
MANUAL PARA LAS BUENAS - Portal de la Conselleria de...
425
Transcript of MANUAL PARA LAS BUENAS - Portal de la Conselleria de...


MANUAL PARA LAS BUENASPRÁCTICAS CLÍNICAS
GUÍA PARA EL INVESTIGADOR DE ENSAYOS CLÍNICOS EN LA COMUNIDAD VALENCIANA

Director y coordinador
Tosca Flores, J. F.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluación delas Organizaciones y Prácticas Sanitarias. Médico Inspector de BPC
Autores
Albiach Domingo, J. C.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluación delas Organizaciones y Prácticas Sanitarias.Andani Cervera, J.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluación delas Organizaciones y Prácticas Sanitarias. Andreu Solsona, PMédica Inspectora de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluaciónde las Organizaciones y Prácticas Sanitarias. Médica Inspectora de BPCBartual Montesinos, M. J.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Médico Inspector de BPCBou de Miguel, E.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Especialista Universitario en Auditoria, Acredita-ción y Evaluación de las Organizaciones y Prácticas Sanitarias. Burriel de San Vicente, CMédico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Facultativo especialista en UrologíaFos Ibáñez, J. A. Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluación delas Organizaciones y Prácticas Sanitarias. Latorre Saiz, F.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad.López Pesquera, B.Facultativo Especialista en Neurología. Agencia Valenciana de Salud. Hospital Clínico Universitario de ValenciaMartínez Blanco, C. Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluación delas Organizaciones y Prácticas Sanitarias. San Ruperto Abella, M. A.Farmacéutica Inspectora de Servicios Sanitarios de la Consellería de Sanidad. Especialista Universitario en Auditoria,Acreditación y Evaluación de las Organizaciones y Prácticas Sanitarias. Farmacéutica Inspectora de BPCTosca Flores, J. F.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluación delas Organizaciones y Prácticas Sanitarias. Médico Inspector de BPCTosca Cuquerella, J.Facultativo Especialista en Aparato Digestivo. Agencia Valenciana de Salud. Hospital Clínico Universitario de ValenciaTrenor Galindo, A.Farmacéutica Inspectora de Servicios Sanitarios de la Consellería de Sanidad. Especialista Universitario en Auditoria,Acreditación y Evaluación de las Organizaciones y Prácticas Sanitarias. Farmacéutica Inspectora de BPCVidal Alamar, A.Médico Inspector de Servicios Sanitarios de la Consellería de Sanidad. Master en Auditoria, Acreditación y Evaluación delas Organizaciones y Prácticas Sanitarias. Vidal Vidal, S.Médica Inspectora de Servicios Sanitarios de la Consellería de Sanidad. Médica Inspectora de BPC
Edita Generalitat Valenciana. Conselleria de SanitatISBN 978-84-482-5207-6Depósito LegalImprime Gráficas Marí Montañana, S.L. - Santo Cáliz, 7 - 46001 Valencia - Tel. 96 391 23 04

ÍNDICE
PRESENTACIÓN.................................................................................................. 12PRÓLOGO........................................................................................................... 12INTRODUCCIÓN.................................................................................................. 12ANTECEDENTES Y JUSTIFICACIÓN...................................................................... 13DEL CÓDIGO DE NÜREMBERG A LA DECLARACIÓN DE HELSINKI ........................ 17EL INFORME BELMONT ...................................................................................... 23OTRAS NORMAS Y DECLARACIONES INTERNACIONALES..................................... 27NORMAS DE BUENA PRÁCTICA CLÍNICA ............................................................. 33NORMATIVA APLICABLE EN ESPAÑA ................................................................... 41NORMATIVA APLICABLE EN LA COMUNIDAD VALENCIANA ................................... 47EL INVESTIGADOR.............................................................................................. 55EL COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA (CEIC)......................................... 59EL PROCESO DE OBTENCIÓN DEL CONSENTIMIENTO INFORMADO...................... 69INTERPRETACIÓN ACTUAL Y VIGENCIA DEL PRINCIPIO “PRIMUM NON NOCERE” .. 75ANEXOS ............................................................................................................. 77LEY 29/2006, DE 26 DE JULIO, DE GARANTÍAS Y USO RACIONAL DE LOSMEDICAMENTOS Y PRODUCTOS SANITARIOS ...................................................... 81REAL DECRETO 223/2004, DE 6 DE FEBRERO, POR EL QUE SE REGULAN LOS ENSAYOS CLÍNICOS CON MEDICAMENTOS. .................................................. 107LEY 41/2002, DE 14 DE NOVIEMBRE, BÁSICA REGULADORA DE LA AUTONOMÍA DEL PACIENTE Y DE DERECHOS Y OBLIGACIONES EN MATERIA DE INFORMACIÓN Y DOCUMENTACIÓN CLÍNICA. ................................................. 143LEY ORGÁNICA 15/1999, DE 13 DE DICIEMBRE, DE PROTECCIÓN DE DATOS DE CARÁCTER PERSONAL................................................................................... 159ORDEN SCO/256/2007, DE 5 DE FEBRERO, POR LA QUE SE ESTABLECEN LOSPRINCIPIOS Y LAS DIRECTRICES DETALLADAS DE BUENA PRÁCTICA CLÍNICA Y LOS REQUISITOS PARA AUTORIZAR LA FABRICACIÓN O IMPORTACIÓN DEMEDICAMENTOS EN INVESTIGACIÓN DE USO HUMANO. ..................................... 189DECLARACION DE HELSINKI DE LA ASOCIACION MÉDICA MUNDIAL .................... 203NORMAS DE LA BUENA PRÁCTICA CLINICA. (CPMP/ICH/135/95) ......................... 2111.GLOSARIO....................................................................................................... 211
3

2.LOS PRINCIPIOS DE LA BPC DE LA ICH............................................................ 2183.COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA (CEIC)........................................... 2194.INVESTIGADOR................................................................................................ 2225.PROMOTOR ..................................................................................................... 2316.PROTOCOLO DEL ENSAYO CLÍNICO Y MODIFICACIONES AL PROTOCOLO ........... 2437.MANUAL DEL INVESTIGADOR (MI) ................................................................... 2468. DOCUMENTOS ESENCIALES PARA LA REALIZACION DE UN ENSAYO CLINICO .. 254CONVENIO PARA LA PROTECCIÓN DE LOS DERECHOS HUMANOS Y LA DIGNIDAD DEL SER HUMANO CON RESPECTO A LAS APLICACIONES DE LA BIOLOGÍA Y LA MEDICINA .................................................................................................. 268NORMATIVA DE LA COMUNIDAD VALENCIANA ..................................................... 278LEY 1/2003, DE 28 DE ENERO, DE DERECHOS E INFORMACIÓN AL PACIENTE DE LA COMUNIDAD VALENCIANA ........................................................................ 282DECRETO 73/2006, DE 5 DE JUNIO, DEL CONSELL, POR EL QUE SE REGULA LA GESTIÓN DE ENSAYOS CLÍNICOS Y ESTUDIOS POST-AUTORIZACIÓNOBSERVACIONALES CON MEDICAMENTOS Y PRODUCTOS SANITARIOS................ 300BIBLIOGRAFÍA.................................................................................................... 321
4

PRESENTACIÓN
La Conselleria de Sanidad de la Generalitat viene apostando por fomentar, en elámbito del Sistema Sanitario Valenciano, la labor investigadora, además de laasistencial, preventiva y docente. Y es en esa labor investigadora donde se incluye,con un elevado peso específico, la investigación relacionada con los medicamentosde uso humano.
Por ello, desde la Consellería de Sanidad, además de prestar la asistencia sanitaria queprecisen los ciudadanos de nuestra Comunidad, prevenir que no lleguen a enfermar a lavez que promocionar estados óptimos de salud y rehabilitar a los que hayan vistomermadas sus posibilidades físicas o psíquicas tras haber padecido procesos deenfermedad o accidentes, debe impulsar y promocionar las funciones docente einvestigadora del sistema sanitario.
En el desarrollo de los ensayos clínicos como elemento esencial de la investigaciónsanitaria y biomédica, es necesario garantizar plenamente los derechos de los ciudadanosincluidos en estos estudios.
Esta actividad investigadora está regulada por leyes y normas nacionales e internacionalestendentes a minimizar los riesgos de los sujetos y a garantizar el respeto de sus derechosy la credibilidad de los datos.
Para esta importante misión, la Consellería de Sanidad dispone de la Inspección deServicios Sanitarios, órgano altamente especializado que, a través del Plan Anual deInspección, desarrolla una labor de evaluación continua de todo el sistema sanitario,incluida la realización de ensayos clínicos con medicamentos, en coordinación conorganismos naciones e internacionales.
Este libro debe suponer una indudable ayuda, entre otros, para los investigadoressanitarios, promotores de ensayos clínicos, comités de investigación, al comentarampliamente y de forma amena la normativa legal y ética, recopilándolas además en unsolo volumen.
5

Si la lectura de este libre permite a todos los agentes implicados en los ensayos clínicoscon medicamentos mejorar sus prácticas clínicas en aras al progreso de la investigación,con el máximo respeto a los principios éticos y como garantía de los derechos de losciudadanos, habrá cumplido plenamente sus objetivos
Manuel Cervera TauletConseller de Sanidad
6

7
PRÓLOGO
La Agencia Valenciana de Salud, como órgano gestor de las prestaciones incluidas en la carterade servicios del sistema sanitario de la Comunidad Valenciana, necesita de una evaluaciónpermanente de sus actividades para detectar tanto las posibles desviaciones de los objetivosque tiene encomendados como las oportunidades de mejora que puedan plantearse.Esta evaluación permanente la proporciona la Inspección de Servicios Sanitarios de laConsellería de Sanidad, dentro del obligado marco de colaboración entre ambos organismos.
En la mencionada cartera de servicios del sistema sanitario, se encuentra la actividadinvestigadora y en ella, la investigación con medicamentos de uso humano, los denominadosensayos clínicos.
El Reglamento de Organización y Funcionamiento de la Inspección de Servicios Sanitarios dela Consellería de Sanidad, aprobado por Decreto del Consell de la Generalitat, prevé entres susfunciones las de inspeccionar y evaluar las actividades de investigación que se realicen en elsistema sanitario, en todo lo relacionado con el cumplimiento de la legislación vigente y elrespeto de los derechos de los sujetos de la investigación.
Así pues, el actual Plan Anual de Inspección de Servicios Sanitarios, dedica el primer programadel Área primera, referida a los derechos de los ciudadanos, a la inspección y evaluación delcumplimiento de las normas de Buena Práctica Clínica en la investigación sanitaria.
El sistema sanitario valenciano en general y la actividad investigadora en particular,precisan cada vez más, de profesionales altamente cualificados como científicosinvestigadores y esta cualificación se refiere tanto a la adquisición de conocimientosteóricos como de técnicas y habilidades. No obstante esto no sería suficiente sin unanecesaria formación en las premisas éticas y morales aceptadas comúnmente por lospaíses firmantes de la Declaración Universal de Derechos Humanos.
Las normas de Buenas Prácticas Clínicas promulgadas por la Conferencia Internacional deArmonización (ICH) en 1997, aceptadas en nuestro entorno europeo y de los EE. UU.,deben ser, por imperativo legal, conocidas por nuestros investigadores y respetadas entodo proyecto de investigación en el que participen seres humanos.

Con la publicación del presente libro, la Inspección de Servicios Sanitarios de laConsellería de Sanidad, no solo nos da las claves para proyectar y desarrollar protocolosde investigación, respetuosos con los derechos de los sujetos, sino que contribuye a laformación de los investigadores en el campo que nos ocupa, llenando un espacio solocubierto hasta este momento por los propios textos legales, dispersos y, en ocasiones, dedifícil comprensión.
Alfonso Bataller Vicent Subsecretario
8

INTRODUCCIÓN
Desde principios del siglo pasado, el conocimiento médico ha avanzado más queen todo el periodo anterior desde la antigüedad. La práctica de la medicina hasta no hacemucho tiempo era considerada un arte más que una ciencia.
La tradición aristotélica de la práctica médica basaba la adquisición de nuevos co-nocimientos en la capacidad de observación de los médicos y en su habilidad para elabo-rar teorías (principio del voluntario indirecto: “todo acto médico realizado en seres humanosper se ha de tener un carácter beneficente y sólo per accidens un carácter investigativo”)
La gran revolución de la medicina viene de la mano del Método Científico comométodo de investigación, a través de diseños experimentales reproducibles, cuyos resul-tados permiten adquirir conocimiento generalizable, de acuerdo con las normas de la es-tadística descriptiva y muestral.
Paulatinamente, la práctica médica va dejando de ser un arte para ser, cada vezmás, una ciencia en la que cada descubrimiento posibilita el siguiente, hasta convertir lasecuencia en exponencial.
Una característica importante de la práctica actual en la investigación biomédicaes que, cada técnica, cada proceso, cada nuevo tratamiento, progresivamente es más agre-sivo aunque también sea más eficaz. Así las técnicas diagnósticas o terapéuticas son cadavez más resolutivas aunque también más invasivas y los tratamientos con medicamentos,a la vez que son más eficaces tienen más efectos adversos.
Por esta razón, la investigación de nuevos medicamentos para uso en humanos,ha devenido en una actividad científica de alto nivel con unas implicaciones éticas que lasociedad debe regular.
En la Comunidad Valenciana, durante los últimos cinco años, se han autorizado901 ensayos clínicos con medicamentos en 1963 centros.
Teniendo en cuenta que un ensayo clínico puede incluir a varios sujetos y que sudesarrollo suele ser plurianual, significa que el número de ciudadanos de la Comunidad
9

que están incluidos en programas de investigación biomédica, de forma permanente, sonvarios millares, admitiendo que el número de nuevos ensayos que se autorizan y el númerode los que finalizan es similar.
Estos ciudadanos pueden ser sujetos de un ensayo clínico sin interés terapéuticopara ellos o, como la gran mayoría, ostentar además la condición de enfermos, puesto queel mayor porcentaje de estas investigaciones están dirigidas a la determinación de la efi-cacia, seguridad o nuevas indicaciones de moléculas en fase avanzada de investigación yel resultado de la misma sí que puede tener interés terapéutico para ellos.
Con independencia de esta diferenciación y de las motivaciones de ambos grupos,el capítulo de los ensayos clínicos es indudable que tiene un interés relevante en la sani-dad valenciana.
Así pues, es una obligación de la Administración Sanitaria Valenciana fomentar lainvestigación sanitaria y biomédica, garantizando los derechos de los sujetos sometidos aestudio y velando por unas buenas prácticas clínicas de los investigadores.
Pilar Viedma Gil de VergaraDirectora General de Ordenación,Evaluación e Investigación Sanitaria
10
José Clérigues BellochDirector General de Farmacia y
Productos Sanitarios

Antecedentes y justificación


Hasta bien avanzado el siglo XX, la práctica médica se ejercía de tal forma que elenfermo rara vez era informado no ya sólo del proceso a seguir para su curación sino que,incluso, se le ocultaba el diagnóstico, sobre todo si era especialmente grave y se sospe-chaba un desenlace fatal. Ni siquiera se informaba a la familia bajo la premisa: “tiempotendrán para llorar”.
A medida que fue avanzando el siglo, esta filosofía se ha ido modificando y, pau-latinamente el enfermo/sujeto ha dejado de ser un organismo alterado en su funciona-miento sobre el que el médico aplicaba sus conocimientos, sin otros límites que su propiaconcepción ética y se ha convertido en un sujeto portador de derechos regulados por lasleyes.
Esta evolución ha sido asumida tanto por el ciudadano como por los médicos y hoydía es impensable la figura del médico que no sigue protocolos de probada solvencia(“cada maestrico ya no tiene su librico”), se ejerce la medicina basada en la evidencia yla práctica de informar y solicitar consentimiento ha devenido en un acto intrínseco delquehacer diario.
Esta misma evolución conceptual ha ocurrido en la investigación biomédica en laque participen seres humanos y, también, en este campo se ha ido dotando al sujeto dederechos que paulatinamente se han incorporado tanto a la concepción de los investiga-dores como a los cuerpos jurídicos de los distintos países.
Esta transformación no ha estado exenta de hechos que han producido escándaloy que han ido determinando la necesidad de regulaciones éticas y, si bien hoy día pode-mos decir que el 100 % de proyectos de investigación cumplen con lo principios éticos ylegales establecidos, es conveniente recordarlos para aprender de la historia.
Sin otra finalidad que la anunciada podemos recordar los siguientes en una expo-sición cronográfica coincidente con el desarrollo de Declaraciones y Códigos éticos.
El principio “primum non nocere” (lo primero no dañar), atribuido a Hipócratesha sido muchas veces menospreciado bajo la coartada de incrementar el conocimiento.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
13

Al respecto, como inciso histórico, cabe apuntar que la anterior frase latina, atri-buida desde siempre a Hipócrates de Cos (460 - 377 a.C.), no se encuentra en el Jura-mento Hipocrático aunque sí que existe una aproximada en el Libro I, Sección II de LasEpidemias (Corpus Hipocrático), en la forma “… para ayudar, o por lo menos no hacerdaño,…“. Según Cedric M. Smith, farmacólogo clínico inglés, en una investigación pre-sentada en Abril de 2005 en el Journal of Clinical Pharmacology, afirma que la expresiónespecífica debe atribuirse al médico Thomas Sydenham (1624–1689).
En el siglo XX, con la finalización de la segunda guerra mundial, a pesar de quela Alemania de la República de Weimar, había dictado una ley relativa a la investigaciónmédica en 1931, el mundo se horrorizó al descubrir lo que los médicos nazis habían es-tado experimentando en los campos de concentración: la resistencia del cuerpo humanoal frío hasta que moría la persona, la resistencia a grandes altitudes, los efectos de la ma-laria, los efectos del gas mostaza, quemaduras por aplicación de fósforo, ablación de mús-culos, castración y esterilización, la observación directa de la muerte del corazón.
No obstante, estos hechos no fueron los únicos que se cometieron en esos años yasí, investigadores de los EE.UU. experimentaron durante la guerra y años posteriores conantídotos contra la malaria en prisioneros y pacientes psicóticos y se realizaron experi-mentos de vacunación contra la gripe en enfermos mentales.
Investigadores Japoneses, en Manchuria, utilizando prisioneros chinos de la se-gunda guerra mundial, ensayaron la resistencia humana al botulismo, ántrax, brucelosis,cólera, disentería, fiebre hemorrágica, sífilis, rayos X y congelamiento, entre otras investi-gaciones.
Durante los mismos años, la universidad de Vanderbilt (Tennessee, Estados Uni-dos) llevó a cabo investigaciones con radiaciones en mujeres pobres embarazadas a las quese les daba dosis 30 veces superiores a las consideradas inocuas.
La actuación de 23 médicos nazis fue juzgada en la ciudad alemana de Nürem-berg y, de ellos, 16 fueron declarados culpables y 7 condenados a muerte. Este juicio dioorigen el Código de Nüremberg, considerado como el primer compendio ético internacio-nal, regulador de la investigación biomédica con seres humanos, si exceptuamos la ley ale-mana de 1931 que no era, en si mismo, un documento de carácter ético.
Con el descubrimiento de la penicilina en el año 1940, se inició una carrera des-enfrenada para encontrar nuevos productos con el fin de incrementar el arsenal terapéu-tico y poder combatir entre otras, las infecciones que comprometían la vida de los
14

pacientes. Ese camino no estaba exento de riesgos como demostró la trágica historia dela Talidomida en la Europa de los años 60, hecho que de nuevo conmocionó al mundo ehizo que empezaran a surgir leyes que garantizaran la seguridad y la eficacia de los nue-vos productos a introducir en el mercado. En esta época se definió el ensayo clínico con-trolado como el mecanismo garante para las evaluaciones de los nuevos productos aensayar en seres humanos.
A pesar de las esperanzas depositadas en que el Código de Nüremberg iba a me-jorar los aspectos éticos de estas prácticas, el 26 de julio de 1972, el New York Times des-cribió “el experimento no terapéutico en seres humanos más largo en la historia clínica”.Se trataba del estudio de Tuskegee sobre la Sífilis, en el cual, a 399 aparceros afroame-ricanos pobres del condado de Macon en Alabama, en un experimento diseñado para do-cumentar la historia natural de la enfermedad, se les negó tratamiento para la sífilis,cuando ya existían tratamientos que se habían mostrado eficaces contra ella.
Los investigadores de este estudio, durante los 40 años que duró el mismo, pu-blicaron 13 artículos en revistas médicas de relevancia científica sin que nadie levantarala voz para denunciar la grave agresión ética que esta investigación suponía. El Senado delos EE.UU. a raíz del escándalo que supuso la publicación del hecho, instruyó una inves-tigación que concluyó que el estudio había sido “injusto éticamente” y lo calificó de atroze intolerable, determinó la indemnización de los supervivientes y no atribuyó responsabi-lidades entre los investigadores.
En 1956, veinte años después de Nüremberg, en Staten Island, se investigó la in-fección deliberada de niños deficientes mentales con virus de hepatitis para encontraruna vacuna.
En 1963 en el Hospital Judío de Enfermos Crónicos de Brooklin, a 22 pacientesancianos se les inyectaron células cancerosas sin su consentimiento para descubrir la ca-pacidad de rechazo.
La lista de violaciones de los códigos éticos en investigaciones biomédicas con se-res humanos es prolija y sigue incrementándose. Es paradójico que, según información deldiario EL PAÍS, de 10 de enero de 2005, en la página 32, en 1997, al tiempo que el Con-sejo Europeo celebraba la Convención sobre Derechos Humanos y Biomedicina y apro-baba la Declaración Bioética de Asturias y las Autoridades reguladoras de la UniónEuropea, EE.UU. y Japón, junto a la Industria farmacéutica de estos mismos países, ela-boraba la Guía ICH de Buenas Prácticas Clínicas, los Institutos Nacionales de la Salud(NIH) de los EE.UU., iniciaron en Uganda un estudio que pretendía valorar hasta que
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
15

punto una sola dosis de un determinado medicamento, cuyos efectos secundarios produ-cen lesiones dermatológicas graves y lesiones hepáticas que llegan a ser mortales, podíafrenar la transmisión del SIDA de madres a hijos. El estudio estaba plagado de errores enel seguimiento de los pacientes que habían recibido el tratamiento. El descontrol era tanabrumador que los encargados de la investigación no sabían con certeza qué madres ha-bían recibido una primera dosis y, en algunos casos, hubo que hacer análisis de sangre alas pacientes para saber qué cantidad de medicamento habían tomado.
A pesar de estos hechos aislados, por nuestra experiencia en Inspecciones de Bue-nas Prácticas Clínicas, podemos afirmar que hoy en día, son anecdóticos los casos detec-tados de incumplimientos graves de las mismas y, en su gran mayoría, están determinadospor la insuficiente formación que en esta materia. se imparte a los investigadores noveles.
Por este motivo pensamos que la recopilación de las mismas en un solo manualpuede ser útil, sin otra pretensión que no sea la de servir de herramienta de consulta.
16

Del Código de Nüremberg a la Declaración de Helsinki


Como respuesta a los crímenes de guerra de los médicos nazis, en 1946 el tribu-nal internacional constituido al efecto por los países vencedores de la 2ª guerra mundial,adoptó el Código de Nüremberg constituyendo así las primeras normas éticas para la in-vestigación en seres humanos.
Se trata de un compendio de 10 puntos que hace especial hincapié en el consen-timiento voluntario y lo cataloga de absolutamente esencial. Así en el punto primero, trasdefinir lo anterior, prosigue diciendo que el sujeto “…debe tener capacidad legal para darsu consentimiento; debe estar situada (la persona) en tal forma que le permita ejercer sulibertad de escoger, sin la intervención de cualquier otro elemento de fuerza, fraude, en-gaño, coacción o algún otro factor posterior para obligar a coercer, y debe tener el sufi-ciente conocimiento y comprensión de los elementos de la materia en cuestión parapermitirle tomar una decisión correcta”.
A pesar de que utiliza la expresión “voluntary consent” y no “informed consent”,indica también que debe contar con todos los elementos necesarios de conocimiento paratomar una decisión correcta y acorde con sus intereses, es decir, establece la necesidadde que se le facilite toda le información relativa a “… la naturaleza, duración y propósitodel mismo (experimento), el método y las formas mediante las cuales se conducirá, todoslos inconvenientes y riesgos que pueden presentarse, y los efectos sobre la salud o la per-sona que pueden derivarse posiblemente de su participación en el experimento”.
No contempla la necesidad de control externo, mediante la existencia de un co-mité que salvaguarde los derechos del sujeto y fía en la honestidad ética del investigador:“… El deber y la responsabilidad para determinar la calidad del consentimiento recaen so-bre el individuo que inicia, dirige, o toma parte del experimento. Es un deber personal yuna responsabilidad que no puede ser delegada a otra persona con impunidad.”
Declara la finalidad y justificación del experimento en conseguir beneficios parala sociedad aunque “… debe ser conducido de manera tal que evite todo sufrimiento ydaño innecesario sea físico o mental”, prohibiéndolo cuando “… hay una razón «a priori»para asumir que puede ocurrir la muerte o daño irreparable: menos, quizás, en aquellos
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
19

experimentos donde los realizadores del mismo también sirvan como sujetos de experi-mentación.” O sea que el interés se justifica en la consecución de conocimiento genera-lizable para el bien de la sociedad y el sufrimiento o daño físico o mental estaríajustificado cuando se considere necesario, salvo que el mismo pueda devenir en muerteo daño irreparable.
Establece la relación riesgo / beneficio pero, mientras el riesgo es para el sujeto,el beneficio se cuantifica “… por la importancia humanitaria del problema a ser resueltopor el experimento”.
Declara que “… el experimento debe ser conducido únicamente por personascientíficamente calificadas”, así como que el sujeto debe tener la libertad de poner fin alexperimento y que “… el científico que lo realiza debe estar preparado para interrumpirloen cualquier momento, si tiene razones para creer, en el ejercicio de su buena fe, habili-dad técnica y juicio cuidadoso, que la continuación del experimento puede resultar en le-sión, incapacidad o muerte para el sujeto bajo experimentación”.
18 años después, en 1964, la Asociación Médica Mundial, promulgó la Declara-ción de Helsinki, ciudad donde se efectuaba la reunión. En sus partes fundamentales, es-tablece pautas éticas para la investigación clínica y no clínica en seres humanos, elconsentimiento informado de los individuos participantes en la investigación y el análisisético de los protocolos de investigación.
Esta Declaración se ha revisado posteriormente.
Las sucesivas revisiones han sido:
29ª Asamblea Médica Mundial. Tokio, Japón, Octubre 1975.
35ª Asamblea Médica Mundial. Venecia, Italia, Octubre 1983.
41ª Asamblea Médica Mundial. Hong Kong, Septiembre 1989.
48ª Asamblea General de la AMM. Somerset West, Sudáfrica, Octubre 1996.
52ª Asamblea General de la AMM. Edimburgo, Escocia, Octubre 2000.
Además ha habido una Nota de Clarificación del Párrafo 29, agregada por la Asam-blea General de la AMM, en la reunión de Washington en 2002 y otra Nota de Clarifica-ción del Párrafo 30, agregada por la Asamblea General de la AMM, en Tokio en 2004.
59ª Asamblea General de la AMM. Seúl, Corea, octubre 2008.
20

