rauscher, F.; Lademann, H. W. A.; Dumsch, I.;
Allard, S.; Scherf, U.; Meerholz, K. Advanced Functional Materials 2012, 22, 397.
(72) http://www.sigmaaldrich.com/catalog/product/aldrich/541443?lang=es®ion=MX.
(73) Mayer, A. C.; Scully, S. R.; Hardin, B. E.; Rowell, M. W.; McGehee, M. D. Materials Today 2007, 10, 28.
(74) Wuister, S. F.; Meijerink, A. Journal of Luminescence 2003, 102-103, 338.

120
(75) Fang, Y.; Chen, L.; Wang, C.-f.; Chen, S. Journal of Polymer Science Part A: Polymer Chemistry 2010, 48,
2170.
(76) Kozhevnikova, N. S.; Demin, A. M.; Krasnov, V. P.; Rempel, A. A. Dokl Chem 2013, 452, 215.
(77) Feng, M.; Zhan, H. Nanoscale 2014.
(78) Li, H.; Powell, D. R.; Hayashi, R. K.; West, R. Macromolecules 1998, 31, 52.
(79) Harrison, N. T.; Hayes, G. R.; Phillips, R. T.; Friend, R. H. Physical Review Letters 1996, 77, 1881.
(80) Thelakkat, M.; Schmidt, H.-W. Advanced Materials 1998, 10, 219.
(81) Li, H.; Zhang, Y.; Hu, Y.; Ma, D.; Wang, L.; Jing, X.; Wang, F. Macromolecular Chemistry and Physics 2004,
205, 247.
(82) Williams, A. T. R.; Winfield, S. A.; Miller, J. N. Analyst 1983, 108, 1067.
(83) Wannier, G. H. Physical Review 1937, 52, 191.
(84) Cabrera, M. J. H., Universidad de la Laguna, 2006.
(85) Hlawiczka, P. Introducción a la electrónica cuantica; Reverte: España, 1977.
(86) Goswami, D. Y. Advance in Solar Energy: An Annual Review of Research and Development; illustrated ed.;
Earthscan, 2007.
(87) Hegedus, A. L. a. S. Handbook of Photovoltaic Science and Engineering; John Wiley and Sons, 2003.
(88) Jana, N. R.; Earhart, C.; Ying, J. Y. Chemistry of Materials 2007, 19, 5074.
(89) Torimoto, T.; Reyes, J. P.; Iwasaki, K.; Pal, B.; Shibayama, T.; Sugawara, K.; Takahashi, H.; Ohtani, B. J Am
Chem Soc 2002, 125, 316.
(90) Hirai, T.; Okubo, H.; Komasawa, I. J Mater Chem 2000, 10, 2592.
(91) Moulder, J. F.; Chastain, J. Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard
Spectra for Identification and Interpretation of XPS Data; Perkin-Elmer Corporatioon, Physical Electronics
Division, 1992.
(92) Kundu, M.; Khosravi, A. A.; Kulkarni, S. K.; Singh, P. Journal of Materials Science 1997, 32, 245.
(93) Koc, K.; Tepehan, F.; Tepehan, G. Nanoscale Research Letters 2012, 7, 610.
(94) Stebbing, S. R.; Hughes, R. W.; Reynolds, P. A. Advances in Colloid and Interface Science 2009, 147–148, 272.
(95) Hill, R.; Edwards, I. A. S. Vacuum 1977, 27, 277.
(96) Dimitrov, R. I.; Moldovanska, N.; Bonev, I. K. Thermochimica Acta 2002, 385, 41.
(97) Torimoto, T.; Yamashita, M.; Kuwabata, S.; Sakata, T.; Mori, H.; Yoneyama, H. The Journal of Physical
Chemistry B 1999, 103, 8799.
(98) Vossmeyer, T.; Katsikas, L.; Giersig, M.; Popovic, I. G.; Diesner, K.; Chemseddine, A.; Eychmueller, A.;
Weller, H. The Journal of Physical Chemistry 1994, 98, 7665.
(99) Pham, M. T.; Möller, D.; Matz, W.; Mücklich, A.; Oswald, S. The Journal of Physical Chemistry B 1998, 102,
4081.
(100) Haase, M.; Alivisatos, A. P. The Journal of Physical Chemistry 1992, 96, 6756.
(101) Tolbert, S. H.; Alivisatos, A. P. Annual Review of Physical Chemistry 1995, 46, 595.
(102) Chen, C.-C.; Herhold, A. B.; Johnson, C. S.; Alivisatos, A. P. Science 1997, 276, 398.
(103) Moffitt, M.; Vali, H.; Eisenberg, A. Chemistry of Materials 1998, 10, 1028.
(104) Seo, S. H.; Chang, J. Y.; Tew, G. N. Angewandte Chemie International Edition 2006, 45, 7526.
(105) Eckert, J.-F.; Nicoud, J.-F.; Nierengarten, J.-F.; Liu, S.-G.; Echegoyen, L.; Barigelletti, F.; Armaroli, N.; Ouali,
L.; Krasnikov, V.; Hadziioannou, G. Journal of the American Chemical Society 2000, 122, 7467.
(106) Armaroli, N. A., G., The Royal Society of Chemistry 2007, 79.
(107) Flores, J. R. Tesis de Doctorado, CIQA, 2012.
(108) Flores, R.; Arias, E.; Moggio, I.; Ponce, A.; Gallardo-Vega, C.; Galembeck, F.; Gouveia, R. F.; Ziolo, R. F.
Journal of Advanced Microscopy Research 2012, 7, 158.