En su versión actual (Seúl), la Declaración consta de (A) Introducción que com-prende los puntos 1 al 10, (B) Principios Básicos para toda Investigación Médica, queabarca desde el punto 11 al 30 y (C) Principios Aplicables cuando la Investigación Mé-dica se combina con la Atención Médica, con los puntos 31 al 35.
Previamente, la Asamblea Médica Mundial, por primera vez, declara anuladas lasversiones anteriores de la Declaración y prohíbe su utilización o su cita excepto para fineshistóricos.
En la Introducción declara que la investigación médica en seres humanos incluyela investigación del material humano y de información identificables.
En los puntos tres y cuatro, declara como deber del médico promover y velar porla salud de los pacientes, “Los conocimientos y la conciencia del médico han de subordi-narse al cumplimiento de ese deber”.
En contraposición con la tradición aristotélica, en el punto cinco reconoce que Elprogreso de la medicina se basa en la investigación que, en último término, debe incluirestudios en seres humanos” y, a continuación, en el punto 7, complementa: “…). Incluso,las mejores intervenciones actuales deben ser evaluadas continuamente a través de la in-vestigación para que sean seguras, eficaces, efectivas, accesibles y de calidad”. En estemismo punto solicita el derecho a participar en la investigación médica de las poblacio-nes que están subrepresentadas.
En el punto 6, por aplicación del art. 2º de la Convención sobre Derechos Huma-nos y Biomedicina del Consejo de Europa de 1997, dispone como principio moral univer-sal que “En investigación médica en seres humanos, el bienestar de la persona queparticipa en la investigación debe tener siempre primacía sobre todos los otros intereses.”
Como hemos visto ya, desde Nüremberg se establece como principio el respeto ala autonomía del paciente entendido, bien como la capacidad del ser humano compe-tente, tras ser informado, de consentir su participación en un proyecto de investigación,bien como el deber de proteger los derechos de las personas y poblaciones no competen-tes para otorgar su consentimiento. Los hechos demostraron que este principio no siem-pre se respetó incluso después de 1946, aun considerando poblaciones libres y con lacapacidad intelectual íntegra.
Se orienta como objetivo de toda la investigación médica, en el punto 7, la adqui-sición de conocimiento y la mejora de las actuaciones tanto preventivas como diagnósti-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
21

cas y terapéuticas, que deben ser evaluadas continuamente “…para que sean seguras, efi-caces, efectivas, accesibles y de calidad”.
En el punto 8 apunta que la práctica de la medicina y la investigación no son ino-cuos sino que entrañan riesgos la mayoría de las actuaciones.
En el punto 9, de la Declaración, se establece la necesidad ética de protecciónespecial de algunas poblaciones vulnerables. Así, dice: “…Algunas poblaciones someti-das a la investigación son particularmente vulnerables y necesitan protección especial. Es-tas incluyen a los que no pueden otorgar o rechazar el consentimiento por sí mismos y alos que pueden ser vulnerables a coerción o influencia indebida.”
Dentro ya del apartado de principios básicos para toda investigación médica, apa-rece el concepto del deber del médico de proteger la vida y la salud, la dignidad, la inte-gridad, el derecho a la autodeterminación, la intimidad y la confidencialidad de lainformación personal de las personas que participan en investigación, así como que hayque prestar atención adecuada a los factores que puedan perjudicar el medio ambiente ycomprometer el futuro de las generaciones futuras. Se debe cuidar también del bienestarde los animales utilizados en los experimentos. Todo ello en concordancia con el CódigoInternacional de Ética Médica de la propia AMM (Sudáfrica, octubre de 2006) y Declara-ción universal sobre bioética y derechos humanos de la Conferencia General de laUNESCO. 33ª sesión de 19 de octubre de 2005.
Igualmente declara la obligación de que “… el proyecto y el método de todo pro-cedimiento experimental en seres humanos debe describirse claramente en un protocolode investigación”. Dicho protocolo tiene que ser revisado y, aprobado, por un comité deevaluación ética especialmente designado, que debe ser independiente del investigador,del patrocinador o de cualquier otro tipo de influencia indebida. En este apartado, la De-claración se ha modificado respecto a la anterior versión en la que se contemplaba que “Di-cho protocolo tiene que ser revisado y, cuando proceda, aprobado por un comité…”. Estamodificación se homologa con las normas de Buena Práctica Clínica de la Conferencia In-ternacional de Armonización.
Los proyectos de investigación con seres humanos, solo podrán llevarse a cabo porpersonas científicamente cualificadas.
Sólo está éticamente justificada una investigación en seres humanos tras una eva-luación de riesgos y una comparación cuidadosa con los beneficios previsibles, de talforma que sólo debe realizarse cuando la importancia de su objetivo es mayor que el riesgo
22

inherente y los costos para el individuo. La investigación médica sólo se justifica si exis-ten posibilidades razonables de que la población sobre la que la investigación se realizapodrá beneficiarse de sus resultados. Así, en el punto 14 dice que “El protocolo debe des-cribir los arreglos (medidas adoptadas) para el acceso después del ensayo a intervencio-nes identificadas como beneficiosas en el estudio o el acceso a otra atención o beneficiosapropiadas”. La Declaración de Helsinki cumple así con estos condicionantes, con dosde los principios universales de la ética de la investigación en humanos: los principios deBeneficencia y de Justicia, que en otro capítulo abordaremos.
A continuación, desarrolla lo apuntado ya en el punto 9 respecto al respeto alprincipio de autonomía y aborda los aspectos relacionados con el consentimiento del su-jeto sometido al proyecto de investigación.
Así, a partir del punto 22 decreta que: “La participación de personas competen-tes en la investigación médica debe ser voluntaria. Aunque puede ser apropiado consul-tar a familiares o líderes de la comunidad, ninguna persona competente debe ser incluidaen un estudio, a menos que ella acepte libremente” y, a continuación recalca el respetoque debe otorgarse al derecho de los sujetos a que se proteja no sólo su integridad física,mental y su personalidad, sino también la intimidad, y la confidencialidad de sus datos.
La información que el sujeto debe recibir para poder ejercer con plenitud su de-recho a decidir, debe comprender: “…objetivos, métodos, fuentes de financiamiento, po-sibles conflictos de intereses, afiliaciones institucionales del investigador, beneficioscalculados, riesgos previsibles e incomodidades derivadas del experimento y todo otro as-pecto pertinente de la investigación y de retirar su consentimiento en cualquier momento,sin exponerse a represalias”. Sólo cuando se tenga la certeza de que el individuo ha en-tendido toda la información debe solicitársele su participación voluntaria e informada, aser posible por escrito, aunque se establecen excepciones: “Si el consentimiento no sepuede obtener por escrito, el proceso para lograrlo debe ser documentado y atestiguadoformalmente”.
La protección a los sujetos, en la Declaración va más allá que el Código de Nü-remberg y destina los puntos 17 al 28 a definir medidas que maximicen esta proteccióna las poblaciones especialmente sensibles, tales como individuos dependientes del mé-dico investigador, en cuyo caso debe ser otro médico que conozca el proyecto de investi-gación pero independiente del mismo quien informe y solicite la participación del sujeto.
Las personas legalmente incapaces o inhábiles física o mentalmente o menoresde edad, sólo deben ser sujetos de un proyecto de investigación si su participación es ne-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
23

cesaria para promover la salud de la población representada y si esta investigación nopuede realizarse en personas legalmente capaces. Si un individuo potencial que participaen la investigación y que es considerado incompetente, es capaz de dar su asentimientoa participar o no en la investigación, el médico debe pedirlo, además del consentimientodel representante legal. El desacuerdo del individuo potencial debe ser respetado.
En el punto 29 declara que “La investigación en individuos que no son capacesfísica o mentalmente de otorgar consentimiento, por ejemplo los pacientes inconscien-tes, se puede realizar sólo si la condición física/mental que impide otorgar el consenti-miento informado es una característica necesaria de la población investigada. En estascircunstancias, el médico debe pedir el consentimiento informado al representante legal.Si dicho representante no está disponible y si no se puede retrasar la investigación, el es-tudio puede llevarse a cabo sin consentimiento informado, siempre que las razones es-pecíficas para incluir a individuos con una enfermedad que no les permite otorgarconsentimiento informado hayan sido estipuladas en el protocolo de la investigación y elestudio haya sido aprobado por un comité de ética de investigación. El consentimientopara mantenerse en la investigación debe obtenerse a la brevedad posible, del individuoo de un representante legal”.
El punto 30, último de este apartado de principios básicos para toda investigaciónmédica, hace hincapié en la obligación de publicar los resultados, tanto los positivos paralos intereses del promotor e investigador como los negativos. Además en la publicaciónse debe citar la fuente de financiación, las afiliaciones institucionales y cualquier posibleconflicto de intereses.
Finalmente, la Declaración de Helsinki dedica los puntos 31 al 35 a los principiosaplicables cuando la investigación médica se combina con la atención médica.
En este supuesto, la población que participa como sujeto de la investigación secaracteriza por dos aspectos diferenciadores, a saber: existe un potencial beneficio di-recto para ellos del resultado de la investigación y se trata de una población con una de-terminada dependencia del médico que le va a proponer participar en la investigación.
El primero de estos aspectos simboliza el paradigma del principio de Justicia,toda vez que la población que soporta la investigación es la que se puede beneficiar desus resultados. Por el segundo, el sujeto a quien se le propone participar en la investiga-ción es fácilmente convertible en sujeto dependiente del médico y debe extremarse lacautela en todo el proceso de información y de solicitud de participación en el proyectode investigación. En este caso debería aplicarse lo aconsejado en el apartado 26.
24

En el punto 32 se contempla el uso como tratamiento comparativo de un placeboo de ningún tratamiento para evaluar los posibles beneficios, riesgos, costos y eficacia detodo procedimiento nuevo. Al respecto dice: “Los posibles beneficios, riesgos, costos yeficacia de toda intervención nueva deben ser evaluados mediante su comparación con lamejor intervención probada existente, excepto en las siguientes circunstancias:
• El uso de un placebo, o ningún tratamiento, es aceptable en estudios para losque no hay una intervención probada existente.
• Cuando por razones metodológicas, científicas y apremiantes, el uso de un pla-cebo es necesario para determinar la eficacia y la seguridad de una intervenciónque no implique un riesgo, efectos adversos graves o daño irreversible para lospacientes que reciben el placebo o ningún tratamiento. Se debe tener muchí-simo cuidado para evitar abusar de esta opción”.
Abundando en el principio de Justicia, el punto 33 remarca lo ya dispuesto en elpunto 14 al establecer que “al final de la investigación, todos los pacientes que partici-pan en el estudio tienen derecho a ser informados sobre sus resultados y compartir cual-quier beneficio, por ejemplo, acceso a intervenciones identificadas como beneficiosas enel estudio o a otra atención apropiada o beneficios.”
Por su parte la legislación española, en el Artículo 29. del Decreto 223/2004,en relación a este asunto dice que, “Una vez finalizado el ensayo, toda continuación enla administración del medicamento en investigación, en tanto no se autorice el medica-mento para esas condiciones de uso, se regirá por las normas establecidas para el usocompasivo ...”.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
25


El Informe Belmont


Tras la profunda conmoción provocada por la publicación del caso “Tuskegee”, en1974 el gobierno de Estados Unidos creó la Comisión Nacional para la Protección de Su-jetos Humanos. En 1978, en la ciudad de Belmont, California, la comisión presentó suinforme, el Informe Belmont: Principios éticos y pautas para la protección de sujetos hu-manos de la investigación biomédica y de comportamiento.
El encargo a la Comisión fue que deliberara sobre (1) la distinción entre la inves-tigación biomédica y de comportamiento y la práctica médica común y aceptada, (2) lafunción que desempeña la evaluación de criterios riesgo/beneficio para determinar si la in-vestigación, incluyendo sujetos humanos es apropiada, (3) directrices adecuadas para laselección de sujetos humanos que habrán de participar en la investigación y (4) la natu-raleza y definición de un consentimiento informado en diferentes situaciones de investi-gación.
Después de cuatro años de trabajo y amplias consultas se presentó el informe queestá estructurado de la siguiente manera:
A. Límites entre práctica e investigación.
B. Principios éticos básicos.
1. Respeto por las personas.
2. Beneficencia.
3. Justicia.
C. Aplicaciones.
1. Consentimiento informado.
2. Valoración de riesgos y beneficios.
3. Selección de los sujetos.
En general el término “práctica” se refiere a intervenciones diseñadas únicamentepara aumentar el bienestar de un individuo y que tienen una expectativa razonable deéxito. El propósito de la práctica médica o del comportamiento es brindar diagnóstico,tratamiento preventivo o terapia a individuos en particular.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
29

En contraste el término “investigación” designa una actividad concebida para pro-bar una hipótesis, para permitir que se saquen conclusiones y, a partir de ellas, desarro-llar o contribuir al conocimiento generalizable (expresado, por ejemplo, en teorías,principios y formulación de relaciones).
La investigación y la práctica pueden llevarse a cabo juntas cuando la primeraestá diseñada para evaluar la seguridad y eficacia de la segunda.
Entre los principios éticos básicos se definen como relevantes en toda actividadde investigación que incluya seres humanos: los principios de autonomía o respeto por laspersonas, de beneficencia y de justicia.
El principio de autonomía contempla a su vez dos aspectos: (1) que los individuosdeben ser tratados como agentes autónomos; y (2) que las personas con autonomía dis-minuida tienen derecho a protección. Dicho de otro modo que existe el deber de respetarla autonomía y el de proteger a quienes la tienen disminuida.
El respeto al principio de autonomía exige que los sujetos acepten voluntaria-mente participar en la investigación, habiendo recibido toda la información disponi-ble. Por otro lado, la protección a los grupos con autonomía disminuida exige que seaproporcional al grado de autonomía que ostenten. El informe contempla específica-mente el caso de la población reclusa, probablemente debido a los abusos que habíantenido lugar en las prisiones de EE. UU. Esta población plantea un problema derivadodel hecho de que por un lado es una población fácil y sutilmente coercionable pero,por otro, no puede negársele el derecho a presentarse voluntariamente como sujetode investigación.
El principio de beneficencia, en investigación, va más allá de los actos de bondadcon que generalmente se identifica el término y está dotado de la condición de exigenciao de deber.
Se refiere a que no sólo se debe respetar la decisión de las personas y no causar-les daño, sino, también, en procurar su bienestar. Esta obligación de buscar el bien debeser una aspiración de todo proyecto de investigación y debe entenderse como que se debeacrecentar al máximo los beneficios esperables y disminuir los posibles daños, es decir,hacer que la relación beneficio / riesgo sea favorable.
Complementariamente a este principio, aunque no está como tal reconocido en elinforme Belmont, se encuentra el principio de no maleficencia, entendido como la obli-
30

gación de no causar daño. Ésta no es una aspiración sino una obligación primaria y por lotanto es anterior a cualquier tipo de información o de consentimiento.
El principio de justicia intenta responder a la doble pregunta ¿Quién debiera re-cibir los beneficios de la investigación y quién soportar sus cargas?. El principio establecela igualdad con respecto a la dignidad de todas las personas, por lo que no pueden jus-tificarse discriminaciones basadas en criterios raciales, religiosos, ideológicos, económi-cos, sexuales, o vinculados a la selección de personas disponibles fácilmente, comoenfermos o presos. El principio de justicia se corresponde con la correcta selección de lossujetos en investigación.
Ocurre injusticia cuando, como en el siglo XIX y comienzos del XX, las cargas deservir como sujetos de investigación recayeron en gran medida en los pacientes de las sa-las de caridad mientras los beneficios del mejor cuidado médico alcanzaban primaria-mente a los pacientes privados que podían costearse los nuevos tratamientos. Tambiénocurre injusticia en el caso Tuskegee, en los campos de concentración nazis y en investi-gaciones efectuadas en el tercer mundo para encontrar nuevos medicamentos destinadosal mundo desarrollado.
Estos tres principios generales de autonomía, beneficencia y justicia se plasmanen la práctica con las aplicaciones siguientes: Consentimiento informado, valoración deriesgos / beneficios y selección de los sujetos.
El consentimiento informado se basa en la tríada Información, comprensión y vo-luntariedad.
Con respecto a la información, el informe no se define en un contenido claramenteidentificado, y propone un estándar del “voluntario razonable”. “Tal vez se debiera pro-poner un estándar del “voluntario razonable”: el alcance y la naturaleza de la informacióndebieran ser tales que las personas, a sabiendas de que el procedimiento no es necesariopara su cuidado ni, quizás, está completamente entendido, puedan decidir si desean par-ticipar en la ampliación del conocimiento. Aun cuando se anticipe algún beneficio directopara ellos, los sujetos deberían entender claramente el rango de riesgos y la naturaleza”.
El informe Belmont plantea un problema especial cuando al dar información sobre unasunto relevante se pueda poner en riesgo la validez del proyecto. En este caso, propone quees suficiente indicar a los sujetos que se les invita a participar en un proyecto de investigacióndel cual no se revelarán algunos puntos hasta que la investigación haya concluido. No contem-pla la posibilidad de delegar la evaluación ética de este comportamiento en un comité.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
31

Sólo justifica este proceder si: “… es claro que: (1) la declaración incompletaes realmente necesaria para lograr los objetivos de la investigación, (2) dentro de la in-formación retenida no existen riesgos que no sean mínimos* para los sujetos y (3) existeun plan adecuado para informar a los sujetos, cuando sea apropiado, y para participara los sujetos los resultados de la investigación”.
La capacidad de comprensión de una persona es una función que depende dela inteligencia, razonamiento, madurez y lenguaje, por tanto, la manera y el contexto enque se presenta la información, por muy completa que ésta sea, puede invalidar el actode informar en sí mismo. Es necesario adaptar la presentación de la información a lascapacidades del sujeto. En cualquier caso se debe presentar de forma organizada, conel tiempo suficiente para que el sujeto pueda asimilarla y hacer las preguntas pertinen-tes. La Comisión redactora del informe, en ocasiones aconseja incluso practicar unaprueba de comprensión.
En los sujetos con la comprensión limitada debe igualmente informárseles y “laoposición de estos sujetos a participar deberá respetarse, a menos que la investigaciónsignifique recibir una terapia que no estaría a su alcance de otra forma”. Esta excepcióncrea evidentemente una nueva contradicción en el informe, pues una vez más, no con-templa como garantía de correcta interpretación la figura de un comité independienteque vele por los derechos del sujeto. El informe continúa proponiendo que la informa-ción se le dé a una tercera persona, que “deberá ser aquella que esté en las mejores con-diciones de entender la situación del sujeto incompetente y actúe en el mejor interés deesa persona”.
La voluntariedad exige ausencia de coerción y de influencia indebida, es decir,de amenaza de daño o promesa de recompensa exagerada. Sin embargo hay una in-fluencia lícita, fundamentalmente en el caso de que investigación y asistencia se denal unísono, pues es difícil diferenciar ya que entre ambas hay un continuo.
La Valoración de riesgos / beneficios es la aplicación del principio de benefi-cencia. Determina lo adecuado del diseño del proyecto de investigación, informa al co-mité evaluador acerca de la justificación de los riesgos y ayuda a decidir al sujeto sobresu participación. Esta evaluación debe reflejar cuando menos las consideraciones si-guientes:
32
* Se entiende por riesgo mínimo el que es similar al propio de la práctica médica habitual para la misma situación.Es decir que no es más probable ni mayor que el que se asocia a los exámenes médicos de rutina.

“(1) El tratamiento brutal o inhumano de sujetos humanos nunca se justifica mo-ralmente.
(2) Los riesgos deben reducirse a aquellos necesarios para lograr el objetivo de lainvestigación. Se debe determinar si es realmente necesario usar sujetos humanos. Talvez el riesgo nunca pueda ser totalmente eliminado, pero con frecuencia puede reducirsemediante el uso de procedimientos alternos estudiados cuidadosamente.
(3) Cuando la investigación involucra un riesgo significativo de deterioro serio, loscomités de inspección deben ser extraordinariamente estrictos en la justificación del riesgo(generalmente estudiando la posibilidad de beneficio para el sujeto o, en algunos casosraros, asegurándose de que la participación sea voluntaria).
(4) Cuando se involucran poblaciones vulnerables, también deberá demostrarseque su participación es justificada. Estas decisiones se componen de un conjunto de va-riables que incluyen la naturaleza y el grado del riesgo, las condiciones de la población par-ticular involucrada y la naturaleza y el nivel de los beneficios previstos.
(5) Los riesgos y beneficios pertinentes deben ser detallados minuciosamente endocumentos y procedimientos usados en el proceso de obtención del consentimiento in-formado”.
Por último el informe Belmont se refiere a la selección de los sujetos de la inves-tigación como expresión del principio de justicia. Aborda este asunto considerando dos ni-veles en la selección: social e individual.
El nivel social de la justicia significa que deben establecerse grupos o poblacio-nes que deben o no deben participar, por ejemplo adultos antes que niños, o grupos quesólo deben ser invitados bajo determinadas circunstancias.
El nivel individual requiere imparcialidad y no preferencias para favorecer a cier-tos individuos si se esperan buenos resultados o hacer participar a personas “indesea-bles” o marginales si los riesgos son elevados. “Un caso especial de injusticia resulta dela participación de sujetos vulnerables. Ciertos grupos, como minorías raciales, los de po-cos recursos económicos, los seriamente enfermos y los institucionalizados, pueden ser re-queridos constantemente como sujetos de investigación debido a su disponibilidad enlugares donde se conducen investigaciones.”
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
33


Otras Normas y Declaraciones Internacionales


La ética médica entendida como el conjunto de principios que regulan la mora-lidad del acto médico individual o colectivo, forma parte de la compilación de principiosde la bioética, entendida ésta como la que regula la moralidad de cualquier acto relacio-nado con la vida y ésta a su vez, forma parte de la ética general.
Por tanto, la ética médica, es cambiante en función de los cambios que se pro-ducen en la sociedad, e incluso es diferente en función de la población en la que el actomédico se encuentre inmerso. La moralidad de un determinado acto médico puede ser di-ferente cuando se realiza en el seno de una sociedad democrática, de tradición liberal, quecuando se realiza en el seno de una sociedad con fuertes tradiciones, escaso desarrolloeconómico o con un determinismo religioso que influya en todos los aspectos del queha-cer diario. Así, la concepción que la sociedad admite sobre la eutanasia o sobre el abortovaría sobremanera, no ya entre los diferentes continentes sino, incluso en países diferen-tes dentro de la propia Unión Europea.
En un intento de homogeneizar posturas y establecer principios universales, dife-rentes organismos nacionales e internacionales, han abordado el asunto, fundamental-mente después de la segunda guerra mundial, influidos por los hechos descubiertos en loscampos de concentración nazis en Europa.
Las Declaraciones y Códigos más relevantes, diferentes de los de Nüremberg, Hel-sinki y del Informe Belmont, han sido: (1) La Declaración Universal de Derechos Huma-nos de la Asamblea General de Naciones Unidas, de 1948. (2) Pautas éticasinternacionales para la investigación biomédica en seres humanos del Consejo de Organi-zaciones Internacionales de las Ciencias Médicas, (CIOMS) en colaboración con la Orga-nización Mundial de la Salud (OMS) en 1982 y revisadas en 1993 y en 2002. (3)Principios de ética médica de la Asamblea General de Naciones Unidas, de 1982. (4) Có-digo de Reglamentos Federales de los EE. UU. de Norteamérica. (5) Convención sobrederechos humanos y biomedicina del Consejo de Europa de 1997. (6) Normas de BuenaPráctica Clínica de la Conferencia Internacional de Armonización de 1997. (7) Guías ope-racionales para Comités de Ética que evalúan investigación biomédica de la OrganizaciónMundial de la Salud (OMS) en 2000. (8) Declaración Universal sobre Bioética y DerechosHumanos, de la UNESCO en 2005.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
37

La Declaración Universal de Derechos Humanos aprobada por la Asamblea Gene-ral de Naciones Unidas el 10 de diciembre de 1948, proclama como ideal común para to-dos los pueblos y naciones el que todos los seres humanos nacen libres e iguales endignidad y derechos y sin distinción alguna fundada en razones de raza, color, sexo,idioma, religión, opinión política o de cualquier otra índole, origen nacional o social, po-sición económica, nacimiento o cualquier otra condición. Además, no se hará distinciónalguna fundada en la condición política, jurídica o internacional del país o territorio decuya jurisdicción dependa una persona.
A partir de esta proclama inicial (art. 1 y 2), la Declaración va desgranando los di-ferentes derechos que ostentan todos los seres humanos.
De entre ellos, cabe destacar a los efectos que nos interesan, los enumerados enel art. 5 que, textualmente, dice: “Nadie será sometido a torturas ni a penas o tratoscrueles, inhumanos o degradantes.” En 1966 la Asamblea General aprobó el AcuerdoInternacional sobre Derechos Civiles y Políticos, cuyo artículo 7 expresa lo siguiente:“Nadie será sometido a tortura o a un tratamiento o castigo cruel, inhumano o degra-dante. En especial, nadie será sometido sin su libre consentimiento a experimentaciónmédica o científica.”
La relevancia de este artículo la manifiesta la propia Asamblea General de las Na-ciones Unidas, cuando en la Resolución 37/194 de 18 de diciembre de 1982, apruebalos “Principios de ética médica aplicables a la función del personal de salud, especial-mente los médicos, en la protección de personas presas y detenidas contra la tortura yotros tratos o penas crueles, inhumanos o degradantes”.
El Consejo de Organizaciones Internacionales de las Ciencias Médicas, (CIOMS)en colaboración con la Organización Mundial de la Salud (OMS) en 1982 (y revisadas en1993 y en 2002), publicaron las Pautas Éticas Internacionales para la Investigación Bio-médica en Seres Humanos. El objetivo era cómo aplicar en forma eficaz los principios éti-cos que deben regir la investigación biomédica en seres humanos, especialmente en lospaíses en desarrollo, dadas sus circunstancias socioeconómicas, leyes, reglamentos y susdisposiciones administrativas.
La versión de 1982 está organizada en una serie de pautas y comentarios a lasmismas. Las pautas están expresadas de manera clara y más o menos precisa, pero loscomentarios, tratan generalmente de las excepciones y están redactados con un len-guaje más ambiguo de forma que, en muchas ocasiones, contradicen a las propiaspautas.
38

En los años inmediatamente siguientes, a finales del siglo XX, el mundo conoce lapandemia VIH/SIDA y se produce una auténtica carrera por realizar ensayos con vacunas ymedicamentos para dicha enfermedad. Es también una época en que aparecen importantesavances en medicina y biotecnología, proliferan los ensayos multicéntricos, multinacionales,controlados, efectuados en países de bajos recursos y en los que se utilizan comparadores dedudosa efectividad (Grupos de población vulnerables y uso de placebos como comparadores)y cuyos patrocinadores e investigadores son externos a los países donde se desarrollan.
La Declaración de Helsinki se revisa seis veces y dos más de forma parcial, (1975,1983, 1989, 1996, 2000, 2002, 2004 y 2008) y las propias pautas CIOMS – OMS, se re-visan en 1993 y en 2002. La versión de 1993, debido a los desacuerdos por los problemaséticos apuntados anteriormente, se empieza a revisar en 1998 y no se consigue un acuerdohasta 2002 en que se publica la versión actual que consta de una introducción, una descrip-ción de instrumentos y pautas anteriores, unos principios éticos generales, un preámbulo y21 pautas.
Su objetivo, como en las versiones anteriores, es orientar, especialmente a los paí-ses en desarrollo y de escasos recursos, en la definición de pautas nacionales sobre ética dela investigación biomédica, aplicando estándares éticos en condiciones locales, y estable-ciendo o redefiniendo mecanismos adecuados para la evaluación ética de la investigación enseres humanos.
Las Pautas no abordan algunas áreas de investigación como la genética humana(aunque se menciona en la pauta 18 al tratar de la confidencialidad en investigación gené-tica) o la de la investigación en productos de la concepción (embriones y fetos, y tejidos fe-tales), debido a la imposibilidad de llegar a acuerdos sobre asuntos tan controvertidos.
En cuanto a los Principios Éticos Generales, desarrolla los conceptos clásicos de Au-tonomía, Beneficencia, No Maleficencia y Justicia.
En el preámbulo diferencia los términos investigación biomédica, práctica de la me-dicina o intervenciones de salud pública e investigación biomédica combinada con la medi-cina asistencial.
La primera se refiere a un tipo de actividad destinada a desarrollar o contribuir al co-nocimiento generalizable. La práctica de la medicina o las intervenciones en salud pública,están diseñadas para contribuir directamente a la salud de los individuos o comunidades. Am-bas pueden darse simultáneamente, en concordancia con el párrafo 35 de la Declaración deHelsinki: “Cuando en la atención de un enfermo las intervenciones probadas han resultado
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
39

ineficaces o no existen, el médico, después de pedir consejo de experto, con el consenti-miento informado del paciente o de un representante legal autorizado, puede permitirse usarintervenciones no comprobadas, si, a su juicio, ello da alguna esperanza de salvar la vida, res-tituir la salud o aliviar el sufrimiento. Siempre que sea posible, tales intervenciones debenser investigadas a fin de evaluar su seguridad y eficacia. …”.
A continuación se detallan 21 Pautas, relativas a:
40
PAUTA MATERIA
1 Justificación ética y validez científica de la investigación biomédica en seres humanos
2 Comités de evaluación ética3 Evaluación ética de la Investigación patrocinada externamente4 Consentimiento informado individual 5 Obtención del CI: Información esencial para potenciales sujetos6 Obtención del CI: Obligaciones de patrocinadores e investigadores7 Incentivos para participar en una investigación8 Beneficios y riesgos de participar en un estudio9 Limitaciones especiales del riesgo cuando se investiga en Individuos
Incapaces de dar su consentimiento informado 10 Investigación en poblaciones y comunidades con recursos limitados11 Elección del control en ensayos clínicos 12 Distribución equitativa de cargas y beneficios en la selección de grupos
de sujetos en la investigación 13 Investigación en que participan personas vulnerables14 Investigación en que participan niños 15 Investigación en que participan individuos cuyos trastornos mentales o
conductuales los incapacitan para dar adecuadamente consentimiento informado16 Las mujeres como sujetos de investigación 17 Mujeres embarazadas como sujetos de investigación18 Protección de la confidencialidad19 Derecho a tratamiento y compensación de sujetos perjudicados20 Fortalecimiento de la capacidad de evaluación ética y científica
y de la investigación biomédica21 Obligación ética de los patrocinadores externos de proporcionar servicios
para la atención de salud

El Código de los Reglamentos Federales de los EE.UU. de Norteamérica, tambiénconocidos como “Regla Común” (The Common Rule), y que son la compilación de regla-mentos promulgados por el poder ejecutivo en desarrollo de disposiciones del Congreso,destina el Título 45 al Bienestar Social. La sección 46 de este título contiene la norma-tiva federal relativa a la protección de sujetos humanos en investigación (45cfr46). En élse exponen las políticas consideradas básicas del gobierno de los EE.UU. para la protec-ción de los sujetos y se aplican a todas aquellas investigaciones en la que participen su-jetos humanos patrocinadas por el gobierno o alguna de las Agencias Federales. Noobstante, los Directores de Departamento o Agencias Federales pueden decidir que a unadeterminada investigación no le sean de aplicación las reglas de este Código.
La Regla Común exige que exista una aprobación previa por un comité de ética,el consentimiento informado y documentado por escrito, que el reclutamiento de los par-ticipantes en la investigación sea equitativo, que se de una protección especial a las po-blaciones o grupos vulnerables y que haya una revisión continua de la investigaciónaprobada.
Como hemos visto, las normas sobre investigación con participación de seres hu-manos, configuran la sección 46 del Título 45 de la Regla Común y están organizadas encuatro apartados:
En el primer apartado, (Inciso A), además de definir la política federal al respecto,regula los Comités Éticos (IRB por sus siglas en Inglés), sus funciones, procedimientos,criterios, registros, etc. y el consentimiento informado.
A continuación en los Incisos B, C y D respectivamente, se establecen proteccio-nes adicionales con respecto a: (B) la investigación, desarrollo y actividades relacionadasque incluyan fetos, mujeres embarazadas y fertilización humana in Vitro, (C) investigacio-nes biomédicas y de comportamiento que incluyan prisioneros como sujetos y (D) niñosincluidos como sujetos de investigación.
Por lo que respecta a la Unión Europea, desde 1965, ha emitido Directivas rela-tivas a la investigación biomédica en humanos, destinadas a que los estados miembros in-corporen sus conceptos a las legislaciones nacionales.
Estas Directivas se caracterizan comparativamente con las Declaraciones, Códigos,Pautas etc., en que no son recomendaciones éticas dirigidas a los investigadores, promo-tores, comités de ética, etc. sino que se orientan a los estados miembros, para su incor-poración al ordenamiento jurídico propio de cada uno de ellos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
41

Los países desarrollados han promulgado leyes relacionadas con los medicamentosdesde los años 60. Así en 1964 legislan Noruega y Suecia, Inglaterra en 1968, Suiza y Ale-mania en 1976, Austria, Bélgica y Grecia lo hacen en 1983 y Japón ha modificado su le-gislación al respecto en varias ocasiones desde 1961. España promulga la Ley delMedicamento el 20 de diciembre de 1990.
En el año 2000, la Organización Mundial de la Salud (OMS), tomando concienciade que todas las guías y declaraciones internacionales vistas hasta ahora, requieren que losproyectos de investigación que se realicen en el campo de la biología y de la medicina conseres humanos, sean revisados en sus aspectos éticos y científicos, en el consentimiento in-formado, así como de la protección apropiada de los sujetos incapaces de consentir, pu-blicó las Guías Operacionales para Comités de Ética que Evalúan Investigación Biomédica.
El propósito de estás guías es asegurar la calidad en la revisión de los aspectos éti-cos, con vocación universal, complementando las leyes, reglas y prácticas existentes y, fun-damentalmente, para que sirvan de base sobre la que los Comités de Ética puedan elaborarsus Procedimientos Normalizados de Trabajo (PNT).
La legislación española incorpora la obligación de los CEIC de contar con estos PNTen el art. 14.2 del R.D, 223/2004, de 6 de febrero, por el que se regulan los ensayos clí-nicos con medicamentos, contemplando como contenido de los mismos todos los aspectosconsiderados en la Guía de la OMS.
A la luz de lo anterior, y resolviendo que es necesario que la comunidad internacio-nal establezca principios universales tal como consta en la Declaración sobre la Ciencia y eluso del saber científico (Declaración de Budapest) de la Conferencia Mundial sobre la cien-cia (UNESCO – ICSU) de julio de 1999, la Conferencia General de la UNESCO, en su 33ªsesión de 19 de octubre de 2005, proclama la Declaración Universal sobre Bioética y De-rechos Humanos, con la finalidad de:
“a) proporcionar un marco universal de principios y procedimientos que sirvan deguía a los Estados en la formulación de legislaciones, políticas u otros instrumentos en elámbito de la bioética.
b) orientar la acción de individuos, grupos, comunidades, instituciones y empresas,públicas y privadas.
c) promover el respeto de la dignidad humana y proteger los derechos humanos, ve-lando por el respeto de la vida de los seres humanos y las libertades fundamentales, de con-formidad con el derecho internacional relativo a los derechos humanos.
42

d) reconocer la importancia de la libertad de investigación científica y las reper-cusiones beneficiosas del desarrollo científico y tecnológico, destacando al mismo tiempola necesidad de que esa investigación y los consiguientes adelantos se realicen en el marcode los principios éticos enunciados en esta Declaración y respeten la dignidad humana,los derechos humanos y las libertades fundamentales.
e) fomentar un diálogo multidisciplinario y pluralista sobre las cuestiones de bioé-tica entre todas las partes interesadas y dentro de la sociedad en su conjunto.
f) promover un acceso equitativo a los adelantos de la medicina, la ciencia y la tec-nología, así como la más amplia circulación posible y un rápido aprovechamiento compar-tido de los conocimientos relativos a esos adelantos y de sus correspondientes beneficios,prestando una especial atención a las necesidades de los países en desarrollo.
g) salvaguardar y promover los intereses de las generaciones presentes y venideras.
h) destacar la importancia de la biodiversidad y su conservación como preocupa-ción común de la especie humana”.
Entre sus principios se reafirman los de respeto a los derechos de autonomía y pro-tección de los sujetos, de beneficencia y de justicia, de privacidad y confidencialidad, derespeto a la diversidad, a la solidaridad, cooperación, responsabilidad social y aprovecha-miento compartido de los beneficios, así como a la protección de las generaciones futu-ras, del medio ambiente, la biosfera y la biodiversidad.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
43


Normas de Buena Práctica Clínica


En un mundo globalizado, la información sobre la seguridad, la calidad y la efica-cia de los medicamentos necesita ser creíble, es decir los intereses científicos, éticos yeconómicos derivados de la investigación biomédica en humanos, no pueden estar some-tidos al albur de datos con niveles de credibilidad no garantizados, por lo que es absolu-tamente necesario establecer herramientas que permitan a las diferentes partes implicadastener la seguridad de que los resultados comunicados de cualquier investigación biomé-dica en humanos cumple con esas premisas.
Para desarrollar estas herramientas, en el año 1997 se acordó la Guía ICH de BPCo Guía de la Conferencia Internacional de Armonización, (por sus siglas anglosajonas) deBuenas Prácticas Clínicas (CPMP/ICH/135/95/Step5).
Para ello, con el objetivo de disponer de una norma unificada que permitiera estaaceptación mutua de resultados, se reunieron las autoridades reguladoras de la Unión Eu-ropea (CPMP), de los Estados Unidos de Norteamérica (FDA) y de Japón (MHW), y lasasociaciones de la industria farmacéutica de estos mismos países, (EFPIA, PhRMA yJPMA). Además como miembros observadores, participaron la OMS, Canadá, la EFTA (Aso-ciación Europea de Libre Comercio, formada por: Noruega, Liechtenstein, Islandia y Suiza)y la Federación Internacional de la Industria Farmacéutica (IFPMA).
Así se consensúa una norma internacional de calidad ética y científica aplicableal diseño, realización, registro y comunicación de los ensayos clínicos en los que partici-pen seres humanos. El cumplimiento de esta norma proporciona una garantía pública dela protección de los derechos, la seguridad y el bienestar de los sujetos del ensayo deacuerdo con los principios de la Declaración de Helsinki, así como también garantiza lacredibilidad de los datos del ensayo clínico.
Esta Guía, se organiza en una introducción y ocho partes:
1. Glosario.
2. Principios de las BPC de la ICH.
3. Comité ético de investigación clínica (CEIC).
4. Investigador.
5. Promotor.
6. Protocolo del ensayo clínico y enmiendas al protocolo.
7. Manual del investigador.
8. Documentos esenciales para la realización de un ensayo clínico.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
47

En su elaboración se han tenido en cuenta las normas anteriores existentes, es-pecialmente las de la Unión Europea, Japón y los Estados Unidos, además de las proce-dentes de Australia, Canadá, Países Nórdicos y la Organización Mundial de la Salud(OMS).
Los Principios de la BPC de la ICH definidos en esta guía son:
1. “Los ensayos clínicos deben realizarse de acuerdo con los principios éticos quetienen su origen en la Declaración de Helsinki, y que sean coherentes con la guíade la BPC y la legislación vigente.
2. Antes de iniciar un ensayo, deberán considerarse los riesgos e inconvenientesprevisibles en relación con el beneficio esperado, tanto para el sujeto individualdel ensayo como para la sociedad. Un ensayo deberá iniciarse y continuarse úni-camente en el caso de que los beneficios previstos justifiquen los riesgos.
3. Los derechos, la seguridad y el bienestar de los sujetos de un ensayo son las con-sideraciones más importantes y deberán prevalecer sobre los intereses de la cien-cia y de la sociedad.
4. La información clínica y no clínica disponible sobre un medicamento en investi-gación deberá ser suficiente para avalar el ensayo clínico propuesto.
5. Los ensayos clínicos deberán estar científicamente justificados y estar descritosen un protocolo claro y detallado.
6. El ensayo se deberá realizar de acuerdo con el protocolo que previamente ha re-cibido un dictamen favorable de un CEIC.
7. El cuidado médico que reciben los sujetos y las decisiones médicas tomadas ensu nombre serán siempre responsabilidad de un médico cualificado o, en su caso,un odontólogo cualificado.
8. Cada individuo implicado en la realización de un ensayo deberá estar cualificado,por su titulación, formación y experiencia, para realizar sus tareas y responsabi-lidades respectivas.
9. Se deberá obtener el consentimiento informado, otorgado de forma libre, de cadasujeto antes de su participación en el ensayo clínico.
10. Toda la información del ensayo clínico deberá ser registrada, manejada y archi-vada de forma que permita su comunicación, interpretación y verificación exac-tas.
11. Se deberá proteger la confidencialidad de los registros que pudieran identificar alos sujetos respetando la privacidad y las normas de confidencialidad de acuerdocon los requisitos legislativos pertinentes.
48

12. Los medicamentos en investigación deberán fabricarse, manejarse y almacenarsede acuerdo con las Normas de Correcta Fabricación (NCF) pertinentes y se de-berán utilizar de acuerdo con el protocolo aprobado.
13. Se implantarán sistemas con procedimientos que aseguren la calidad de cada as-pecto del ensayo.”
Respecto de estos principios cabe decir que supedita los principios éticos en larealización de los ensayos clínicos a los contenidos de la Declaración de Helsinki. Te-niendo en cuenta que el texto de la Guía de BPC es de 1997, resulta que desde ese añola Declaración de Helsinki se ha modificado en Edimburgo en el año 2000, se han efec-tuado dos Notas de clarificación en 2002 y 2004 respectivamente y existe una nueva ver-sión de octubre de 2008 aprobada por la Asamblea General de la AMM en Seúl, por tanto,si la nueva redacción de la Declaración de Helsinki fuera contradictoria con los postula-dos de la Guía de BPC, estos deberían supeditarse a los principios allí contenidos.
Respecto del Comité Ético de Investigación Clínica, se enumeran las responsabi-lidades, composición, funciones, actividades, procedimientos, documentos y archivo de losmismos.
Entre las responsabilidades, la primera es salvaguardar los derechos, la seguri-dad y el bienestar de todos los sujetos del ensayo, poniendo especial cuidado en los en-sayos que incluyan sujetos de poblaciones vulnerables.
Enumera los documentos que deben ser revisados por el CEIC y todas sus modifi-caciones relevantes y, tras recomendar la revisión indica la necesidad de informar en untiempo razonable, identificando los documentos revisados con expresión de versión y fecha.
El CEIC debe emitir juicio acerca de la idoneidad del investigador y determinar uncalendario de seguimiento. Aun cuando la legislación no determine esta circunstancia, elCEIC tiene la obligación ética de establecer el calendario aludido, con tanta más frecuen-cia en los controles como riesgo presente la investigación.
En beneficio de los derechos de los sujetos, el comité tiene la potestad de hacersugerencias al protocolo si éstas mejoran la protección de los mismos. Si se da esta cir-cunstancia estaremos ante una autorización condicionada al cumplimiento de las mismasy el investigador tiene la obligación de seguirlas, puesto que en caso contrario debe en-tenderse que no hay autorización para el ensayo. Igualmente puede decidir proponer la fi-nalización el ensayo en el centro o retirar la autorización.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
49

En lo que se refiere al investigador deberá estar cualificado como científico e in-vestigador, justificándolo mediante un curriculum vitae que además, demuestre conoci-miento de las normas de BPC.
Respecto al medicamento en investigación, el investigador debe conocer a fondotoda la información disponible en el Manual del investigador, ficha técnica etc.
El investigador principal es responsable de que los investigadores colaboradorestengan la cualificación adecuada para las funciones que les van a ser delegadas y de queconocen el protocolo y el manual del investigador a la perfección.
Debe demostrar mediante datos retrospectivos que es capaz de reclutar el nº desujetos propuesto en el protocolo en el tiempo previsto para el estudio y disponer de lasinstalaciones apropiadas. En relación a este punto es importante tener en cuenta que lacobertura del seguro generalmente especifica el nº de sujetos y el periodo de validez dela póliza, por lo que una modificación en el reclutamiento o en el periodo del ensayo, nocorrelacionada con la modificación de la cobertura de la póliza del seguro, posiblementederivaría hacia el investigador, la institución y el promotor, la garantía financiera estable-cida en la legislación española.
En relación a la asistencia médica del sujeto, la responsabilidad será siempre deun médico (u odontólogo) tanto durante el ensayo como después, en aquellas situacionesen que ésta se necesite como consecuencia directa o indirecta del ensayo.
La relación entre investigador y CEIC se ha visto modificada tras la publicación delDecreto 223/2004, puesto que tanto la solicitud inicial de autorización con toda la docu-mentación pertinente como las que se produzcan posteriormente ante modificaciones re-levantes, las debe efectuar el promotor.
Si el CEIC, en uso de las atribuciones que le asigna el artículo 17.f del RD,223/2004 y las normas de BPC, determina un calendario de seguimiento mediante laemisión de informes periódicos por el investigador, éste tiene la obligación de cumpli-mentarlos. En caso contrario, al incumplir las condiciones de la autorización, ésta dejaríade tener validez.
Si así lo determina el CEIC, deberá informar de los acontecimientos adversos gra-ves y de las muertes acaecidas, además de la comunicación al promotor. Igualmente, debeinformar a los sujetos del ensayo en caso de finalización prematura del mismo por partedel promotor y garantizar la continuidad del tratamiento y seguimiento.
50

Si la finalización prematura es decisión del investigador, deberá informar al promotor,a la institución y al CEIC mediante informe razonado emitido a la mayor brevedad posible.
Caso de que el ensayo se finalice por recomendación del CEIC, el investigador de-berá informar al promotor y a la institución.
El protocolo es un compromiso que el investigador ha firmado y cuyo cumplimientoestricto debe respetarse en todos los casos, salvo que sea necesario para garantizar la se-guridad de los sujetos de la investigación.
Una desviación frecuente consiste en incumplimientos en la frecuencia de las vi-sitas protocolizadas, más o menos justificados por la dificultad de compaginar éstas conla actividad ordinaria de los investigadores. Esta práctica constituye una desviación delprotocolo que condiciona falta de credibilidad en los datos de la investigación, puesto queéstos no se corresponden con la respuesta esperada en un determinado momento, trasuna dosis o dosis acumulada del tratamiento sino que son la respuesta tras una situacióndiferente, lo que debería conllevar la desestimación de los datos del sujeto en cuestióndel resultado final del ensayo.
La custodia, conservación y dispensación del medicamento en investigación en Es-paña, según el art. 2.6 de la Ley 29/2006, corresponde a los Servicios de Farmacia delhospital o de las estructuras de Atención Primaria, pero la responsabilidad del investiga-dor no está delegada sino que, en todo caso, es compartida.
Deben seguirse los procedimientos estrictos de aleatorización y, en los estudiosciegos, documentar cualquier desenmascaramiento efectuado en el ensayo.
La obtención del consentimiento informado constituye uno de los aspectos ética-mente más importantes de todo el proceso de realización de un ensayo clínico. Constituyela máxima expresión del respeto al derecho a la autonomía del sujeto.
En la fase de reclutamiento de cada sujeto debe proporcionársele toda la informa-ción disponible, adaptando el lenguaje y las explicaciones a la capacidad de comprensiónde cada uno, informándole que la negativa a participar en el ensayo no va a significar enningún caso una discriminación negativa para él y de la posibilidad de retirarse en cual-quier momento, No se pueden ofrecer ni expectativas exageradas en la obtención de re-sultados ni pérdidas de beneficios si no acepta la participación, pues ambas situacionesserían coacciones intolerables para los postulados éticos de la Declaración de Helsinki ydemás normas éticas internacionalmente aceptadas. Incluso, si el sujeto es muy depen-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
51

diente del investigador, la inclusión en el ensayo debe ser planteada por otro miembro enque no se dé dicha circunstancia.
Debe prestarse especial atención en el proceso de obtención del consentimiento informado,cuando la población diana del ensayo tiene la autonomía limitada para decidir.
Así, en el caso de sujetos menores de edad o enfermos mentales graves con la ca-pacidad intelectual alterada, u otros colectivos cuyo reclutamiento sólo pueda ser efec-tuado a través del representante legal, el sujeto deberá ser informado en la medida quesu capacidad permita y, si es capaz, deberá firmar también el consentimiento informado.
Si se trata de menores de edad cuyos representantes legales sean los padres, anteel posible desconocimiento de la situación legal de ellos como pareja, se deberá solicitarla firma de ambos padres, excepto que se atestigüe mediante resolución judicial o admi-nistrativa que quien otorga el consentimiento es el único que ostenta la guarda y custo-dia o la patria potestad del menor.
Cuando el ensayo carezca de beneficio terapéutico directo para los sujetos de es-tos colectivos con la capacidad disminuida, sólo podrán realizarse siempre que se cum-plan las siguientes condiciones:
Que no se puedan obtener los objetivos del ensayo mediante un ensayo con suje-tos que pueden dar su consentimiento informado personalmente, que estos objetivos seande interés para la población que se investiga y que los sujetos estén afectados por algunaenfermedad relacionada con el estudio en cuestión.
Que el riesgo previsible para el sujeto sea mínimo, (es decir, no superior al exis-tente en la práctica médica asistencial ordinaria) y que sus intereses prevalezcan sobre losde la ciencia y los de la sociedad.
Que el ensayo no esté prohibido por la ley.
Que se solicite expresamente el dictamen favorable del CEIC para la inclusión detales sujetos y que por tanto, el dictamen favorable cubra este aspecto.
Que el consentimiento informado se obtenga según lo visto anteriormente.
En mujeres gestantes o en periodo de lactancia, sólo se podrán realizar ensayosclínicos sin beneficio directo para ellas cuando el Comité Ético de Investigación Clínica
52

concluya que no suponen ningún riesgo previsible para su salud ni para la del feto o niño,y que se obtendrán conocimientos útiles y relevantes sobre el embarazo o la lactancia.
Cuando exista la posibilidad de que en un ensayo se recluten enfermos en situa-ción de urgencia que puedan no estar en condiciones de otorgar su consentimiento nitampoco sus representantes legales, esta circunstancia deberá estar explicitada en el pro-tocolo y autorizada por el CEIC.
En todos estos casos de otorgamiento del consentimiento por representante le-gal, cuando la decisión del mismo pueda suponerse contraria a los intereses del menor odiscapacitado, deberá informarse de esta circunstancia a la autoridad competente segúndisposición de la legislación civil. (Artículo 9.3 de la Ley 1/2003, de 28 de enero, de de-rechos e información al paciente de la Comunidad Valenciana.
Tras haber informado al posible sujeto y haberle facilitado la documentación per-tinente, debe dejarse transcurrir el tiempo necesario para que pueda meditar sin la pre-sión psicológica de la consulta y de la presencia de la persona que le ha informado. Noes admisible que se le exija una decisión en ese mismo momento.
La información debe contener todos los aspectos enumerados en la legislación envigor. Entre esta legislación debe tenerse en cuenta, entre otras, la Ley de protección dedatos de carácter personal.
Es obligación del investigador garantizar la exactitud de los datos de la investiga-ción y la trazabilidad de los mismos con sus documentos fuente. Cualquier modificaciónen los datos anotados debe ser efectuada de forma que no se enmascare lo anotado pre-viamente y debe ser fechada, firmada y explicada si procede.
El investigador es responsable de la custodia de toda la documentación del en-sayo, durante el tiempo establecido por la ley, y debe garantizar que la misma no esté so-metida a riesgos de destrucción accidental. Esta obligación en todo caso es compartidacon la Institución donde se realiza el ensayo.
En cuanto al promotor, también delimita sus responsabilidades en lo que se re-fiere a: control y garantía de calidad, organización de investigación por contrato (CRO), pe-ricia médica, diseño del ensayo, dirección del ensayo, manejo de datos y preparación dedocumentos, selección del investigador, asignación de las responsabilidades y funciones,indemnización a los sujetos e investigadores, financiación, notificación/solicitud a las au-toridades reguladoras, confirmación de la evaluación por el CEIC, información sobre los
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
53

medicamentos, fabricación, envasado, etiquetado y codificación de los medicamentos eninvestigación, suministro y manejo de los mismos, acceso a los documentos, informaciónde seguridad, notificación de reacciones adversas medicamentosas, monitorización, audi-toria, incumplimiento, finalización prematura o suspensión de un ensayo, informes delensayo, ensayos multicéntricos.
En el apartado 6 se estructura el contenido del protocolo y en el 7 el del manualdel investigador, detallando los apartados con que deben constar cada uno de ellos.
En los ensayos clínicos con medicamentos ya comercializados en cualquier Estadomiembro de la Unión Europea, cuando el medicamento se utilice en las condiciones deuso autorizadas, el manual del investigador podrá reemplazarse por la ficha técnica auto-rizada (artículo 2.ñ de la Orden SCO/256/2007, de 5 de febrero y artículo 8.2 de la Di-rectiva 2005/28/CE de la Comisión de 8 de abril de 2005). Este reemplazo no será posiblesi el objeto del ensayo es demostrar nuevas indicaciones o si el MI se utiliza en condicio-nes distintas a las contempladas en la ficha técnica.
Por último, en el apartado 8 se indican los documentos esenciales del ensayo, esdecir, aquellos “… que individualmente y colectivamente permiten la evaluación del des-arrollo de un ensayo y la calidad de los datos producidos. Estos documentos sirven parademostrar el cumplimiento del investigador, promotor y monitor con las normas de BuenaPráctica Clínica y con todos los requisitos reguladores pertinentes”.
Son también aquellos que “…inspeccionan las autoridades reguladoras comoparte del proceso realizado para confirmar la validez del desarrollo del ensayo y la integri-dad de los datos recogidos”.
El cumplimiento de sus principios, además de ser una exigencia legal en todos lospaíses firmantes que las han incorporado a su ordenamiento jurídico, es un compromisoético y filosófico de todos aquellos que intervienen en cualquier investigación biomédicacon participación de seres humanos. Es un compromiso ético con la Declaración Univer-sal de Derechos Humanos y con los principios universales de respeto a la Autonomía delsujeto, de Beneficencia y de Justicia. Es un compromiso ético para anteponer los intere-ses del sujeto de la investigación a los de la ciencia, de la sociedad y de los legítimos delinvestigador, del promotor, etc.
54

Normativa aplicable en España


España, a su ingreso en la CEE en 1986, incorporó las Directivas, en la ley25/1990, de 20 de diciembre, del medicamento, dedicando todo el Título III (artículos59 al 69 ambos inclusive) a los ensayos clínicos.
Si las leyes de los años sesenta exigían seguridad, el escándalo de la Talidomidacondicionó modificaciones cualitativas y además de seguridad, a partir de ese momento,se exige también eficacia demostrada mediante la realización de ensayos clínicos contro-lados. A partir de los años 90, además de lo anterior, se exigen condiciones para un usoracional de los medicamentos.
Como dice la propia ley 25/1990 en su preámbulo: “El objetivo primordial de laLey es contribuir a la existencia de medicamentos seguros, eficaces y de calidad, correc-tamente identificados y con información apropiada.
Para conseguirlo, determina:
A) El principio de intervención pública, sometiendo la comercialización de medi-camentos a autorización sanitaria y registro previos, que a estos efectos tienencarácter constitutivo y que determina que los medicamentos sean legalmentereconocidos y no clandestinos.
B) Una lista cerrada de las categorías de medicamentos legales.
C) Las condiciones a las que se debe ajustar la investigación de medicamentos,especialmente en personas.
D) Los criterios que deben regir el proceso de evaluación, previo a la autorización, dela especialidad farmacéutica para comprobar que se puede poner en el mercado:
1) Alto nivel técnico, garantizado con la realización y firma por expertos califi-cados de los estudios y protocolos; definición de procedimientos correctosde laboratorio y clínicos y normas de correcta fabricación.
2) Producto seguro, eficaz, de calidad, correctamente identificado, con infor-mación apropiada y actualizado según el progreso técnico, lo cual se garan-tiza con estudios analíticos, farmacológicos, toxicológicos y clínicos,controles de calidad, denominaciones, etiquetado, envase, ficha técnica y
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
57

prospecto reglados y autorización de validez quinquenal, todo ello conformea las Directivas Comunitarias.
3) Empresa con capacidad, garantizada con la oportuna autorización.
4) Actuación administrativa responsable, ágil, neutral, rigurosa, cierta y flexi-ble, asegurada con la atribución de competencias a un órgano especializado;un procedimiento detallado, licencias y fichas técnicas normalizadas, orga-nizadas en un registro e informatizadas y con singularidades cuando venganexigidas por las circunstancias del producto.
D) La Ley regula también las condiciones de la fabricación y del tráfico exteriorcon instrumentos tales como la Real Farmacopea Española, el Formulario Na-cional y las normas de correcta fabricación.
D) El sistema de intervención pública prosigue una vez que el medicamento espuesto a disposición de los profesionales sanitarios y del público. Se regula lavigilancia de reacciones adversas, ratificando el deber de declararlas y dandolas normas básicas de funcionamiento del sistema español de farmacovigilan-cia como elemento integrador de los planes y programas realizados por las di-ferentes Administraciones Públicas y profesionales sanitarios de una parte y porla industria farmacéutica de otra.
D) También establece la revisión de medicamentos para ajustar los ya disponiblesa los requisitos de la Ley”.
Posteriormente desarrolló el Real Decreto 561/1993 de 16 de abril, por el quese establecen los requisitos para la realización de ensayos clínicos con medicamentos.Éste actualiza y sustituye al Real Decreto 944/1978, de 14 de abril, y la Orden ministe-rial de 3 de agosto de 1982.
Este Real Decreto, al dictarse de acuerdo a la directiva 91/507/CEE, de 19 de ju-lio, que modifica el anexo de la directiva 75/318/CEE, de 20 de mayo, introduce las Nor-mas de Buena Práctica Clínica, como requisito imprescindible en la realización de todoslos ensayos clínicos en todas las fases, incluyendo aquellos de biodisponibilidad y bioe-quivalencia. Estas normas pretenden garantizar que los ensayos clínicos sean diseñados,realizados y comunicados de modo que se asegure que los datos sean fiables y que seprotejan los derechos de los sujetos.
Como vemos, en este momento de la historia de la investigación biomédica, apa-rece una nueva necesidad consistente en la fiabilidad de los datos y de los resultados, ne-cesaria para el mutuo reconocimiento entre las Administraciones Sanitarias de losdiferentes Estados con respecto a los resultados de los ensayos clínicos realizados.
58

Así, se definen las Normas de Buena Práctica Clínica como el conjunto de normasinternacionales de calidad ética y científica aplicables al diseño, realización, registro ycomunicación de los ensayos clínicos en los que participen seres humanos. El cumpli-miento de estas normas proporciona una garantía pública de la protección de los derechos,la seguridad y el bienestar de los sujetos del ensayo de acuerdo con los principios de laDeclaración de Helsinki, así como también garantiza la credibilidad de los datos del en-sayo clínico.
Paralelamente, en el año 1997, el Consejo de Europa, propone a los estadosmiembros, con la posibilidad de adhesión de terceros estados, el Convenio de Asturias deBioética, Convenio para la protección de los Derechos Humanos y la dignidad del ser hu-mano con respecto a las aplicaciones de la Biología y la Medicina.
Como aspectos relevantes del mismo, cabe destacar la definición sobre la prima-cía del ser humano al expresar en el artículo 2 que “el interés y el bienestar del ser hu-mano deberán prevalecer sobre el interés exclusivo de la sociedad o de la ciencia.”.También introduce el derecho al respeto a la vida privada, a la no discriminación de unapersona a causa de su patrimonio genético y limita las pruebas e intervenciones sobre elgenoma humano. Se prohíbe la constitución de embriones humanos con fines de experi-mentación.
Se regula la extracción de órganos y de tejidos de donantes vivos para trasplantesy se prohíbe el lucro en la utilización del cuerpo humano y sus partes, así como el uso deuna parte del cuerpo humano con una finalidad distinta de aquella para la que hubiera sidoextraída.
Obliga a reparar el daño injustificado que una persona haya podido sufrir como re-sultado de una intervención e indica a los estados miembros y a los terceros que puedanadherirse que “...deberán prever sanciones apropiadas para los casos de incumplimientode lo dispuesto en el presente Convenio”.
Posteriormente, en el año 2001, el Parlamento Europeo y el Consejo, apruebanla Directiva 2001/20/CE, de 4 de abril, sobre la aplicación de Buenas Prácticas Clínicasen la realización de ensayos clínicos de medicamentos de uso humano. En España, el ar-tículo 125 de la Ley 53/2002, de 30 de diciembre, de medidas fiscales, administrativasy del orden social, introduce diversas modificaciones en el título III de la Ley 25/1990,de 20 de diciembre, del Medicamento. Por otra parte, en el panorama sanitario español,se publica la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía delpaciente y de derechos y obligaciones en materia de información y documentación clí-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
59

nica. La Comunidad Valenciana, a su vez, aprueba la ley 1/2003, de 28 de enero, de de-rechos e información al paciente de la Comunidad Valenciana.
El quehacer médico siempre ha estado marcado (Juramento de Hipócrates) por elsecreto profesional, considerándose como tal la protección de datos de carácter sanitarioy personal, pero esta obligación se acentúa en España con los requerimientos contenidosen la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter per-sonal.
Esta ley, en lo que concierne al tratamiento de los datos personales, tiene por ob-jeto garantizar y proteger, las libertades públicas y los derechos fundamentales de las per-sonas físicas, y especialmente de su honor e intimidad personal y familiar.
Declara datos especialmente protegidos aquellos que se refieren a la salud de laspersonas y ordena que sólo puedan ser recabados, tratados y cedidos cuando, por razonesde interés general, así lo disponga una Ley o el afectado consienta expresamente (art. 7).Sólo podrán ser comunicados a un tercero para el cumplimiento de fines directamente re-lacionados con las funciones legítimas del cedente y del cesionario con el previo consen-timiento del interesado (art. 11). Todo tratamiento por cuenta de terceros deberá estarregulado en un contrato que deberá constar por escrito o en alguna otra forma que per-mita acreditar su celebración y contenido (art. 12). Así pues, toda cesión de datos no di-sociados, de carácter sanitario, que se produzca en la realización de un ensayo, debe estarexpresamente autorizada por el sujeto.
Con cierta frecuencia, observamos que el investigador del ensayo, remite da-tos directamente a la Organización de Investigación por Contrato (CRO), al amparo deun acuerdo establecido entre ésta y el promotor. Aún cuando este acuerdo sea legal yesté autorizado por el sujeto, (incluido en la documentación para el consentimiento in-formado), si expresamente no consta la participación del investigador en la cesión delos datos, éste debe remitir la información al promotor y no utilizar puentes por eco-nomía del proceso, que le harían incurrir en la comisión de una falta muy grave, tipi-ficada en el art. 44.4.b) de la mencionada Ley de protección de datos de carácterpersonal.
Por último, como aspecto relevante, dado lo habitual de ensayos multicéntri-cos internacionales, cabe resaltar lo dispuesto en el artículo 33 que, textualmentedice: “No podrán realizarse transferencias temporales ni definitivas de datos de carác-ter personal que hayan sido objeto de tratamiento o hayan sido recogidos para some-terlos a dicho tratamiento con destino a países que no proporcionen un nivel de
60

protección equiparable al que presta la presente Ley, salvo que, además de haberse ob-servado lo dispuesto en ésta, se obtenga autorización previa del Director de la Agen-cia Española de Protección de Datos, que sólo podrá otorgarla si se obtienen garantíasadecuadas.”
Todos estos cambios en la legislación hacen necesaria la actualización del RD561/1993, por lo que éste es sustituido por el Real Decreto 223/2004, de 6 de febrero,por el que se regulan los ensayos clínicos con medicamentos.
En él, se han tenido en cuenta los principios básicos para la realización de ensa-yos clínicos con seres humanos reflejados en la Declaración de Helsinki y en el Conveniode Asturias sobre los derechos humanos y la biomedicina.
Igualmente se recogen aspectos relativos a la inspección por parte de las Autori-dades Sanitarias, del cumplimiento de las Normas de Buena Práctica Clínica, que garan-tice que la información, los datos y los documentos han sido producidos, recogidos ypublicados correctamente, así como el cumplimiento de las normas de correcta fabrica-ción en la elaboración, importación y etiquetado de los medicamentos en investigación,la vigilancia de la seguridad de estos medicamentos y las comunicaciones entre las auto-ridades competentes en la materia.
En el año 2006, la propia ley del medicamento es sustituida por la Ley 29/2006,de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios,en vigor actualmente.
Ésta, que como su propio nombre indica, es una ley de garantías, entre su articu-lado destinado a los ensayos clínicos considera:
• Garantías de idoneidad, mediante el control por parte de administración: Régi-men de autorización previa, posibilidad de interrupción del ensayo o de exigirmodificaciones en su desarrollo, definición de las facultades inspectoras, exigen-cia del cumplimiento de las Normas de BPC, control de los acontecimientos ad-versos graves, exigencia de minimizar los riesgos al imponer como patrón decomparación el mejor disponible, exigencia de adhesión al protocolo y conoci-miento de resultados.
• Garantías de respeto a los postulados éticos, exigiendo el cumplimiento de losprincipios básicos de respeto al derecho de autonomía de los sujetos de la in-vestigación, respeto al principio de beneficencia y respeto al principio de jus-ticia, a los principios de la declaración de Helsinki, a la aceptación previa por
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
61

un CEIC.
• Garantía de asunción de responsabilidades, determinando la existencia de un se-guro u otra garantía financiera y definiendo la responsabilidad solidaria del pro-motor, el investigador y el centro donde se realice el ensayo, en aquellosaspectos no cubiertos por éstas.
• Garantía de transparencia, al establecer un registro nacional público y libre deensayos clínicos, imputar al promotor la obligación de publicar los resultados,sean positivos o no a sus intereses e indicando los fondos obtenidos para la in-vestigación y las fuentes de financiación.
Por último, la normativa aplicable en España, se completa, hasta la fecha actual,con la publicación por el Ministerio de Sanidad y Consumo del R.D. 1344/2007, de 11de octubre, por el que se regula la farmacovigilancia de medicamentos de uso humano,que persigue agilizar la tramitación de los estudios post-autorización observacionales conmedicamentos y productos sanitarios, de interés para la salud pública y de la OrdenSCO/256/2007, de 5 de febrero, por la que se establecen los principios y las directricesdetalladas de Buena Práctica Clínica y los requisitos para autorizar la fabricación o impor-tación de medicamentos en investigación de uso humano.
Esta Orden desarrolla algunos aspectos del RD. 223/2006 e incorpora las Direc-tivas de la Unión Europea, 2003/94/CE de la Comisión y 2005/28/CE de 8 de abril.
Entre su articulado es de destacar la regulación que realiza en su artículo 2 deprincipios y directrices de Buena Práctica Clínica; en su art. 6 del archivo de la documen-tación del ensayo y regula las inspecciones de Buena Práctica Clínica por las AutoridadesSanitarias en los artículos 7 al 11 ambos inclusive.
62

Normativa aplicable en la Comunidad Valenciana


La Comunidad Valenciana, en uso de las competencias que le otorga la Cons-titución Española y todas las Normas concordantes, ha venido legislando desde laasunción de competencias en materia de Sanidad en el año 1988 y, en el momentoactual, son de aplicación, además de las normas básicas de ámbito estatal, las si-guientes disposiciones:
a) Ley 1/2003, de 28 de enero, de derechos e información al paciente de laComunidad Valenciana
b) Decreto 73/2009, de 5 de junio, del Consell, por el que se regula la ges-tión de ensayos clínicos y estudios post-autorización observacionales conmedicamentos y productos sanitarios.
La ley 1/2003, de derechos e información al paciente, dictada al amparo del ar-tículo 38.1 del Estatuto de Autonomía, que atribuye a la Generalitat Valenciana el desarro-llo legislativo de la legislación básica del Estado en materia de sanidad interior y queincorpora, entre otras, la Directiva 95/46, de la Unión Europea, de 24 de octubre, sobrelos derechos de los ciudadanos a la intimidad en la información relacionada con su salud,el Convenio Europeo sobre Derechos Humanos y Biomedicina, de 4 de abril de 1997, co-nocido como Convenio de Asturias, la Ley Orgánica 15/1999, de 13 de diciembre, de pro-tección de datos de carácter personal, así como las recomendaciones del Dictamen deExpertos del Ministerio de Sanidad y Consumo, incorpora a la doctrina jurídica de la Co-munidad Valenciana, una clara definición de los derechos y obligaciones de los pacientes.
En el Título I define el objeto de la ley como el de reconocer y garantizar los de-rechos y obligaciones que en materia sanitaria tienen los pacientes en el ámbito territo-rial de la Comunidad Valenciana, tanto en los centros públicos como privados.
En el Título II recoge la relación de derechos, nuevos o ya definidos en otros tex-tos legales como la propia Constitución Española, pero que, con el paso del tiempo, ha-cía aconsejable agrupar en el mismo texto.
El Título III articula lo relativo al derecho a la información y el Título IV desarro-lla el derecho al consentimiento informado y, como medida complementaria para poder
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
65

conformar el criterio que ayude al paciente a tomar una decisión informada y libre de lainfluencia legítima que pueda ejercer el médico responsable de la asistencia que le faci-lita la información y recaba su consentimiento, se establece el derecho del paciente a ob-tener una segunda opinión. En este mismo Título IV, se declara el derecho al “testamentovital” o declaración de voluntades anticipadas.
El título V regula la historia clínica, en todos los aspectos relativos a su contenido,tratamiento, propiedad y custodia, así como los derechos de acceso a la misma, por losprofesionales e instituciones, los pacientes, o, en los supuestos que proceda, los familia-res, allegados o representantes legales.
En el título VI se recoge el derecho a la intimidad y en el Título VII se tratan losderechos de participación de los pacientes y de los ciudadanos, como ejercicio de respon-sabilidad y solidaridad.
El Título VIII crea el Consejo Asesor de Bioética, adscrito a la Conselleria de Sa-nidad, y los Comités de Bioética Asistencial, con el objeto de proteger los derechos de lospacientes y asesorar en la adopción de decisiones complejas.
Por último, el Título IX recoge las obligaciones de los pacientes.
Esta Ley, que en su origen está dedicada a los derechos y obligaciones de lossujetos enfermos en relación con el sistema sanitario público o privado que les atienda,se convierte así en supletoria de todos aquellos aspectos relacionados con los ensayosclínicos en seres humanos que no estén contemplados expresamente en la normativaespecífica.
Específicamente, en lo que concierne a los objetivos de este trabajo, interesan losderechos enumerados en el art. 3 siguientes:
“1. Al respeto de su dignidad, sin que pueda sufrir discriminación por razones deraza, sexo, económicas, sociales, ideológicas o de edad.
4. A la confidencialidad de los datos sobre su salud, sin que nadie, sin su autori-zación, pueda acceder a ellos, salvo en los casos previstos en la legislación vigente. El se-creto profesional estará garantizado en todo momento.
7. A recibir información sanitaria en la forma más idónea para su comprensión y,especialmente, en la lengua oficial de la comunidad autónoma y asegurarse que aquellasea inteligible para los pacientes…
66

10. A no ser sometido a procedimientos diagnósticos o terapéuticos de eficacia nocomprobada, salvo si, previamente advertido de sus riesgos y ventajas, da su consenti-miento por escrito y siempre de acuerdo con lo legislado para ensayos clínicos. Este con-sentimiento podrá ser revocado en cualquier momento del procedimiento, debiendo quedarconstancia en su historia clínica”.
Del artículo 4, cabe destacar el apartado 2 en relación a la capacidad inspectorade la Administración Sanitaria: “La autoridad sanitaria velará por el derecho de los ciuda-danos a recibir información sanitaria clara, veraz, relevante, fiable, actualizada, de cali-dad y basada en evidencia científica, que posibilite el ejercicio autónomo y responsablede la facultad de elección y la participación activa del ciudadano en el mantenimiento orecuperación de su salud”.
La ley presta especial dedicación en el artículo 9 a las poblaciones vulnerable (ni-ños e incapacitados legalmente), y determina que: “El consentimiento informado se otor-gará por sustitución en los siguientes supuestos:
1. Por los familiares o miembro de unión de hecho, y en su defecto por las per-sonas allegadas, cuando el paciente esté circunstancialmente incapacitado para to-marlas. En el caso de los familiares, tendrá preferencia el cónyuge no separadolegalmente; en su defecto, el familiar de grado más próximo y, dentro del mismogrado, el de mayor edad. Si el paciente hubiera designado previamente una persona,a efectos de la emisión en su nombre del consentimiento informado, corresponderáa ella la preferencia.
2. Cuando el paciente sea menor de edad o se trate de un incapacitado legal-mente, el derecho corresponde a sus padres o representante legal, el cual deberá acredi-tar de forma clara e inequívoca, en virtud de la correspondiente sentencia deincapacitación y constitución de la tutela, que está legalmente habilitado para tomar de-cisiones que afecten a la persona menor o incapacitada por él tutelada. En el caso de me-nores emancipados, el menor deberá dar personalmente su consentimiento. No obstante,cuando se trate de un menor y, a juicio del médico responsable, éste tenga el suficientegrado de madurez, se le facilitará también a él la información adecuada a su edad, for-mación y capacidad.
En los supuestos legales de interrupción voluntaria del embarazo, de ensayos clí-nicos y de prácticas de reproducción asistida, se actuará según lo establecido con carác-ter general por la legislación civil y, si procede, por la normativa específica que le sea deaplicación.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
67

Como medida adicional de protección, añade:
3. Cuando la decisión del representante legal pueda presumirse contraria a los in-tereses del menor o incapacitado, deberán ponerse los hechos en conocimiento de la au-toridad competente en virtud de lo dispuesto en la legislación civil.
En relación a la Historia Clínica, máximo exponente de documento fuente dondese contienen los datos de cualquier ensayo clínico con participación de seres humanos,es de destacar la definición del artículo 21 que dice: “La historia clínica es el conjuntode documentos en los que está contenida toda la información obtenida en todos los pro-cesos asistenciales del paciente. La historia clínica tiene como fin principal facilitar laasistencia sanitaria, dejando constancia de todos aquellos datos que permitan el conoci-miento veraz y actualizado del estado de salud del paciente, acumulando toda la informa-ción generada en cada episodio asistencial”.
Según esta exigencia, cualquier dato o información obtenida, tanto en la prácticaasistencial como en el desarrollo de un ensayo clínico, debe constar en la historia clínicaaun cuando el protocolo indique que debe recogerse sólo en el CRD.
Del mismo modo, es una práctica contraria a esta ley, la recogida de datos y de-más información sólo en el CRD o en historias paralelas propuestas por el propio proto-colo del ensayo. Los CEIC no deberían autorizar estas prácticas pues, si bien laautorización no libera a los investigadores del cumplimiento de sus obligaciones, sí quees motivo de confusión, dando pie a situaciones de incumplimiento manifiesto y grave.
En cuanto a la conservación de la misma se fija un tiempo de cinco años desdeel último episodio asistencial del sujeto en cuestión. En todo caso, los documentos es-pecialmente relevantes se conservarán indefinidamente o por el tiempo que fije la norma-tiva vigente al respecto. Esta conservación deberá garantizar la preservación de lainformación y no necesariamente del soporte original.
En el artículo 24, se regula el derecho de acceso a la historia clínica de los pro-fesionales sanitarios, limitándolo, como norma general, a aquellos implicados en el diag-nóstico o el tratamiento del enfermo.
No obstante, reconoce que se puede acceder a la historia clínica, con finalidadesepidemiológicas, información estadística sanitaria, actividades relacionadas con el controly evaluación de la calidad asistencial, las encuestas oficiales y los programas oficiales deinvestigación o docencia, con sujeción a lo establecido en la Ley orgánica 15/1999, de
68

13 de diciembre, de Protección de Datos de Carácter Personal, y la Ley 14/1986, de 25de abril, General de Sanidad, y las disposiciones concordantes. En estos casos el accesoa la historia clínica obliga a preservar los datos de identificación personal del paciente, quesiempre tendrán que estar separadas de las de carácter clínico asistencial, salvo si el pa-ciente, previamente, ha dado su consentimiento.
Por último, además de a la autoridad judicial, se autoriza el acceso, con absolutagarantía del derecho a la intimidad personal y familiar, a efectos de inspección sanitaria,para las actividades de evaluación, acreditación y comprobación del cumplimiento de losderechos del paciente, y otras debidamente motivadas por la autoridad sanitaria y quetengan por finalidad contribuir a la mejora de la calidad asistencial; el acceso a la histo-ria clínica en este caso, estará limitado a la información relacionada con tales fines.
En cuanto al derecho a la intimidad, el artículo 26 indica que toda persona tienederecho a que se respete la confidencialidad de los datos referentes a su salud y que na-die que no esté autorizado podrá acceder a ellos si no es al amparo de la legislación vi-gente. Los centros sanitarios serán garantes de este derecho y deben adoptar las medidasde estructura y de organización necesarias a este fin.
El Decreto 73/2009, de 5 de junio, del Consell, por el que se regula la gestión deensayos clínicos y estudios post-autorización observacionales con medicamentos y pro-ductos sanitarios, deroga la Orden de 6 de julio de 1994, de la Conselleria de Sanidad yConsumo por la que se regulaban los ensayos clínicos en la Comunidad Valenciana y la Or-den de 8 de marzo de 2004, por la que se crea la Comisión Asesora para la evaluación delos estudios post-autorización observacionales prospectivos.
El importante cambio legislativo en el panorama nacional y europeo, hacen nece-saria la ordenación y actualización del marco normativo valenciano, desarrollando un mo-delo en el que se crea el Programa de Estudios Clínicos en medicamentos y productossanitarios de la Comunidad Valenciana (PECHE) y se constituye el Comité Autonómico deEstudios Clínicos de medicamentos y productos sanitarios (CAEC), el cual, además defunciones de evaluación clínica, se configura como un “órgano consultivo encargado deasistir y asesorar en todo aquello relacionado con el desarrollo de los procesos de evalua-ción clínica en medicamentos y productos sanitarios”.
El objeto del Decreto, como define el artículo 1, es establecer la estructura orga-nizativa y requisitos para la realización de ensayos clínicos y estudios post-autorización detipo observacional con medicamentos y productos sanitarios de uso humano en la Comu-nidad Valenciana.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
69

Así, se crea el Programa de Estudios Clínicos de Medicamentos y Productos Sa-nitarios (PECHE), que, además de coordinar lo dispuesto en la propia Orden, lo hace tam-bién en materia de ensayos clínicos, de productos sanitarios y en materia defarmacovigilancia de medicamentos de uso humano.
Los objetivos quedan definidos en el artículo tercero, apartado 2:
“a) Promover la correcta realización de ensayos clínicos y estudios post-autoriza-ción observacionales de medicamentos y productos sanitarios tanto en la aplicación de lanormativa vigente como desde el punto de vista científico, así como el adecuado conoci-miento de los aspectos éticos y metodológicos por parte de todos los agentes implicadosen la realización y evaluación de dichos ensayos y estudios.
b) Garantizar que los miembros de los CEIC acreditados en la Comunidad Valen-ciana están capacitados para evaluar los protocolos de los ensayos clínicos y estudios post-autorización observacionales en sus aspectos metodológico, ético y legal.
c) Establecer y organizar sistemas de información adecuados y fluidos entre laAdministración Sanitaria autonómica, la Agencia Española del Medicamento, Centro Co-ordinador de CEICs, los CEIC y los promotores e investigadores de los ensayos clínicos”.
Para ello se crean y se definen las competencias de los siguientes órganos:
a) El Consejo de Dirección y la Comisión Delegada del PECHE.
b) El Coordinador del PECHE.
c) El Comité Autonómico de Estudios Clínicos de Medicamentos y Productos Sa-nitarios de la Comunidad Valenciana (CAEC).
d) El Comité Autonómico de Estudios Post-autorización Observacionales de Me-dicamentos y Productos sanitarios de la Comunidad Valenciana (CAEPO).
e) Los CEIC locales y corporativos acreditados en el ámbito de la Comunidad Va-lenciana (CEIC).
A lo largo del articulado del Decreto, se continúa desarrollando la estructura yfunciones de cada uno de ellos, así como el Procedimiento y los requisitos de acreditaciónde los CEIC, su organización, la obligación de contar con unos Procedimientos Normali-zados de trabajo (PNT) adecuados a las condiciones de funcionamiento desarrolladas enla orden, la incompatibilidad de ser miembro de un CEIC y mantener intereses de cual-quier clase derivados de la fabricación y venta de medicamentos y productos sanitarios.
70

Continúa con la definición de las funciones de la dirección del centro o estable-cimiento sanitario con CEIC acreditado en la Comunidad Valenciana.
En el artículo decimotercero, desarrolla los aspectos legales y económicos de losensayos y estudios con medicamentos y productos sanitarios debiendo quedar plasmadostodos ellos en un contrato entre el promotor y cada centro donde se vaya a realizar el en-sayo con el fin de conseguir la mayor transparencia económica de los pactos existentes.
La Dirección General de Farmacia y Productos Sanitarios de la Agencia Valen-ciana de Salud dispondrá de un portal web de trabajo exclusivo de PECHE, que facilite lacomunicación entre los CEIC y sirva de fuente común de información en todo lo relacio-nado con las materias en cuestión sirviendo también de puente de comunicación con laAGEMED y PS.
El PECHE impulsará las actividades periódicas de formación y jornadas de normali-zación de criterios, entre los profesionales que integran los CEIC y promoverá jornadas de co-ordinación con todos los agentes implicados, evaluadores, promotores e investigadores con lacolaboración del centro coordinador de los Comités Éticos de Investigación Clínica y el Comitéde Coordinación de Estudios Post-autorización de la Agencia Española del Medicamento.
Se regula la Inspección por parte de las Autoridades Sanitarias para verificar elcumplimiento de lo dispuesto en la normativa vigente en la materia, haciendo hincapié enverificar la observancia de las Normas de Buena Práctica Clínica en los ensayos clínicosy estudios post-autorización, disponiendo que del informe de Inspección se remitirá co-pia al PECHE y a la Agencia Española del Medicamento para su introducción en la basede datos europea de ensayos clínicos EUDRACT de acuerdo con las directrices de la Co-misión Europea.
En el artículo diecisiete, remite el régimen sancionador al previsto en el CapítuloII del Título VIII de la Ley 29/2006, de 26 de julio, de garantías y uso racional del medi-camentos y productos sanitarios.
La disposición final por último, faculta al Conseller de Sanidad a dictar las nor-mas necesarias para el desarrollo y aplicación del Decreto.
Para ello, debe desarrollarse lo relativo a la regulación de los procedimientos, do-cumentación y plazos a observar en la presentación y modificaciones en los procesos re-lacionados con la investigación clínica de medicamentos y productos sanitarios y debeigualmente regularse el modelo de contrato que ha de suscribirse entre la gerencia de un
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
71

centro sanitario, el promotor y los investigadores, para la realización de un ensayo clínicoo estudios de investigación con medicamentos y productos sanitarios en las organizacio-nes de los servicios sanitarios de la Conselleria de Sanidad de la Comunidad Valenciana.
La realización de ensayos clínicos con medicamentos de uso humano, es una ac-tividad compleja que conlleva derechos y obligaciones de diversos tipos, entre varias par-tes, entre otras promotor, investigador, centro donde se realiza el ensayo y en algunoscasos fundaciones para la investigación u organizaciones de investigación por contrato(CRO). En la mayoría de casos, además intervienen otros actores como investigadores co-laboradores u otros servicios internos de los centros como Servicio de Farmacia, personalde enfermería etc.
Todos los derechos y obligaciones de todos estos actores, deben estar regulados ydocumentados mediante contratos.
Así, el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensa-yos clínicos con medicamentos, en su artículo 30, en relación con los aspectos económi-cos declara: “Todos los aspectos económicos relacionados con el ensayo clínico quedaránreflejados en un contrato entre el promotor y cada uno de los centros donde se vaya a re-alizar el ensayo” y a continuación dispone que: “Las Administraciones sanitarias compe-tentes para cada servicio de salud establecerán los requisitos comunes y condiciones definanciación, así como el modelo de contrato de conformidad con los principios generalesde coordinación que acuerde el Consejo Interterritorial del Sistema Nacional de Salud”.
La existencia de contratos diferentes en función del centro de que se trate, del tipode ensayo, de contratos entre promotor y centro por un lado, promotor e investigador porotro, acuerdos no escritos de compensación a los servicios colaboradores etc. unido a laparticipación de las fundaciones para la investigación de algunos centros, dificulta sobre-manera la gestión administrativa de todo el proceso siendo causa, en muchas ocasiones,de retrasos por no hablar de situaciones de opacidad no deseables.
Se hace necesaria la existencia de un contrato único que será suscrito entre el pro-motor, la gerencia y los investigadores de un ensayo clínico o proyecto de investigación conmedicamentos y productos sanitarios, en cualquier organización de servicios sanitarios dela Agencia Valenciana de Salud. Además en el caso de los centros que gestionan sus ac-tividades de investigación mediante fundación de derecho privado, constituida al amparode lo previsto en la Ley 50/2002, de 26 de diciembre, de Fundaciones, el contrato debeser firmado por el representante legal de la misma y la gestión de los cobros y los pagosderivados de la realización del ensayo clínico, deberá tramitarse a través de la fundación.
72

Como anexos al mismo se adjuntará la Memoria Técnica, la Memoria económicay la Relación del equipo investigador con expresión de los siguientes datos de cada inves-tigador colaborador: Nombre y apellidos, D.N.I., categoría profesional, centro / organismo,servicio, función a realizar, y firma. Éste último anexo deberá ser sustituido cada vez quecambie alguno de los miembros del equipo o se modifiquen las funciones a realizar o cual-quier otra circunstancia. El documento sustituido deberá quedar en el archivo del inves-tigador (al menos) a efectos de inspección o auditorías.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
73

74

El Investigador


Investigador es el profesional o persona que ejerce una profesión reconocida parallevar a cabo investigaciones en razón de su formación científica y de su experiencia en laatención sanitaria requerida. El investigador es responsable de la realización del ensayoclínico en un centro. Si es un equipo el que realiza el ensayo en un centro, el investiga-dor es el responsable del equipo y puede denominarse investigador principal.
Si el ensayo es multicéntrico, se denomina Investigador coordinador al investiga-dor responsable de la coordinación de los investigadores de todos los centros que partici-pan en el ensayo clínico.
El investigador ostenta responsabilidades por disposición legal pero también porimperativo ético.
La investigación mediante ensayos clínicos comparte con la realizada en otras áreasde conocimiento el afán de avanzar en el saber consolidado que se tiene. Sin embargo, pre-senta la particularidad de ser aplicada. Eso significa que la búsqueda emprendida ofrecerasgos propios: tiene una finalidad concreta y necesita respuestas que puedan ser aplica-das de inmediato. Además, sin poderse desprender de lo anterior, destaca que su realiza-ción se lleva a cabo sobre seres humanos con voluntad y capacidad de decisión propias.
Las responsabilidades por disposición legal, contenidas en el RD 223/2004 son:
a) Estar de acuerdo y firmar junto con el promotor el protocolo del ensayo.
b) Conocer a fondo las propiedades de los medicamentos en investigación.
c) Garantizar que el consentimiento informado se recoge de conformidad a lo es-tablecido en las normas éticas y legales en vigor.
d) Recoger, registrar y notificar los datos de forma correcta y garantizar su vera-cidad.
e) Notificar inmediatamente los acontecimientos adversos graves o inesperados alpromotor.
f) Garantizar que todas las personas implicadas respetarán la confidencialidadde cualquier información acerca de los sujetos del ensayo, así como la protec-ción de sus datos de carácter personal.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
77

g) Informar regularmente al Comité Ético de Investigación Clínica de la marchadel ensayo. Esta obligación incluye la notificación de inicio del ensayo, infor-mando de la fecha de inclusión del primer sujeto.
h) Corresponsabilizarse con el promotor de la elaboración del informe final delensayo, dando su acuerdo con su firma.
Además, como hemos apuntado antes, el investigador ostenta también responsa-bilidades de tipo ético, contempladas, tanto en Normas y Declaraciones relativas a la in-vestigación en la que participen seres humanos (tales como la Declaración de Helsinki,cuya última versión es la de Seúl 2008, las Normas de Buenas Prácticas Clínicas de laICH, etc.) como a las de ámbito más amplio como la Declaración Universal de DerechosHumanos, de la Asamblea General de la ONU, la Declaración Universal sobre el GenomaHumano y los Derechos Humanos de la Conferencia General de la UNESCO, la Declara-ción Universal sobre Bioética y Derechos Humanos del mismo organismo, etc.
Sin ánimo de exaustividad cabría decir:
1. Cualificación.
El investigador principal y los colaboradores deberán justificar su cualificaciónmediante un curriculum vitae actualizado, con los datos más relevantes acerca de su for-mación y de investigación. Igualmente deberá conocer y respetar las normas de BuenaPráctica Clínica y la Declaración de Helsinki en su última versión.
2. Capacidad de reclutamiento.
Antes de aceptar la realización de un ensayo debe valorar, con datos retrospecti-vos, la capacidad de reclutamiento para el nº de sujetos previstos, en el tiempo estipuladoen el protocolo.
3. Delegación de competencias.
Podrá delegar parte de sus responsabilidades en otros investigadores colaboradores,siempre que la relación con los mismos esté legalmente definida y consten documental-mente las funciones delegadas en cada uno de ellos, bien en el propio protocolo, en el con-trato entre investigador y promotor, en el contrato con la institución etc. A estos efectos ypara no tener que repetir el contrato cada vez que uno de los colaboradores deje de serlo ose incorpore uno nuevo, se recomienda que se incluya dicha relación con la delegación decompetencias en un “Adenda” al contrato, con lo que sólo será necesario cambiar la misma
78

si se dan las circunstancias mencionadas. Las “Adendas” que dejen de estar en vigor, de-ben conservarse y pasar a formar parte de la documentación esencial del ensayo.
Si la relación entre estos investigadores colaboradores y el investigador principales ajena a la propia institución donde se realiza el ensayo, (personal contratado expresa-mente), la actuación sanitaria de estos investigadores colaboradores sobre los sujetos (má-xime si además son pacientes de la institución), deberá ajustarse a la legalidad en vigor.Quiere esto decir que, salvo autorización expresa de la Dirección, no pueden realizar nin-guna actividad sanitaria ni tener acceso a información sanitaria confidencial o de datospersonales de ningún paciente. En cualquier caso, sobre estos extremos debe informarseal sujeto y figurar esta circunstancia en la hoja de información.
4. Respeto al protocolo.
Es obligación del investigador respetar en todos sus extremos los términos del pro-tocolo aceptado por el CEIC y autorizado por la Agencia Española del Medicamento y Pro-ductos Sanitarios. No debe realizar ninguna desviación del protocolo sin el acuerdo delpromotor y el dictamen favorable del CEIC a la enmienda en cuestión. Sólo podrá incumplirel protocolo sin los requisitos anteriores para salvaguardar al sujeto de la investigación deun riesgo grave e inminente. Posteriormente deberá justificar y documentar tal decisión.
5. Obtención del consentimiento informado. (Ver capítulo correspondiente).
6. Fiabilidad e integridad de los datos.
Debe garantizar la exactitud e integridad de los datos y su consistencia (trazabi-lidad) con los datos de los documentos fuente.
El investigador es responsable de la custodia de su archivo por el tiempo que es-tablezca la legislación. Esta responsabilidad no puede ser transferida aun cuando, en vir-tud de un contrato legal, se transfiera la guarda física de los mencionados archivos. Estaresponsabilidad incluye no sólo la garantía de que no se van a destruir los documentosesenciales del ensayo sino también la garantía del mantenimiento de la intimidad de lossujetos y confidencialidad de sus datos.
7. Acontecimientos adversos.
Deberá informar inmediatamente al promotor de todos aquellos acontecimientosadversos graves que sucedan, excepto los que expresamente indique lo contrario el proto-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
79

colo. Estas notificaciones no eximen al investigador de la declaración pertinente al Sistemade Farmacovigilancia de la Comunidad Autónoma correspondiente.
Igualmente deberá informar al CEIC de cuantos controles de seguridad le sean exigi-dos por éste, sin que al respecto pueda alegar que esta tarea es responsabilidad del promotor.
8. Seguridad de los sujetos de la investigación.
Ante la finalización prematura o suspensión de un ensayo clínico, promovida porel investigador, por el promotor, por el CEIC o por la Autoridad Sanitaria competente, elinvestigador es responsable de informar puntualmente a los sujetos del ensayo y garanti-zar la continuidad en su asistencia sanitaria, de tal forma que se minimicen al máximo losriesgos que la mencionada finalización prematura o suspensión puedan significar paraaquellos. De la misma manera, ante la finalización ordinaria del ensayo, debe promover lonecesario para que la población que se ha sometido a la experimentación, disponga de losbeneficios demostrados en la misma.
9. En relación con el Medicamento en fase de investigación.
Es responsabilidad del investigador, informar adecuadamente al sujeto acerca delas condiciones de uso (frecuencia, dosis, etc.) del medicamento en investigación.
La legislación española , en el artículo 2.6 de la ley 29/2006, de 26 de julio, degarantías y uso racional de los medicamentos y Productos Sanitarios, encomienda la cus-todia, conservación y dispensación de los medicamentos, sin excluir a los medicamentosen fase de investigación, a los Servicios de Farmacia de los Hospitales o de las estructu-ras de Atención Primaria del SNS, pero el hecho de que estas fases de la gestión de losmedicamentos estén encomendadas a dichas estructuras, no libera al investigador de serel responsable último de su gestión y contabilidad.
En determinados casos en los que existan dificultades serias para el cumplimientodel punto anterior, podrá aceptarse que la custodia, conservación y dispensación de losmedicamentos la realice directamente el investigador.
El investigador no debe olvidar que la investigación clínica debe tener valor (im-portancia social, científica o clínica), es decir, que sus resultados deben tener la probabi-lidad de promover mejoras en la salud, el bienestar o el conocimiento de la población. Esterequisito de que la investigación clínica sea valiosa asegura a los sujetos de investigaciónque no serán expuestos a riesgos sin la probabilidad de algún beneficio personal o social.
80

El Comité Ético de InvestigaciónClínica (CEIC)


El Comité Ético de Investigación Clínica es el organismo independiente, consti-tuido por profesionales sanitarios y miembros no sanitarios, encargado de velar por la pro-tección de los derechos, la seguridad y el bienestar de los sujetos que participen en unensayo clínico, durante todo el tiempo de permanencia como sujetos del ensayo y de ofre-cer garantía pública al respecto, mediante un dictamen sobre el protocolo del ensayo, laidoneidad de los investigadores y la adecuación de las instalaciones, así como los méto-dos y los documentos que vayan a utilizarse para informar a los sujetos del ensayo con elfin de obtener su consentimiento informado, etc.
Entre otras tareas está velar por el respeto a los aspectos éticos del ensayo, in-cluyendo en esta categoría, no sólo el garantizar que se respetan los derechos del su-jeto en cuanto a que ha recibido una información adecuada y ha manifestado suconsentimiento a participar en los términos establecidos por la legislación, etc. sino queademás, el sujeto está suficientemente protegido por una garantía financiera adecuadaen caso de sufrir algún daño inherente al proceso experimental, que no se trata de su-jetos pertenecientes a poblaciones que precisen protección adicional, en cuyo caso de-berá extremar las cautelas del proceso de autorización, que el ensayo es pertinente enfunción de los objetivos perseguidos, que la ponderación entre riesgos y beneficios esaceptable, que los criterios de inclusión y exclusión están adecuadamente formulados,que el investigador está suficientemente cualificado y, además de ostentar calificacióncientífica suficiente tiene formación específica en Buenas Prácticas Clínicas y conocela Declaración de Helsinki en su última versión, así como el Convenio sobre los DerechosHumanos y la Biomedicina, firmado en Oviedo, en 1997 y ratificado por el Reino de Es-paña en 2000 y cuantas normas de origen ético y legal sean aplicables. Igualmentedebe pronunciarse acerca de los aspectos financieros del ensayo, de la idoneidad de lasinstalaciones, etc.
Para ello, debe efectuar una adecuada revisión de la documentación relativa al en-sayo, así como de las modificaciones que se planteen durante el desarrollo del mismo y,en función de las circunstancias locales, puede plantear modificaciones tanto al protocolopresentado para su autorización, como a la información contenida en la hoja de informa-ción al sujeto, establecer un plan de seguimiento del ensayo que incluya la obligación delinvestigador y/o promotor de emitir informes periódicos de seguridad o del desarrollo del
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
83

ensayo, de la situación del reclutamiento, etc. suplementarios a los inicialmente estable-cidos y con tanta mayor frecuencia cuanto mayores sean los potenciales riesgos para lossujetos de la investigación. Esta facultad de los CEIC, en determinadas circunstanciasdebe ser una obligación ética de ejercicio de su propia responsabilidad, pues la garantíade tutor de los derechos del sujeto no acaba con la valoración inicial y previa a la autori-zación, sino que se mantiene en el tiempo hasta tanto no finaliza el ensayo.
El establecimiento de estas modificaciones, confiere a la autorización para la re-alización del ensayo, la cualidad de una autorización condicional, de tal forma que el in-cumplimiento por el promotor y/o el investigador dejaría sin efecto la autorización y nopodría realizarse el ensayo. El CEIC debe informar a la Dirección de la Institución del es-tablecimiento de estas condiciones, quien, a la vista de las mismas, valorará la conve-niencia de no autorizar el ensayo en el centro hasta tanto no se cumplan las mismas. Estamedida no es necesaria en la Comunidad Valenciana, puesto que el Director del centro esmiembro del CEIC.
Los CEIC deben estar acreditados por la Autoridad Sanitaria competente de cadaCC.AA. y constar en el Registro Oficial que a tal efecto mantiene el Centro Coordinador delos CEIC, adscrito al Ministerio de Sanidad y Consumo y Política Social.
Para ello, los responsables de los centros, servicios y establecimientos sanitariosde la Comunidad Valenciana autorizados por la Conselleria de Sanidad, podrán solicitar laacreditación/reacreditación de los CEIC de dichos centros ante la Dirección General de Far-macia y Productos Sanitarios de la Agencia Valenciana de Salud, a través del Coordinadordel PECME. Esta acreditación será temporal (3 años) y deberá renovarse al final de cadaperiodo.
La Dirección General de Farmacia y Productos Sanitarios de la Agencia Valen-ciana de Salud podrá revocar la acreditación concedida a cualquier CEIC que no cumplalos requisitos mínimos exigidos en la normativa aplicable.
Los requisitos que, en la Comunidad valenciana, debe reunir un CEIC para poderser acreditado son:
Los requisitos mínimos para la acreditación de un CEIC serán los siguientes:
a) Estar formado como mínimo por once miembros, y en todo caso formar parte de él:
84

• El director del centro.
• Médicos:
Un farmacólogo clínico
Dos médicos especialistas con labor asistencial activa en un centro hospitalario
Un médico con labor asistencial activa en un Centro de Atención Primaria
Un pediatra
• Farmacéuticos:
Un farmacéutico de Hospital
Un farmacéutico de Atención Primaria
• Un diplomado en Enfermería.
• Dos miembros ajenos a las profesiones sanitarias, debiendo ser uno de ellos li-cenciado en Derecho. Uno de estos miembros será una persona independiente dela organización asistencial, entendiendo por independiente la desvinculación la-boral en relación a los centros en los que se lleven a cabo los proyectos de inves-tigación que requieran la evaluación ética por parte del Comité, a ser posiblepropuesto por el Consejo de Salud.Tener garantía explícita, por parte del titulardel centro, de que el comité cuenta con los medios necesarios para poder reali-zar su cometido
b) Tener garantía explícita, por parte del titular del centro, de que el CEIC cuenta conlos medios necesarios para poder realizar su cometido. Los medios básicos a dis-poner son:
• Instalaciones específicas que permitan la realización de su trabajo, en condicio-nes que garanticen la confidencialidad. Deberán disponer de un espacio apro-piado para la secretaría del comité, para la realización de las reuniones y para elmanejo y archivo de documentos confidenciales.
• Equipamiento informático con capacidad suficiente para manejar toda la infor-mación generada por el comité y disponibilidad de un sistema rápido de transmi-sión de información.
• Personal administrativo y técnico que permita al comité poder ejercer de maneraapropiada sus funciones.
c) Disponer de los correspondientes procedimientos normalizados de trabajo.
Los miembros de los CEIC serán nombrados por el Director General de Farmaciay Productos Sanitarios de la Agencia Valenciana de Salud, a propuesta de la Dirección de
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
85

la institución de que se trate, después de que el Coordinador del PECME haya emitido elinforme preceptivo.
La pertenencia a un CEIC es incompatible con el mantenimiento de intereses liga-dos a la fabricación y venta de medicamentos y sus miembros deben firmar al inicio de suactividad como tales, una declaración de intereses.
La organización y funcionamiento de un CEIC debe estar recogida en unos procedi-mientos Normalizados de Trabajo (PNT).
Como mínimo éstos deben contemplar:
a) La composición y requisitos que deben cumplir sus miembros.
1) Formación y capacitación en metodología de la investigación, excepto losmiembros legos.
2) Experiencia en investigación (curriculum).
3) Compromiso escrito de aceptación.
4) Compromiso escrito de garantía de confidencialidad.
5) Declaración escrita de intereses.
6) Obligación de renuncia tras un número de ausencias no justificadas a juiciodel CEIC.
b) La periodicidad de las reuniones, que al menos deberá ser mensual con un mí-nimo de once anuales.
c) El procedimiento para convocar a sus miembros.
d) Los aspectos administrativos, incluyendo la documentación que debe presentarse.
e) Los casos en que se pueda realizar una revisión rápida de la documentación co-rrespondiente a un ensayo clínico y el procedimiento que debe seguirse en estoscasos.
f) La evaluación inicial de los protocolos y sistema de seguimiento de los ensayos.
g) Los mecanismos de toma de decisiones.
h) La preparación y aprobación de las actas de las reuniones.
i) El archivo y conservación de la documentación del comité y de la relacionada conlos ensayos clínicos evaluados.
1) Documentación relacionada con su propia acreditación y reacreditaciones pos-teriores.
86

2) Correspondencia con las Autoridades Sanitarias u otras instancias superioresde la administración.
3) Correspondencia con CEIC de referencia de los ensayos realizados en el cen-tro.
4) Correspondencia con el Centro Coordinador de los CEIC.
5) PNT.
6) Lista de miembros, curriculum de cada uno, compromiso escrito de aceptación,de confidencialidad, declaración de intereses, de renuncias en su caso.
7) Registros financieros del CEIC.
8) Actas e informes del CEIC.
9) Protocolos evaluados junto a la documentación generada por el CEIC en cadacaso.
10) Correspondencia con la Dirección de la Institución.
11) Correspondencia con otros servicios de la Institución (Farmacia, laboratorios,etc.).
12) Correspondencia con promotores, monitores, CRO, etc.
Los PNT deben ser públicos y estar aprobados por el propio CEIC y, en desarrollo delos puntos anteriores, deben contemplar, al menos:
• Nº de miembros y composición, con la titulación y/o cualificación y puesto de tra-bajo de cada uno de ellos.
• Ámbito de actuación autorizado del CEIC.
• Declaración de principios básicos por los que rse rige el comité.
• Funciones del presidente, Vicepresidente, secretario y vocales.
• Criterios para la selección, renovación o sustitución de los miembros del comité.
• Periodicidad de renovación de los miembros.
• Periodicidad de las reuniones y mínimo mensual y anual de reuniones.
• Condiciones de las convocatorias y Quórum necesario para dar validez a las decisio-nes adoptadas, que nunca será inferior al 50 % de sus miembros, con presenciaobligada del Presidente o Vicepresidente y del Secretario. En la Comunidad valen-ciana, en segunda convocatoria, será suficiente con la presencia del Presidente o Vi-cepresidente y del Secretario y, al menos, tres miembros más.
• Requisitos mínimos del quórum. (cualificación de los miembros, etc).
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
87

• Previsión de asistencia, con voz y sin voto de expertos en los temas propios del en-sayo, en el manejo de poblaciones incluidas en el ensayo susceptibles de ser espe-cialmente protegidas (por ejemplo, Pediatras) y del propio investigador ocolaboradores, sin voto, aun cuando sean miembros del CEIC.
• No delegabilidad del voto por ausencia de algún miembro.
• Procedimientos para apelar sus decisiones/opiniones.
• Contenido de las actas con, al menos, la siguiente información: Fecha y lugar de lareunión, miembros asistentes, protocolos que se han evaluado debidamente identi-ficados, los aspectos evaluados y la decisión adoptada para cada protocolo, protoco-los no aprobados y motivos por los que el comité no los ha aprobado, aspectosponderados en cada protocolo, versión y fecha de los documentos evaluados. (Se re-comienda incluir la información contenida en el modelo adjunto).
• Documentación necesaria a aportar para evaluar un ensayo.
• Plazos de entrega de la documentación, para efectuar la evaluación y para la emi-sión del dictamen.
• Condiciones especiales para cuando se actúa como CEIC de referencia
• Condiciones para la emisión de dictámenes sobre modificaciones de ensayos ya au-torizados y de revisión rápida.
• Procedimiento para el seguimiento de ensayos aprobados tales como, definición decasos en que se precisará una evaluación de seguimiento, intervalos de las evalua-ciones de seguimiento, circunstancias en las que el CEIC debe decidir la revocaciónde autorización y proponer la finalización del estudio, etc.
• Procedimiento para el archivo de la documentación generada por el comité.
• Modo de acceso y recuperación de la documentación y personas autorizadas a ello.
• Definición de documentación a archivar. Plazos de archivo.
En el dictamen el CEIC, según recomienda el Centro Coordinador de CEIC del Minis-terio de Sanidad y Consumo, dejará constancia expresa de que se han evaluado los siguientesextremos:
1. La pertinencia del ensayo clínico, teniendo en cuenta el conocimiento disponible.
2. La pertinencia de su diseño para obtener conclusiones fundamentadas con el nú-mero adecuado de sujetos en relación con el objetivo del estudio.
3. Los criterios de selección y retirada de los sujetos del ensayo, así como la selecciónequitativa de la muestra.
88

4. La justificación de los riesgos e inconvenientes previsibles en relación con los be-neficios esperables para los sujetos del ensayo, para otros pacientes y para la co-munidad.
5. La justificación del grupo control (ya sea placebo o un tratamiento activo).
6. Las previsiones para el seguimiento del ensayo.
7. La idoneidad del investigador y de sus colaboradores.
8. La idoneidad de las instalaciones.
9. La idoneidad de la información escrita para los sujetos del ensayo y el procedi-miento de obtención del consentimiento informado, y la justificación de la investi-gación en personas incapaces de dar su consentimiento informado.
10. El seguro o la garantía financiera previstos para el ensayo.
11. Las cantidades y, en su caso, previsiones de remuneración o compensación paralos investigadores y sujetos del ensayo y los aspectos relevantes de cualquieracuerdo entre el promotor y el centro.
12. El plan previsto para el reclutamiento de los sujetos.
Como modelo orientativo de acta de autorización, se propone la siguiente:
ACTA Nº /
INFORME DE EVALUACIÓN DEL PROTOCOLO DEL ENSAYO CLÍNICO
Nº de protocolo Nº Eudrac
Asistentes:
Apellidos y nombre Profesión Cargo en el CEIC Conflicto de intereses SINO SI NO SI NO SINO SI NO SI NO SINO SI NO SI NO SINO SI NO SI NO
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
89

En , a de , de , siendo las horas, se reúne el Comité Éticode Investigación Clínica del , con la asistencia de los miembros relacionados al ini-cio.
Constatada la existencia de quórum, según los PNT del comité, se inicia la sesión.
Se revisan los documentos siguientes:
Protocolo del ensayo clínico referenciado, versión en español , de fecha .Manual del investigador, versión en español , de fecha .Hoja de información para la obtención del Consentimiento Informado, versión , defecha .Póliza de seguro o garantía financiera de referencia (detallar aseguradora, importeasegurado total y por sujeto, nº de sujetos y periodo de vigencia).Condiciones económicas del contratoCurriculum Vitae del investigador y de los investigadores colaboradores.
Tras la revisión efectuada se acuerda:
El diseño de la investigación se ajusta a las normas para ensayos clínicos en los que par-ticipan seres humanos.Los objetivos del ensayo son científicamente pertinentes y no se trata de un ensayo repe-titivo ni por razones que no se ajusten al interés científico.Los riesgos para los sujetos de la investigación son aceptables y la relación en cuanto alos beneficios esperados es favorable.[Los riesgos para los sujetos son elevados aún cuando la relación en cuanto a los benefi-cios esperados es favorable, por lo que se acuerda monitorizar el desarrollo del ensayomediante las siguientes medidas de seguimiento: (enumerar)] (1)
[Los riesgos para los sujetos son inaceptables por: (justificar)] (1)
El método de comparación utilizado en el grupo de control es el mejor existente a la luzde los conocimientos actualesLa hoja de información a los sujetos para solicitar el Consentimiento Informado se ajustaa lo dispuesto en la legislación en vigor y en las Normas de Buena Práctica Clínica.[La hoja de información a los sujetos para solicitar el Consentimiento Informado debe sermodificada en los siguientes términos: (enumerar)] (1)
Los criterios de inclusión y exclusión son médicamente correctos para los fines que sepersiguen en el ensayo.Las instalaciones del centro en el que se va a desarrollar el ensayo se consideran adecua-
90

das.Analizados los Curriculum Vitae del investigador y de los investigadores colaboradores seconsideran con la suficiente formación y experiencia para la realización del ensayo.Las medidas propuestas para el tratamiento médico necesario en caso de acontecimien-tos adversos graves se estima adecuado.La póliza de seguro o garantía financiera es específica para el ensayo evaluado y el centrodonde se va a desarrollar. Cubre al 100 % de los sujetos propuestos en la previsión delreclutamiento y la cuantía de dicha cobertura se ajusta a lo establecido en el RD223/2004 de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentosen España.La remuneración económica a los investigadores se considera adecuada.Las compensaciones previstas para los sujetos participantes son adecuadas y se consideraque no suponen un incentivo suficiente para condicionar la aceptación de participar en elensayo.
A rellenar en caso de poblaciones vulnerables. (1)
Se trata de un ensayo con interés terapéutico para la población en cuestión.El conocimiento generalizable que se pretende obtener no es obtenible con otro tipo depoblación no vulnerable.Los riesgos a los que se verán sometidos los sujetos durante la investigación son mínimos,es decir, no mayores que los correspondientes a la práctica médica habitual para la pato-logía de que se trate.La obtención del Consentimiento Informado se ajusta a lo dispuesto en el RD 223/2004de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos en Españay en las Normas de BPC.
A rellenar en caso de que el comparador sea un placebo (1)
Para efectuar la comparación con el grupo control no existe ninguna intervención cientí-ficamente probada, o bien …Por razones metodológicas, científicas y apremiantes, el uso del placebo es necesario paradeterminar la eficacia y la seguridad del medicamento en investigación y no implicariesgo, efectos adversos graves o daño irreversible para los pacientes que reciben el pla-cebo.
Tras todo lo anterior, este CEIC AUTORIZA (NO AUTORIZA)(1) por unanimidad (mayoría)(1),que el ensayo clínico referenciado al inicio sea llevado a cabo en el centro , bajo la
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
91

dirección de (apellidos, nombre, categoría profesional, servicio o especialidad) comoInvestigador principal.
Del presente informe se remite copia a:Dirección del centro investigadorCoordinador del PICMEPromotorInvestigador principalServicio de Farmacia (si procede)
Siendo las horas del día indicado al inicio, se da por finalizada la reunión levan-tándose la presente Acta de lo acordado.Doy fe.
EL SECRETARIO
Vº Bº EL PRESIDENTE
Fdo:
Fdo:
VOCALES
Fdo: Fdo: Fdo:
Fdo: Fdo: Fdo:
Fdo: Fdo: Fdo:
92

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
93


El proceso de obtención del Consentimiento Informado


En cualquier investigación en la que participen como sujetos de experimentación se-res humanos, la máxima expresión del respeto a la autonomía de los mismos, entendida comola capacidad de éstos de decidir acerca de lo más conveniente para sus intereses, es la solici-tud de su consentimiento libre e informado para participar como sujetos de un ensayo clínico.
El origen de esta obligación ética de cualquier investigador, se encuentra contempladaen el Código de Nüremberg, cuando dice que el sujeto, posible participante en una investiga-ción, “… debe tener capacidad legal para dar su consentimiento; debe estar situada (la per-sona) en tal forma que le permita ejercer su libertad de escoger, sin la intervención de cualquierotro elemento de fuerza, fraude, engaño, coacción o algún otro factor posterior para obligar acoercer, y debe tener el suficiente conocimiento y comprensión de los elementos de la mate-ria en cuestión para permitirle tomar una decisión correcta”.
De esta definición cabe destacar que el sujeto debe ser capaz, legal e intelectualmentede otorgar su consentimiento y que no es admisible ningún tipo de fraude, engaño o coacción.
Son poblaciones o grupos de población vulnerables aquellas que no tienen dicha ca-pacidad legal y/o intelectual para otorgar su consentimiento, tales como niños y personas dis-capacitadas y también aquellas otras cuyas circunstancias las conviertan en fácilmentecoercionables, tales como residentes en instituciones benéficas, residencias de ancianos (so-bre todo ancianos dependientes), prisiones, o sujetos que, a su vez, ostenten la condición deenfermos y sean atendidos por el investigador que les propone su participación en el ensayo,siempre que la dependencia de éste sea tal que pueda influir en la decisión autónoma del su-jeto.
Por último, a pesar de que el Código de Nüremberg utiliza la expresión “voluntaryconsent” y no “informed consent”, reconoce que para que el posible sujeto de la investi-gación pueda tomar la decisión correcta debe tener el suficiente conocimiento y compren-sión de aquello para lo que se le propone su participación.
Con el devenir del tiempo y la evolución de los conceptos del código mencionado,se han implementado medidas, unas de carácter legal y otras de carácter ético, para sal-vaguardar los derechos de estas poblaciones o grupos vulnerables.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
97

La normativa en vigor dispone la obligación de recabar el consentimiento, previa lainformación pertinente, del representante legal o persona o personas que ostenten la patriapotestad del menor. En este último caso de menores de edad, debe tenerse en cuenta quesi la patria potestad es compartida por ambos progenitores, deben ser ambos quienes otor-guen el consentimiento. En el caso de que la ostente sólo uno de los padres, se debe soli-citar el consentimiento sólo a éste, previa constatación documental de dicha circunstancia.
El consentimiento deberá reflejar la presunta voluntad del menor y podrá retirarseen cualquier momento sin perjuicio alguno para él. Cuando el menor tenga 12 o más años,deberá prestar además su consentimiento para participar en el ensayo.
Es obligación del promotor poner en conocimiento del Ministerio Fiscal las auto-rizaciones de los ensayos clínicos cuya población incluya a menores.
Por otro lado, en las investigaciones planteadas en este tipo de poblaciones, las normaséticas (Declaración de Helsinki, Seúl 2008, punto 17.) dicen que “la investigación médica enuna población o comunidad con desventajas o vulnerable sólo se justifica si la investigación res-ponde a las necesidades y prioridades de salud de esta población o comunidad y si existen po-sibilidades razonables de que la población o comunidad, sobre la que la investigación se realiza,podrá beneficiarse de sus resultados”. Es decir que solo debe realizarse este tipo de investiga-ción si, para obtener el conocimiento buscado, es absolutamente necesario realizarlo en esta po-blación y no puede obtenerse utilizando grupos de población no vulnerable.
Del mismo modo, la planificación de la investigación será tal que los sujetos estén ex-puestos sólo a riesgos mínimos y tanto la evaluación para la autorización inicial como el segui-miento del ensayo se llevarán a cabo por el CEIC de forma minuciosa y monitorizando el proceso.
En cualquier caso, estos ensayos sin interés terapéutico en poblaciones vulnera-bles sólo podrán plantearse siempre:
a) Que no puedan obtenerse los objetivos del ensayo mediante otro con sujetosque pueden dar su consentimiento informado personalmente.
b) Que el riesgo previsible para el sujeto sea mínimo.
c) Que el impacto negativo sobre el bienestar del sujeto sea bajo y además quesea el menor posible.
d) Que el ensayo no esté prohibido por la ley.
e) Que se solicite expresamente el dictamen favorable del CEIC para la inclusiónde tales sujetos y que por tanto el dictamen favorable cubra este aspecto.
98

La legislación y las normas de aplicación europeas solo permiten la inclusión desujetos en proyectos de investigación sin la obtención previa del Consentimiento Infor-mado, otorgado por el propio sujeto o por su representante legal, ante situaciones de ur-gencia, por imposibilidad física o psíquica pasajeras y con el dictamen previo de un CEIC.
En la normativa española, el RD 223/2004 dispone que, cuando el ensayo clínico tengaun interés específico para la población en la que se realiza la investigación y lo justifiquen ra-zones de necesidad en la administración del medicamento en investigación, podrá someterse aun sujeto a un ensayo clínico sin obtener el consentimiento previo en los siguientes casos:
a) Si existe un riesgo inmediato grave para la integridad física o psíquica del su-jeto, se carece de una alternativa terapéutica apropiada en la práctica clínica y no es po-sible obtener su consentimiento o el de su representante legal. En este caso, siempre quelas circunstancias lo permitan, se consultará previamente a las personas vinculadas a élpor razones familiares o de hecho.
b) Si el sujeto no es capaz para tomar decisiones debido a su estado físico o psí-quico y carece de representante legal. En este caso, el consentimiento lo prestarán las per-sonas vinculadas a él por razones familiares o de hecho.
En ambos casos, esta eventualidad y la forma en que se procederá debe hallarseprevista en la documentación del ensayo aprobada por el Comité Ético de Investigación Clí-nica, y el sujeto o su representante legal será informado en cuanto sea posible y deberáotorgar su consentimiento para continuar en el ensayo si procediera.
No obstante, la legislación de algunos países contempla la posibilidad de realiza-ción de ensayos clínicos exentos del Consentimiento previo, libre e informado de los su-jetos. Así, en el Código de Reglamentos Federales de los EEUU de Norteamérica(45CFR46), se permite dicha excepción, siempre que este extremo esté supervisado porun comité de ética local, cuando se considere que la información que debe dársele al su-jeto puede poner en riesgo la obtención del conocimiento perseguido en la investigacióny siempre que se den los cuatro criterios siguientes:
a) La investigación no debe suponer más que un riesgo mínimo para el sujeto.
b) La exención no afectará negativamente ni los derechos ni el bienestar de losmismos.
c) La investigación no pueda ser realizada sin la exención.
d) En el momento apropiado, los participantes recibirán la información pertinente.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
99

De las condiciones anteriores, llama la atención la aparente contradicción exis-tente en el apartado b), pues, según la concepción europea (Normas de Buena PrácticaClínica) es contradictorio el ocultamiento de la información con el respeto a los derechosindividuales de los sujetos. El informe Belmont, intenta resolver el conflicto proponiendoque se indique a los sujetos la circunstancia de que algunos aspectos del ensayo no se lesvan a revelar hasta que haya concluido la investigación, con lo que éstos, si acceden,otorgan su consentimiento al ocultamiento de dicha información.
Al recabar el Consentimiento Informado de un posible participante en el ensayo,si éste es paciente del investigador y pueda existir entre ambos una cierta relación de de-pendencia, el sujeto pasa a pertenecer a un “grupo de población vulnerable”, en cuyo casoel investigador debe delegar la solicitud de participación en otro profesional cualificado,incluso ajeno al proyecto de investigación, el cual deberá informar sin ninguna duda delderecho que asiste al paciente a no participar sin que esto suponga ningún menoscabo nien su asistencia ni en su relación con su médico habitual.
En cuanto a la última parte de la definición vista al inicio, contenida en el Códigode Nüremberg, en relación a que el sujeto “… debe tener el suficiente conocimiento ycomprensión de los elementos de la materia en cuestión para permitirle tomar una deci-sión correcta”, hay que tener en cuenta que la capacidad de comprensión de una personadepende de la inteligencia, de la capacidad de razonamiento, de la madurez y del domi-nio del lenguaje, por tanto, si al transmitir la información no se tienen en cuenta estas cir-cunstancias, diferentes para cada sujeto, el procedimiento de obtención delconsentimiento puede quedar invalidado.
La información, teniendo en cuenta lo anterior, debe ser facilitada por escrito yoralmente y debe referirse como mínimo a los siguientes aspectos:
a) Que el ensayo representa una investigación.
b) El propósito del ensayo.
c) Los tratamientos del ensayo y la probabilidad de asignación aleatoria para cadatratamiento.
d) Los procedimientos a seguir en el ensayo, incluyendo todos los procedimien-tos invasivos.
e) Las responsabilidades del sujeto.
f) Aquellos aspectos del ensayo que son experimentales.
g) Los riesgos o inconveniencias razonablemente previsibles para el sujeto y, ensu caso, para el embrión, feto o niño lactante.
100

h) Los beneficios razonablemente esperados. Se deberá informar claramente al su-jeto en aquellos casos en que no se pretende ningún beneficio clínico específicopara él.
i) Los procedimientos o tratamientos alternativos disponibles para el sujeto y sus po-sibles beneficios y riesgos más importantes.
j) La indemnización y/o tratamiento disponible para el sujeto en caso de cualquierperjuicio relacionado con el ensayo.
k) El prorrateo previsto de pago, si lo hay, al sujeto por su participación en el ensayo.
l) Los gastos previsibles, si los hay, al sujeto por su participación en el ensayo.
m) Que la participación del sujeto en el ensayo es voluntaria y que el sujeto puedenegarse a participar o retirarse del ensayo en cualquier momento, sin ningunapenalización ni pérdida de los beneficios a los que hubieses tenido derecho deotro modo.
n) Que los monitores, auditores, CEIC, y las autoridades competentes tendrán accesodirecto a la historia clínica original del sujeto para la verificación de los procedi-mientos y/o datos del ensayo clínico, sin violar la confidencialidad del sujeto,dentro de lo permitido por la normativa pertinente y que, al firmar el consenti-miento informado, el sujeto o su representante legal está autorizando el accesoa estos datos.
o) Que los registros que identifican al sujeto serán confidenciales y, según lo permi-tido por las leyes y/o regulaciones pertinentes, no estarán a disposición pública. Sise publican los resultados del ensayo, la identidad del sujeto será confidencial.
p) Que se informará al sujeto o al representante legal del sujeto en todo momento sise dispone de nueva información que pueda modificar su decisión de continuar enel ensayo.
q) Las personas de contacto para obtener información adicional del ensayo y de losderechos de los sujetos participantes, y con quien contactar en caso de lesiones re-lacionadas con el mismo.
r) Las circunstancias y/o razones previsibles bajo las cuales puede finalizar la parti-cipación del sujeto en el ensayo.
s) La duración esperada de la participación del sujeto en el ensayo.
t) El número aproximado de sujetos implicados en el ensayo.
Quien solicite el consentimiento del sujeto, debe confirmar que éste ha compren-dido toda la información facilitada. El informe Belmont incluso recomienda practicar unaprueba escrita para evidenciar lo anterior.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
101

A medida que avance la investigación y se obtenga nueva información, deberá po-nerse en conocimiento de los sujetos y recabar de nuevo su consentimiento.
El respeto a la autonomía del sujeto obliga también a que medie un cierto pe-riodo de tiempo, no inferior a 24 horas, entre el momento de facilitar la información y elde documentar (firmar) el otorgamiento del consentimiento informado y libre.
Por último cabe reseñar que el respeto a la autonomía del sujeto, cuya máxima ex-presión como hemos visto, consiste en recabar por parte del investigador, el consenti-miento informado, entendido, voluntario y libre de los sujetos participantes en un ensayoclínico, entraña una responsabilidad no menos importante desde el punto de vista ético,consistente en la obligación de proteger los derechos de las poblaciones o grupos vulne-rables sobre los que pueda plantearse una investigación, convirtiéndose así en garantes deesta circunstancia.
102

Interpretación actual y vigencia del principio “Primum non Nocere”


La Declaración de Helsinki, en su punto 8 de la Introducción dice que “En lapráctica de la medicina y de la investigación médica, la mayoría de las intervenciones im-plican algunos riesgos y costos.”
En realidad, como veíamos al inicio de este trabajo, en la práctica actual de la me-dicina y de la investigación biomédica, cada técnica, cada proceso, cada nuevo trata-miento es, progresivamente, más agresivo aunque también sea más eficaz.
El Institute of Medicine de Estados Unidos publicó en el año 2000 un texto lla-mado “Errar es humano. Construyendo un sistema de salud más seguro”, en el que seda cuenta de entre 45.000 y 98.000 muertes anuales por errores médicos. Se estimaque más del 13% de los ingresos en un hospital se deben a efectos adversos del diag-nóstico o del tratamiento y que más del 50 % de las complicaciones iatrogénicas sonprevenibles.
La aplicación estricta del principio “Primum non nocere” produciría la parálisisde la práctica médica. Colocar este compromiso “sobre todo”, “antes que nada” y “pri-mero que nada” hace énfasis en lo paradójico que resulta que una profesión que tiene elpropósito de hacer el bien pueda resultar dañina, y que a veces resultaría preferible ni si-quiera intentar hacer el bien si al hacerlo se pudiera generar daño.
Llegamos así a considerar el doble efecto de los actos médicos: uno deseable(extirpación de un órgano o miembro por un tumor) y otro indeseable (impotencia funcio-nal, mutilación, dolor, dependencia farmacológica de sustitución etc.).
Este principio del doble efecto señala que es lícito realizar una acción de la quese siguen dos efectos, uno bueno y otro malo, siempre y cuando se satisfagan cuatro con-diciones:
1) La acción en cuestión ha de ser buena o, al menos, no mala; es decir, indife-rente o permitida.
2) No se desea el mal resultado; no entra en la intención del médico causar mal al-guno.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
105

3) El buen resultado no es consecuencia del mal, es decir, no se usa un mal comomedio para obtener el fin (bueno), sino que aquel es un hecho advenido nadamás.
4) Lo bueno tiene que ser proporcionado, es decir, en el resultado final el bien ob-tenido debe superar al mal accidental acumulado.
La posición de los médicos al respecto, puede oscilar entre dos extremos, a saber:la de aquellos que opinan que lo importante es salvar la vida sin importar que se causesufrimiento o daño iatrogénico y en el otro extremo se ubican los partidarios de intervenirlo menos posible para permitir actuar a las “fuerzas de la naturaleza”. Los primeros tien-den a producir daños por comisión y los segundos por omisión.
Sin embargo, la gran mayoría de médicos se sitúan en posiciones intermedias,generalmente lejos de ambos extremos y en la actualidad, el precepto “Primum non no-cere” lo que considera es una auténtica ponderación del cociente beneficio/daño o bene-ficio/riesgo, es decir, que en algunos casos vale la pena correr el riesgo de producir un dañopuesto que se obtendrá un beneficio considerable y, en todo caso, siempre intentando mi-nimizar tanto el riesgo como la magnitud del daño mismo.
Por todo ello, teniendo en cuenta como valor ético universal el respeto a la auto-nomía del paciente o sujeto de experimentación, entendido como el derecho que le asistea decidir de forma voluntaria e informada y también el principio del doble efecto de losactos médicos, el rol del médico, tanto en su faceta asistencial como investigadora, debeevolucionar desde la actitud paternalista de no hace muchas décadas hacia la figura deasesor cualificado que ayuda a tomar decisiones, haciéndose corresponsable de las mis-mas y aportando al proceso elegido por el sujeto/enfermo, todo su bagaje ético, sus cono-cimientos médicos y sus habilidades técnicas.
106

Anexos


LEY 29/2006, DE 26 DE JULIO, DE GARANTÍAS Y USO RACIONAL DE LOS MEDICAMENTOS
Y PRODUCTOS SANITARIOS
• EXPOSICIÓN DE MOTIVOS
• TÍTULO III De las garantías de la investigación de los medicamentos de uso humano
• TÍTULO VIII Régimen sancionador
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
109


LEY 29/2006, DE 26 DE JULIO, DE GARANTÍAS Y USO RACIONAL DE LOS MEDICAMENTOS Y PRODUCTOS SANITARIOS
EXPOSICIÓN DE MOTIVOS
I
La Ley 25/1990, de 20 de diciembre, del Medicamento pretendía, según se se-ñala en su exposición de motivos, dotar a la sociedad española de un instrumento insti-tucional que le permitiera esperar confiadamente que los problemas relativos a losmedicamentos fueran abordados por cuantos agentes sociales se vieran involucrados ensu manejo, (industria farmacéutica, profesionales sanitarios, poderes públicos y los pro-pios ciudadanos), en la perspectiva del perfeccionamiento de la atención a la salud. Losquince años transcurridos desde la aprobación de la citada Ley permiten afirmar que seha alcanzado en gran parte el objetivo pretendido consagrándose la prestación farmacéu-tica como una prestación universal.
La prestación farmacéutica comprende los medicamentos y productos sanitariosy el conjunto de actuaciones encaminadas a que los pacientes los reciban y utilicen deforma adecuada a sus necesidades clínicas, en las dosis precisas según sus requerimien-tos individuales, durante el período de tiempo adecuado, con la información para su co-rrecto uso y al menor coste posible.
Es necesario hacer una valoración positiva de lo que son y representan los medi-camentos y los productos sanitarios para el Sistema Nacional de Salud, por lo que la po-lítica farmacéutica desarrollada durante este periodo se ha orientado en la dirección deasegurar su disponibilidad para cubrir las necesidades de los pacientes. A lo largo de es-tos años se ha completado la descentralización sanitaria prevista en la Ley General de Sa-nidad de 1986 y así, desde comienzos del año 2002, todas las Comunidades Autónomashan asumido las funciones que venía desempeñando y los servicios que venía prestandoel Instituto Nacional de Salud, lo que supone una descentralización completa de la asis-tencia sanitaria del Sistema Nacional de Salud, incluida la de la prestación farmacéutica.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
111

En los últimos años el papel de los profesionales del sector ha sido fundamentalen estos logros. El médico es una figura central en las estrategias de impulso de la cali-dad en la prestación farmacéutica dado el papel que se le atribuye en el cuidado de la sa-lud del paciente y, por tanto, en la prevención y el diagnóstico de la enfermedad, así comoen la prescripción, en su caso, de tratamiento con medicamentos. El trabajo que los far-macéuticos y otros profesionales sanitarios realizan en los procedimientos de atención far-macéutica también tiene una importancia esencial ya que asegura la accesibilidad almedicamento ofreciendo, en coordinación con el médico, consejo sanitario, seguimientofarmacoterapéutico y apoyo profesional a los pacientes.
El desafío actual es asegurar la calidad de la prestación en todo el Sistema Na-cional de Salud en un marco descentralizado capaz de impulsar el uso racional de los me-dicamentos y en el que el objetivo central sea que todos los ciudadanos sigan teniendoacceso al medicamento que necesiten, cuando y donde lo necesiten, en condiciones deefectividad y seguridad.
II
Durante estos años, la modificación de la configuración jurídica y la composiciónde las estructuras de la Unión Europea, obligada por los nuevos retos y necesidades emer-gentes, ha afectado a la regulación, entre otros, del sector farmacéutico, obligando a nues-tro país a revisar la normativa interna vigente. Por este motivo se incorporan a través deesta Ley a nuestro ordenamiento jurídico la Directiva 2004/27/CE del Parlamento Euro-peo y del Consejo, de 31 de marzo de 2004, que modifica la Directiva 2001/83/CE, porla que se establece un código comunitario sobre medicamentos de uso humano, y la Di-rectiva 2004/28/CE del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, quemodifica la Directiva 2001/82/CE por la que se establece un código comunitario sobre me-dicamentos veterinarios. Además, también se asegura la armonización de nuestra norma-tiva con el Reglamento (CE) n.º 726/2004, por el que se establecen los procedimientoscomunitarios para la autorización y el control de los medicamentos de uso humano y ve-terinario y por el que se crea la Agencia Europea de Medicamentos.
La experiencia derivada de la aplicación de la Ley 25/1990 ha puesto en eviden-cia la necesidad de intensificar la orientación de la reforma en torno a dos ideas-fuerza:la ampliación y reforzamiento de un sistema de garantías que gire en relación a la autori-zación del medicamento y la promoción del uso racional del mismo. Es de señalar que lacitada Ley se refería ya a la primera de ellas al establecer la exigencia de garantía de ca-lidad, seguridad y eficacia de los medicamentos. Sin embargo, el desarrollo tecnológico,la globalización y el acceso a la información así como la pluralidad de agentes que pro-
112

gresivamente intervienen en el ámbito de la producción, distribución, dispensación y ad-ministración de medicamentos aconsejan en estos momentos, además de intensificar di-chas garantías, ampliarlas a la transparencia y objetividad de las decisiones adoptadasasí como al control de sus resultados.
La transferencia de competencias a las Comunidades Autónomas en materia desanidad iniciada con anterioridad a la Ley 25/1990, de 20 de diciembre, se ha ampliadoy extendido a todas las Comunidades Autónomas con posterioridad a la entrada en vigorde la citada Ley. La gestión de las Comunidades Autónomas en materia de sanidad com-prende un amplio espectro de políticas en cuanto a prioridades en el tratamiento de losproblemas de salud, introducción de nuevas tecnologías y nuevos tratamientos, promociónde las alternativas más eficientes en los procesos diagnósticos y terapéuticos desarrolla-dos por los profesionales de las respectivas Comunidades Autónomas, así como en políti-cas de rentas que afectan a los sistemas retributivos y de incentivos económicos aprofesionales y centros sanitarios, todo ello dentro del amplio margen que corresponde alejercicio de las competencias asumidas en el marco de los criterios establecidos por la Ley16/2003, de Cohesión y Calidad del Sistema Nacional de Salud, y demás normativa es-tatal sobre la materia.
Además, el Plan Estratégico de Política Farmacéutica para el Sistema Nacional deSalud establece diversas estrategias que se incorporan en esta Ley para intensificar el usoracional de los medicamentos, entre las que se pueden señalar las orientadas a ofrecer unainformación de calidad, periódica e independiente a los profesionales, a garantizar una for-mación sobre uso racional de los medicamentos a los profesionales sanitarios, al refuerzode la exigencia de la receta médica como documento imprescindible para la seguridad delpaciente o las referidas a la modificación de los prospectos de los medicamentos para ha-cerlos inteligibles a los ciudadanos, ayudando a la consecución de la necesaria adheren-cia al tratamiento para que pueda alcanzarse el éxito terapéutico previsto por el médicocon la imprescindible cooperación del farmacéutico.
Es necesario que nuestro Sistema garantice a los profesionales sanitarios que lainformación, la formación y la promoción comercial de los medicamentos tengan como ele-mentos centrales de su desarrollo el rigor científico, la transparencia y la ética en la prác-tica de estas actividades.
Aunque los medicamentos han contribuido decisivamente a la mejora de la espe-ranza y al aumento de la calidad de vida, en ocasiones plantean problemas de efectividady de seguridad que han de ser conocidos por los profesionales por lo que cobra especialrelevancia el protagonismo que esta Ley otorga al sistema español de farmacovigilancia del
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
113

Sistema Nacional de Salud, con un enfoque más innovador, que incorpora el concepto defarmacoepidemiología y gestión de los riesgos, y la garantía de seguimiento continuado delbalance beneficio/riesgo de los medicamentos autorizados.
Los próximos años dibujan un panorama con un sensible aumento de la población,un marcado envejecimiento de la misma y, por tanto, unas mayores necesidades sanita-rias derivadas de este fenómeno así como de la cronificación de numerosas patologías. Es-tas necesidades tienen que garantizarse en un marco riguroso en cuanto a las exigenciasde seguridad y eficacia de los medicamentos en beneficio de la calidad asistencial paralos ciudadanos.
El crecimiento sostenido de las necesidades en materia de prestación farmacéuticatendrá, por tanto, que enmarcarse necesariamente en estrategias de uso racional de los me-dicamentos y de control del gasto farmacéutico, que permitan seguir asegurando una pres-tación universal de calidad contribuyendo a la sostenibilidad del Sistema Nacional de Salud.
En este sentido, la Ley considera necesario que la financiación selectiva y no in-discriminada de medicamentos se realice en función de la utilidad terapéutica de los mis-mos y de su necesidad para mejorar la salud de los ciudadanos.
Se modifica también en esta Ley el sistema de precios de referencia para posibi-litar los necesarios ahorros al Sistema Nacional de Salud y asegurar la previsibilidad, laestabilidad y la gradualidad en el impacto para la industria farmacéutica, afectando a to-dos los medicamentos en fase de madurez en el mercado.
La aparición en estos años de los medicamentos genéricos, productos de eficaciaclínica demostrada y más económicos al haber expirado el periodo de exclusividad de da-tos del medicamento original, asegura idénticas condiciones de calidad, seguridad y efi-cacia a menor precio. Por ello, en este objetivo de sostenibilidad, las medidas incorporadasen esta Ley pretenden eliminar los obstáculos que dificultan una mayor presencia de es-tos medicamentos en el mercado, equiparando la situación española con la de otros paí-ses de nuestro entorno.
Esta Ley aborda todos estos elementos e incorpora a la prestación farmacéuticalas novedades pertinentes, las más relevantes de las cuales se señalan a continuación.
III
El título I aborda las disposiciones generales de la Ley, definiendo con precisiónsu ámbito de aplicación, extensivo tanto a los medicamentos de uso humano como vete-
114

rinario, y las garantías de abastecimiento y dispensación que han de procurar laboratoriosfarmacéuticos, almacenes mayoristas, oficinas de farmacia y demás agentes del sector.Particularmente novedosa es la regulación de las garantías de independencia de los pro-fesionales del sector, que se traduce básicamente en una más precisa definición de los su-puestos en que pueden surgir conflictos de intereses, de la que es fiel reflejo la prohibiciónde conceder cualquier tipo de incentivo, bonificación, descuento no permitido, prima u ob-sequio por parte de quien tenga intereses directos o indirectos en la producción, fabrica-ción y comercialización de medicamentos y productos sanitarios. Como reconoce laDirectiva 2001/83/CE, no debe permitirse otorgar, ofrecer o prometer a las personas fa-cultadas para prescribir o dispensar medicamentos y en el marco de la promoción de losmismos frente a dichas personas, primas, ventajas pecuniarias o ventajas en especie. Eltítulo se cierra con normas relativas a la defensa y protección de la salud pública y a lacolaboración y participación Interadministrativa.
IV
El título II, «De los medicamentos», contempla a lo largo de seis capítulos la re-gulación de todos los aspectos relacionados con los mismos.
El capítulo I aborda una serie de modificaciones que traen causa de las directi-vas mencionadas, entre las que cabe destacar el abandono del concepto de especialidadfarmacéutica sobre el que ha venido asentándose la normativa española, y que afecta a ladefinición de los medicamentos legalmente reconocidos, la nueva definición de medica-mento de uso humano, el concepto de genérico armonizado en la Unión Europea y la in-corporación de la definición de medicamento de uso veterinario.
El capítulo II incorpora criterios europeos de protección de la innovación, inves-tigación y desarrollo, para colaborar en el fomento de la competitividad del sector en Es-paña. Particular importancia reviste el nuevo sistema de exclusividad de datos, diverso yplenamente respetuoso con la necesaria protección de la propiedad intelectual e indus-trial asociadas a la innovación, al tiempo que promueve la disponibilidad rápida de gené-ricos en el mercado. De acuerdo con el mismo, el solicitante de un producto genéricopuede presentar la solicitud de autorización transcurridos ocho años como mínimo desdeque se autorizó el medicamento de referencia en cualquier Estado miembro de la UniónEuropea, lo que permitirá ir realizando la evaluación y tramitación administrativa para suautorización, si bien se garantiza el cumplimiento del periodo armonizado de exclusividadde los datos de la innovación al establecer que no podrá comercializar el medicamentohasta transcurridos diez años, u once si obtiene una indicación adicional con beneficio clí-nico significativo en comparación con las terapias existentes.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
115

Este régimen de protección se completa, como no podía ser de otra manera, con laincorporación, con fines aclaratorios, mediante la oportuna modificación de la Ley de Paten-tes, de la denominada «cláusula o estipulación Bolar», según la cual no se considera viola-ción del derecho de patente la realización con fines experimentales de los estudios y ensayosnecesarios para la autorización de medicamentos genéricos. Además, se incorpora la habi-litación para, reglamentariamente, abrir la posibilidad de introducir en el mercado los me-dicamentos genéricos con marca, como consecuencia de la nueva regulación europea.
Es importante también la incorporación del concepto de «autocuidado de la sa-lud» relacionado con la calificación de medicamentos como medicamentos no sujetos aprescripción médica. Se trata de adaptar la redacción de la Ley a la realidad social del si-glo XXI, en la que cada vez tiene más importancia el uso de medicamentos sin prescrip-ción en las condiciones que se establecen, pero que debe ser realizada en el marco de unuso racional del medicamento, a cuyos fines es imprescindible el papel del farmacéuticoen todo lo relacionado con una automedicación responsable.
El capítulo III regula los medicamentos veterinarios, lo que constituye una autén-tica novedad de la Ley, que acoge una concepción de estos medicamentos alejada de laconsideración de medicamentos especiales. Se regulan como medicamentos dotados de ca-racterísticas propias, toda vez que por efecto de la normativa comunitaria europea de apli-cación, los citados medicamentos han de cumplir las garantías generales de calidad,seguridad y eficacia para la salvaguardia de la salud y el bienestar de los animales, así comola salud pública. Por este motivo se ha considerado necesario incorporar a la Ley un capí-tulo donde se regulen específicamente los aspectos fundamentales de estos medicamentos.
Asimismo, se incorpora como regla general la exigencia de prescripción veterina-ria previa a la dispensación de medicamentos destinados a los animales productores dealimentos y se regula el sistema español de farmacovigilancia veterinaria, necesario porcuanto la Directiva 2001/82/CE impone el necesario refuerzo de los sistemas de farma-covigilancia.
El capítulo IV se refiere a las garantías sanitarias de las fórmulas magistrales y pre-parados oficinales. Las primeras son preparadas con sustancias de acción e indicación re-conocidas legalmente en España en las oficinas de farmacia y servicios farmacéuticoslegalmente establecidos que dispongan de los medios necesarios para su preparación. Lospreparados oficinales deberán cumplir determinadas condiciones, entre las que destacala necesidad de presentarse y dispensarse bajo principio activo, denominación común in-ternacional (DCI) o, en su defecto, denominación común o científica y en ningún casobajo marca comercial.
116

El capítulo V regula las garantías sanitarias de los medicamentos especiales, en-tendiendo como tales a aquellos medicamentos que por sus características particulares re-quieren una regulación específica. En esta categoría se incluyen las vacunas y demásmedicamentos biológicos, los medicamentos de origen humano, los medicamentos de te-rapia avanzada, los radiofármacos, los medicamentos con sustancias psicoactivas con po-tencial adictivo, los medicamentos homeopáticos, los de plantas medicinales y los gasesmedicinales. De entre todos ellos, debe destacarse a los medicamentos de terapia celu-lar. El texto pretende aclarar que la Ley y la normativa europea relativa a garantías y con-diciones de autorización serán aplicables sólo a los que se fabriquen industrialmente; elresto de medicamentos, que no estén destinados a la producción industrial, aún cuandoconcurran en ellos las características y condiciones establecidas en las definiciones de«medicamento de terapia génica» o de «medicamento de terapia celular somática», ten-drán la regulación que reglamentariamente se determine.
En materia de farmacovigilancia, tanto de medicamentos de uso humano como deuso veterinario, el capítulo VI regula las actividades de salud pública tendentes a la iden-tificación, cuantificación, evaluación y prevención de los riesgos del uso de los medica-mentos una vez comercializados, permitiendo así el seguimiento de sus posibles efectosadversos, siendo de destacar el sistema español de farmacovigilancia, en el que las Ad-ministraciones sanitarias han de realizar lo necesario para recoger, elaborar y, en su caso,procesar toda la información útil para la supervisión de medicamentos y, en particular, lainformación sobre reacciones adversas a los mismos, así como para la realización de cuan-tos estudios se consideren necesarios para evaluar su seguridad.
V
El título III regula, bajo la rúbrica «de las garantías de la investigación de los me-dicamentos de uso humano», los ensayos clínicos con medicamentos. Destaca como no-vedad, y como garantía de transparencia, la posibilidad de que la Administración sanitariapueda publicar los resultados de los ensayos clínicos cuando dicha publicación no se hayallevado a cabo por el promotor del mismo en plazo y siempre que los citados resultadospermitan concluir que el producto presenta modificaciones de su perfil de eficacia o deseguridad; y ello, porque se toma en especial consideración el interés que, tanto para lospacientes que han participado en el ensayo como para los médicos y para la población engeneral, reviste el poder conocer los resultados del mismo, si de éstos se deriva que el me-dicamento plantea problemas de eficacia o de seguridad. Además, se mantiene el régimende autorización administrativa previa, respetando los derechos fundamentales de la per-sona y los postulados éticos que afectan a la investigación biomédica, y la necesidad deque se cumplan las normas de buena práctica clínica como requisitos indispensables para
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
117

garantizar la idoneidad del ensayo. Del mismo modo se mantienen las garantías de in-demnización para los sujetos que pudieran verse perjudicados por su participación en losensayos clínicos mediante la exigencia del aseguramiento previo de los daños y perjuiciosque pudieran derivarse de aquéllos.
Por otra parte, la Ley faculta al Ministerio de Sanidad y Consumo para el desarro-llo de acciones que permitan que los Comités Éticos de Investigación clínica acreditadospor las Comunidades Autónomas puedan compartir estándares de calidad y criterios deevaluación adecuados y homogéneos en el conjunto del Sistema Nacional de Salud.
VI
Las novedades introducidas en el capítulo I del título IV se orientan, conformeexige la normativa comunitaria de aplicación, a garantizar la calidad de los medicamen-tos de uso humano y veterinario disponibles en el mercado, exigiendo que se respeten losprincipios relativos a las prácticas correctas de fabricación de estos medicamentos.
Por ello, se incorpora en esta Ley la autorización administrativa a las entidades que fa-briquen medicamentos para su exportación y a las que fabriquen productos intermedios, asícomo la obligación para los fabricantes de principios activos utilizados como materias primasde cumplir las normas de correcta fabricación de las mismas. Por su parte, los laboratorios de-berán utilizar únicamente, como materias primas, principios activos fabricados de conformidadcon las directrices detalladas en las normas de correcta fabricación de dichas materias primas.
El capítulo II refuerza las obligaciones de los almacenes mayoristas, en especialen el ámbito del abastecimiento. La Ley permite la utilización de estos intermediarios queposibilitan la llegada del medicamento a cualquier parte del territorio en un tiempo mí-nimo, lo que permite garantizar el acceso del ciudadano al medicamento cuando lo nece-site. Precisamente por este motivo deben asumir una serie de obligaciones con el SistemaNacional de Salud, entre las que debe destacar el tenerlo continuamente abastecido. Ade-más, deben disponer de locales y medios precisos, garantizar la observancia de las con-diciones generales o particulares de conservación de los medicamentos, mantener unasexistencias mínimas, asegurar plazos de entrega, frecuencia mínima de repartos, cumplirservicios de guardia y prevención de catástrofes, etc.
VII
El título V está dedicado a las garantías sanitarias del comercio exterior de medi-camentos, un ámbito que en un marco cada vez más globalizado va adquiriendo una ma-
118

yor relevancia. Se regulan en este título las importaciones y exportaciones y el régimen delos medicamentos destinados al tratamiento de los viajeros.
VIII
El título VI está dedicado al uso racional de los medicamentos, principio que seconcreta en medidas como una nueva regulación de la receta médica o la prohibición deque las actividades relacionadas con el proceso de puesta en el mercado de un medica-mento tengan por finalidad aumentar las capacidades físicas de los deportistas. En rela-ción con la receta médica, es destacable la previsión que contiene la Ley, que atribuye almédico u odontólogo en exclusiva la facultad de prescribir medicamentos, tendente a erra-dicar prácticas no deseables. La receta médica se configura como una auténtica garantíade servicio profesional para el paciente, por lo que el farmacéutico dispensará con recetaaquellos medicamentos que la requieran y no podrá prescribir por sí mismo un medica-mento que precise de receta médica, pero sí colaborar en el seguimiento farmacoterapéu-tico de los tratamientos prescritos, a través de los procedimientos de la atenciónfarmacéutica.
La Ley contiene una precisa y concreta regulación de las obligaciones de trazabi-lidad. Se impone la obligación, tanto a laboratorios como a almacenes mayoristas y ofici-nas de farmacia, de garantizar la adecuada trazabilidad de los medicamentos, comomedida que coadyuva tanto a evitar un eventual desabastecimiento como a suministrar unaprecisa información sobre el destino último de los medicamentos comercializados en Es-paña. Como reconoce la Directiva 2004/27/CE, es necesario controlar el conjunto de la ca-dena de distribución de medicamentos, desde su fabricación o su importación hasta sudespacho al público, de forma que quede garantizado que los medicamentos se conser-van, transportan y manipulan en condiciones adecuadas. Las disposiciones que convieneadoptar con este objetivo facilitarán considerablemente la retirada del mercado de produc-tos defectuosos y permitirán luchar más eficazmente contra las imitaciones fraudulentas.
Por otra parte, se adoptan medidas que pretenden reforzar la política de promo-ción de medicamentos genéricos conforme a lo establecido en el Plan Estratégico de Po-lítica Farmacéutica.
IX
El título VII está dedicado a la financiación pública de los medicamentos. Regulael régimen de fijación y revisión de precios industriales y de márgenes de distribución ydispensación, incorporando, como criterio para la fijación de precio, la valoración de la uti-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
119

lidad terapéutica del medicamento y el grado de innovación, consecuencia de la aplica-ción efectiva del principio de financiación selectiva de medicamentos, principio que debeinspirar la incorporación de todo medicamento al Sistema Nacional de Salud.
Para garantizar la máxima objetividad en la fijación de precios, se tendrán en conside-ración los informes sobre utilidad terapéutica de los medicamentos que elabore la Agencia Es-pañola de Medicamentos y Productos Sanitarios, con la colaboración de una red de expertosindependientes de reconocido prestigio científico propuestos por las Comunidades Autónomas.
Una de las novedades de este título es la modificación del sistema de precios dereferencia. Este sistema de control del gasto farmacéutico es común en los países de nues-tro entorno. Sin embargo, en la experiencia de su implantación en nuestro país, en los úl-timos años se han venido detectando deficiencias en su diseño que con este modelo seintentan paliar. Incorpora como novedades más relevantes la gradualidad en su impacto,la objetividad, al afectar a todos los medicamentos con más de diez años en el mercadou once si han tenido alguna nueva indicación, y su previsibilidad, lo que determina laconfiguración de un marco predecible para la industria farmacéutica en nuestro país.
Por último, se dispone la aplicación de la normativa sobre medicamentos a los pro-ductos sanitarios que, financiados con fondos públicos, se dispensen, a través de recetaoficial del Sistema Nacional de Salud, en territorio nacional.
X
El título VIII de la Ley está dedicado al régimen sancionador en materia farmacéu-tica, adaptado a las circunstancias actuales del sector y pendiente de revisión desde 1990.Por ello, con base tanto en la normativa comunitaria de reciente aprobación como en lainterna de procedimiento administrativo común y en la experiencia acumulada, se consi-dera necesario adaptar el catálogo de infracciones, modificando la calificación de algunasconductas y creando nuevos tipos de conductas sancionadas.
Entre las modificaciones más importantes, se incorporan algunas nuevas comovender medicamentos sujetos a prescripción médica a través de internet, falsificar medi-camentos, no comunicar, por parte de los laboratorios farmacéuticos, almacenes mayoris-tas y oficinas de farmacia, a las Administraciones sanitarias competentes, las unidades demedicamentos vendidas para su dispensación en territorio nacional; incumplir los requi-sitos que, para la realización de la visita médica, establezca la normativa de las Comuni-dades Autónomas; incumplir el promotor o investigador de un ensayo clínico, lasobligaciones establecidas en la legislación vigente o en las normas de buena práctica clí-
120

nica, así como la realización de un ensayo clínico sin ajustarse al protocolo aprobado o elincumplimiento por parte del titular de la autorización de comercialización de la presen-tación de los informes periódicos de seguridad.
Otras infracciones ven agravada su tipificación, como ocurre con la modificación,por parte del titular de la autorización, de cualquiera de las condiciones por las que seotorgó la misma; el ofrecimiento directo o indirecto y la aceptación de cualquier tipo deincentivo, bonificación, descuento no permitido, prima u obsequio, efectuado por quientenga intereses directos o indirectos en la producción, fabricación y comercialización demedicamentos, a/de los profesionales sanitarios, con motivo de la prescripción, dispensa-ción y administración de los mismos, o a sus parientes y personas de su convivencia o nodisponer las entidades de distribución y dispensación de las existencias de medicamen-tos adecuadas para la normal prestación de sus actividades o servicios.
XI
El título IX incorpora la acción de cesación, medida procesal importada, por ex-presa exigencia de la Directiva 98/27/CE, de 19 de mayo de 1998, del derecho comuni-tario europeo, y concebida para aquellos casos en los que la publicidad de unmedicamento de uso humano sea contraria al contenido de la Ley o de sus disposicionesde desarrollo, afectando a intereses colectivos o difusos de los consumidores y usuarios.El objetivo fundamental de la medida es obtener el cese de la actividad contraria a las nor-mas citadas y prohibir su reiteración futura.
XII
El título X recoge la regulación de las tasas correspondientes a los servicios sumi-nistrados por la Administración en el ámbito material de la Ley. Este título debe su modifi-cación a la necesidad de adecuarlo a lo previsto por la Ley 4/2004, de 29 de diciembre, demodificación de tasas y de beneficios fiscales de acontecimientos de excepcional interés.
XIII
Por último, el texto regula, en su disposición adicional sexta, las aportaciones alSistema Nacional de Salud por parte de los laboratorios, calculadas en función de su vo-lumen de ventas. Tales aportaciones se destinan a la investigación en el ámbito de la bio-medicina y al desarrollo de políticas de cohesión sanitaria y programas de formación parafacultativos médicos y farmacéuticos y de educación sanitaria de la población, para favo-recer el uso racional o responsable de medicamentos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
121

TÍTULO III De las garantías de la investigación de los medicamentos de uso humano
Artículo 58. Ensayos clínicos.
1. A los efectos de esta Ley, se entiende por ensayo clínico toda investigaciónefectuada en seres humanos, con el fin de determinar o confirmar los efectos clínicos,farmacológicos, y/o demás efectos farmacodinámicos, y/o de detectar las reacciones ad-versas, y/o de estudiar la absorción, distribución, metabolismo y eliminación de uno o va-rios medicamentos en investigación con el fin de determinar su seguridad y/o su eficacia.
Todos los ensayos clínicos, incluidos los estudios de biodisponibilidad y bioequi-valencia, serán diseñados, realizados y comunicados de acuerdo con las normas de «buenapráctica clínica» y con respeto a los derechos, la seguridad y el bienestar de los sujetosdel ensayo, que prevalecerán sobre los intereses de la ciencia y la sociedad.
2. Las autoridades sanitarias deberán facilitar la realización de los ensayos clíni-cos en el Sistema Nacional de Salud, tanto en el ámbito de la atención primaria como dela hospitalaria. Las condiciones de desarrollo de los ensayos clínicos en los servicios sa-nitarios del Sistema Nacional de Salud se establecerán en virtud de los acuerdos que seestablezcan entre el promotor y los servicios de salud de las Comunidades Autónomas concriterios de transparencia, y según lo establecido en esta Ley. Dichos acuerdos incluirántodos los aspectos necesarios para la correcta realización del ensayo, incluidos los profe-sionales participantes, los recursos implicados y las compensaciones que se establezcan.
3. No están sometidos a lo dispuesto en este capítulo los estudios observaciona-les. A los efectos de esta Ley, se entiende por estudio observacional el estudio en el quelos medicamentos se prescriben de la manera habitual, de acuerdo con las condicionesestablecidas en la autorización. La asignación de un paciente a una estrategia terapéuticaconcreta no estará decidida de antemano por el protocolo de un ensayo, sino que estarádeterminada por la práctica habitual de la medicina, y la decisión de prescribir un medi-camento determinado estará claramente disociada de la decisión de incluir al paciente enel estudio. No se aplicará a los pacientes ninguna intervención, ya sea diagnóstica o deseguimiento, que no sea la habitual de la práctica clínica, y se utilizarán métodos epide-miológicos para el análisis de los datos recogidos.
Artículo 59. Garantías de idoneidad.
1. Los ensayos clínicos con medicamentos en investigación estarán sometidos arégimen de autorización por la Agencia Española de Medicamentos y Productos Sanitarios,conforme al procedimiento reglamentariamente establecido.
122

2. La Agencia Española de Medicamentos y Productos Sanitarios podrá interrum-pir en cualquier momento la realización de un ensayo clínico o exigir la introducción demodificaciones en su protocolo, en los casos siguientes:
a) Si se viola la Ley.
b) Si se alteran las condiciones de su autorización.
c) Si no se cumplen los principios éticos recogidos en el artículo 60 de esta Ley.
d) Para proteger la salud de los sujetos del ensayo, o
e) En defensa de la salud pública.
3. Las Administraciones sanitarias tendrán facultades inspectoras en materia deensayos clínicos, pudiendo investigar incluso las historias clínicas individuales de los su-jetos del ensayo, guardando siempre su carácter confidencial. Asimismo, podrán realizarla interrupción cautelar del ensayo por cualquiera de las causas señaladas en el punto an-terior, comunicándolo de inmediato a la Agencia Española de Medicamentos y ProductosSanitarios.
4. Las Administraciones sanitarias velarán por el cumplimiento de las normas de«buena práctica clínica», realizando las inspecciones oportunas, con personas de la de-bida cualificación y formación universitaria en medicina, farmacia, farmacología, toxico-logía u otras materias pertinentes.
5. A los efectos previstos en el apartado 2, el investigador de un ensayo deberánotificar inmediatamente al promotor todos los acontecimientos adversos graves, salvocuando se trate de los señalados en el protocolo como acontecimientos que no requierencomunicación inmediata. El promotor a su vez notificará, en el menor plazo posible, a laAgencia Española de Medicamentos y Productos Sanitarios las reacciones adversas gra-ves e inesperadas que surjan a lo largo del ensayo y adicionalmente enviará informes pe-riódicos de seguridad. Asimismo el promotor deberá llevar un registro detallado de todoslos acontecimientos adversos que le sean notificados, cuya comunicación a las Adminis-traciones sanitarias y al Comité Ético de Investigación Clínica deberá realizarse en los tér-minos y plazos que reglamentariamente se establezcan.
6. El método de los ensayos clínicos deberá ser tal que la evaluación de los resul-tados que se obtengan con la aplicación de la sustancia o medicamento objeto del ensayoquede controlada por comparación con el mejor patrón de referencia, en orden a asegu-rar su objetividad, salvo las excepciones impuestas por la naturaleza de su propia inves-tigación.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
123

7. La realización del ensayo deberá ajustarse en todo caso al contenido del pro-tocolo de investigación de cada ensayo, de acuerdo con el cual se hubiera otorgado la au-torización, así como a sus modificaciones posteriores.
8. Los resultados favorables o desfavorables de cada ensayo clínico, tanto si éstellega a su fin como si se abandona la investigación, deberán ser comunicados a la Agen-cia Española de Medicamentos y Productos Sanitarios, sin perjuicio de su comunicacióna las Comunidades Autónomas en las que se hayan realizado dichos ensayos clínicos.
Artículo 60. Garantías de respeto a los postulados éticos.
1. Los ensayos clínicos deberán realizarse en condiciones de respeto a los dere-chos fundamentales de la persona y a los postulados éticos que afectan a la investigaciónbiomédica en la que resultan afectados seres humanos, siguiéndose a estos efectos loscontenidos en la Declaración de Helsinki.
2. No podrá iniciarse ningún ensayo clínico en tanto no se disponga de suficien-tes datos científicos y, en particular, ensayos farmacológicos y toxicológicos en animales,que garanticen que los riesgos que implica en la persona en que se realiza son admisibles.
3. Con el fin de evitar investigaciones obsoletas o repetitivas, sólo se podrán ini-ciar ensayos clínicos para demostrar la eficacia y seguridad de las modificaciones terapéu-ticas propuestas, siempre que sobre las mismas existan dudas razonables.
4. El sujeto del ensayo prestará su consentimiento libremente, expresado por es-crito, tras haber sido informado sobre la naturaleza, importancia, implicaciones y riesgosdel ensayo clínico. Si el sujeto del ensayo no está en condiciones de escribir, podrá dar,en casos excepcionales, su consentimiento verbal en presencia de, al menos, un testigomayor de edad y con capacidad de obrar. El sujeto participante en un ensayo clínico o surepresentante podrá revocar, en todo momento, su consentimiento sin expresión de causa.
En el caso de personas que no puedan emitir libremente su consentimiento, éste de-berá ser otorgado por su representante legal previa instrucción y exposición ante el mismo delalcance y riesgos del ensayo. Será necesario, además, la conformidad del representado si suscondiciones le permiten comprender la naturaleza, importancia, alcance y riesgos del ensayo.
5. Lo establecido en el apartado anterior se entenderá sin perjuicio de lo previstoen el apartado 2 del artículo 9 de la Ley 41/2002, de 14 de noviembre, reguladora de laautonomía del paciente y de derechos y obligaciones en materia de información y docu-mentación clínica, en los términos que reglamentariamente se determinen.
124

6. Ningún ensayo clínico podrá ser realizado sin informe previo favorable de unComité Ético de Investigación Clínica, que será independiente de los promotores e in-vestigadores y de las autoridades sanitarias. El Comité deberá ser acreditado por el ór-gano competente de la Comunidad Autónoma que corresponda, el cuál asegurará laindependencia de aquél. La acreditación será comunicada a la Agencia Española de Me-dicamentos y Productos Sanitarios por el órgano competente de la respectiva Comuni-dad Autónoma.
7. Los Comités Éticos de Investigación Clínica estarán formados, como mínimo,por un equipo interdisciplinar integrado por médicos, farmacéuticos de atención prima-ria y hospitalaria, farmacólogos clínicos, personal de enfermería y personas ajenas a lasprofesiones sanitarias de las que al menos uno será licenciado en Derecho especialistaen la materia.
8. El Comité Ético de Investigación Clínica ponderará los aspectos metodológicos,éticos y legales del protocolo propuesto, así como el balance de riesgos y beneficios anti-cipados dimanantes del ensayo.
9. Los Comités Éticos de Investigación Clínica podrán requerir información com-pleta sobre las fuentes y cuantía de la financiación del ensayo y la distribución de los gas-tos en, entre otros, los siguientes apartados: reembolso de gastos a los pacientes, pagospor análisis especiales o asistencia técnica, compra de aparatos, equipos y materiales,pagos debidos a los hospitales o a los centros en que se desarrolla la investigación por elempleo de sus recursos y compensación a los investigadores.
10. Reglamentariamente se establecerá el procedimiento para la designación delComité Ético de referencia y para la obtención del dictamen único con validez en todo elterritorio, con el objetivo de impulsar la investigación clínica en el Sistema Nacional deSalud. El Ministerio de Sanidad y Consumo desarrollará acciones que permitan que los Co-mités Éticos de Investigación Clínica acreditados puedan compartir estándares de calidady criterios de evaluación adecuados y homogéneos.
Artículo 61. Garantías de asunción de responsabilidades.
1. La realización de un ensayo clínico exigirá que, mediante la contratación de unseguro o la constitución de otra garantía financiera, se garantice previamente la coberturade los daños y perjuicios que, para la persona en la que se lleva a efecto, pudieran deri-varse de aquél.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
125

2. Cuando, por cualquier circunstancia, el seguro no cubra enteramente los da-ños causados, el promotor del ensayo, el investigador responsable del mismo y el hospi-tal o centro en que se hubiere realizado responderán solidariamente de aquéllos, aunqueno medie culpa, incumbiéndoles la carga de la prueba. Ni la autorización administrativani el informe del Comité Ético de Investigación Clínica les eximirán de responsabilidad.
3. Se presume, salvo prueba en contrario, que los daños que afecten a la saludde la persona sujeta al ensayo, durante la realización del mismo y durante el plazo de unaño contado desde su finalización, se han producido como consecuencia del ensayo. Sinembargo, una vez concluido el año, el sujeto del mismo está obligado a probar el daño ynexo entre el ensayo y el daño producido.
4. Es promotor del ensayo clínico la persona física o jurídica que tiene interés en surealización, firma la solicitud de autorización dirigida al Comité Ético de Investigación Clínicay a la Agencia Española de Medicamentos y Productos Sanitarios y se responsabiliza de él.
5. Es investigador principal quien dirige la realización del ensayo y firma en unióndel promotor la solicitud, corresponsabilizándose con él. La condición de promotor y la deinvestigador principal pueden concurrir en la misma persona física.
Artículo 62. Garantías de transparencia.
1. Los ensayos clínicos autorizados por la Agencia Española de los Medicamen-tos y Productos Sanitarios formarán parte de un registro nacional de ensayos clínicos pú-blico y libre que será accesible en las condiciones que reglamentariamente se determine.
2. El promotor está obligado a publicar los resultados del ensayo clínico, sean po-sitivos o no. La publicación se realizará, previa supresión de cualquier información comer-cial de carácter confidencial, preferentemente en revistas científicas y, de no ser ello posible,a través de los medios y en los plazos máximos que se establezcan reglamentariamente. Enla publicación se mencionará el Comité Ético de Investigación Clínica que los informó.
3. Cuando se hagan públicos estudios y trabajos de investigación sobre medica-mentos dirigidos a la comunidad científica, se harán constar los fondos obtenidos por elautor por o para su realización y la fuente de financiación.
4. En caso de no publicarse los resultados de los ensayos clínicos y cuando losmismos permitan concluir que el medicamento presenta modificaciones de su perfil de efi-cacia o seguridad, la Agencia Española de Medicamentos y Productos Sanitarios hará pú-blicos los resultados.
126

5. Toda la información sobre el ensayo clínico deberá registrarse, tratarse y conser-varse de forma que pueda ser comunicada, interpretada y comprobada de manera precisa, pro-tegiendo al mismo tiempo el carácter confidencial de los registros de los sujetos del ensayo.
6. El Gobierno, previo informe del Consejo Interterritorial del Sistema Nacional deSalud, y con carácter básico, regulará los requisitos comunes para la realización y finan-ciación de los ensayos clínicos, asegurando la buena práctica clínica y las condiciones desu realización. Los centros, servicios, establecimientos y profesionales sanitarios partici-parán en la realización de ensayos clínicos de acuerdo con estos requisitos comunes ycondiciones de financiación y los que en su desarrollo puedan establecer las Administra-ciones sanitarias competentes.
TÍTULO VIII. Régimen sancionador.
CAPÍTULO I. Inspección y medidas cautelares
Artículo 98. Inspección.
1. Corresponde a las Administraciones sanitarias en el ámbito de sus competen-cias la realización de las inspecciones necesarias para asegurar el cumplimiento de lo pre-visto en esta Ley.
2. Corresponde a la Administración General del Estado la realización de la funcióninspectora en los siguientes casos:
a) Cuando se trate de las actuaciones necesarias para las oportunas autorizacioneso registros que, de acuerdo con esta Ley, corresponden a la Administración Ge-neral del Estado.
b) En todo caso, cuando se trate de inspecciones a realizar en el territorio de las Co-munidades Autónomas que no ostenten competencias de ejecución de la legis-lación de productos farmacéuticos o no hubieren recibido los correspondientestraspasos.
c) Cuando se trate de medicamentos, productos o artículos destinados al comer-cio exterior o cuya utilización o consumo pudiera afectar a la seguridad pública.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
127

3. El personal al servicio de las Administraciones públicas que desarrolle las fun-ciones de inspección, cuando ejerza tales funciones y acredite su identidad, estará auto-rizado para:
a) Entrar libremente y sin previa notificación, en cualquier momento, en todo cen-tro o establecimiento sujeto a esta Ley.
b) Proceder a las pruebas, investigaciones o exámenes necesarios para compro-bar el cumplimiento de esta Ley y de las normas que se dicten para su de-sarrollo.
c) Tomar o sacar muestras, en orden a la comprobación del cumplimiento de loprevisto en esta Ley y en las disposiciones para su desarrollo.
d) Realizar cuantas actuaciones sean precisas en orden al cumplimiento de lasfunciones de inspección que desarrollen.
Artículo 99. Medidas cautelares.
1. En el caso de que exista o se sospeche razonablemente la existencia de unriesgo inminente y grave para la salud, las autoridades sanitarias podrán adoptar las si-guientes medidas cautelares en el ámbito de esta Ley:
a) La puesta en cuarentena, la retirada del mercado y la prohibición de utilizaciónde medicamentos, fórmulas magistrales y preparados oficinales, así como lasuspensión de actividades, publicidad y la clausura provisional de estableci-mientos, centros o servicios.
La puesta en cuarentena supondrá el bloqueo inmediato en el establecimientofarmacéutico en que se encuentren o al que se destinen, en caso de transporteno concluido, por el tiempo que se determine o hasta nueva orden, a cargo desu responsable.
b) La suspensión de la elaboración, prescripción, dispensación y suministro demedicamentos en investigación.
2. La duración de las medidas a que se refiere el apartado anterior, que se fija-rán para cada caso, sin perjuicio de las prórrogas sucesivas acordadas por resolucionesmotivadas, no excederá de lo que exija la situación de riesgo inminente y grave que lajustificó.
3. La Agencia Española de Medicamentos y Productos Sanitarios deberá ser in-formada de modo inmediato por la autoridad sanitaria que adoptó la medida cautelar.
128

4. De las medidas cautelares la Agencia Española de Medicamentos y ProductosSanitarios dará conocimiento por los medios idóneos y con la rapidez adecuada a cadacaso, a los servicios sanitarios, entidades responsables o público en general, según proceda.
5. El coste de las medidas cautelares será sufragado por la persona física o jurí-dica que hubiese dado lugar a su adopción.
CAPÍTULO II. Infracciones y sanciones.
Artículo 100. Disposiciones generales.
1. Las infracciones en materia de medicamentos serán objeto de las sanciones ad-ministrativas correspondientes, previa instrucción del oportuno expediente, sin perjuiciode las responsabilidades civiles, penales o de otro orden que puedan concurrir.
2. La instrucción de causa penal ante los Tribunales de Justicia suspenderá latramitación del expediente administrativo sancionador que hubiera sido incoado por losmismos hechos y, en su caso, la eficacia de los actos administrativos de imposición de san-ción. Las medidas administrativas que hubieran sido adoptadas para salvaguardar la sa-lud y seguridad de las personas se mantendrán en tanto la autoridad judicial se pronunciesobre las mismas.
3. En ningún caso se impondrá una doble sanción por los mismos hechos y en fun-ción de los mismos intereses públicos protegidos, si bien deberán exigirse las demás res-ponsabilidades que se deduzcan de otros hechos o infracciones concurrentes.
4. Con respecto al régimen sancionador y en lo no previsto por esta Ley será deaplicación lo establecido por el título IX de la Ley de Régimen Jurídico de las Administra-ciones Públicas y del Procedimiento Administrativo Común.
Artículo 101. Infracciones.
1. Las infracciones se calificarán como leves, graves y muy graves atendiendo alos criterios de riesgos para la salud, cuantía del eventual beneficio obtenido, gravedad dela alteración sanitaria y social producida, generalización de la infracción y reincidencia.
2. Constituirán faltas administrativas y serán sancionadas en los términos previs-tos en el artículo siguiente, las infracciones que a continuación se tipifican:
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
129

a) Infracciones leves:
1. No aportar, las entidades o personas responsables, los datos, declaracio-nes así como cualesquiera información que estén obligados a suministrarpor razones sanitarias, técnicas, económicas, administrativas y financieras.
2. Incumplir el deber de colaborar con la administración sanitaria en la eva-luación y control de medicamentos.
3. No disponer, los establecimientos obligados a ello, de acceso a la Real Far-macopea Española y al Formulario Nacional.
4. Dificultar la labor inspectora mediante cualquier acción u omisión que per-turbe o retrase la misma.
5. No proporcionar, los laboratorios farmacéuticos, a los facultativos sanita-rios en ejercicio que lo soliciten la ficha técnica de medicamentos antesde su comercialización.
6. Realizar publicidad de fórmulas magistrales o de preparados oficinales.
7. Incumplir los requisitos que, para la realización de la visita médica, esta-blezca la normativa de las Comunidades Autónomas.
8. No cumplimentar correctamente los datos y advertencias que deben con-tener las recetas normalizadas.
9. Dispensar medicamentos transcurrido el plazo de validez de la receta.
10. Realizar la sustitución de un medicamento, en los casos que ésta sea po-sible, incumpliendo los requisitos establecidos en esta Ley.
11. Incumplir los requisitos, obligaciones o prohibiciones establecidas en esta Leyy disposiciones que la desarrollan de manera que, en razón de los criterioscontemplados en este artículo, tales incumplimientos merezcan la calificaciónde leves o no proceda su calificación como faltas graves o muy graves.
12. No incluir en los envases de los medicamentos la información en alfabetobraille para su correcta identificación por las personas invidentes y condiscapacidad visual, conforme a lo dispuesto en el apartado 5 del artículo15 de esta Ley.
b) Infracciones graves:
1. No realizar en la elaboración, fabricación, importación, exportación y dis-tribución de medicamentos los controles de calidad exigidos en la legisla-ción sanitaria o efectuar los procesos de fabricación o control medianteprocedimientos no validados.
130

2. Elaborar, fabricar, importar, exportar, dispensar y distribuir medicamentosy productos sanitarios por personas físicas o jurídicas que no cuenten conla preceptiva autorización.
3. Impedir la actuación de los inspectores debidamente acreditados, en loscentros en los que se elaboren, fabriquen, distribuyan y dispensen medi-camentos.
4. Preparar individualizadamente vacunas y alérgenos en establecimientosdistintos de los autorizados.
5. Prescribir y preparar fórmulas magistrales y preparados oficinales incum-pliendo los requisitos legales establecidos.
6. Modificar por parte del titular, sin autorización previa, cualquiera de lascondiciones de autorización del medicamento.
7. No disponer, un laboratorio farmacéutico o almacén mayorista, de direc-tor técnico o del resto del personal exigido en cada caso.
8. Incumplir, el director técnico y demás personal, las obligaciones que com-peten a sus cargos.
9. Incumplir, el promotor o investigador de un ensayo clínico, las obligacio-nes establecidas en la legislación vigente o en las normas de buena prác-tica clínica, así como realización de un ensayo clínico sin ajustarse alprotocolo aprobado, cuando el hecho, en razón de los criterios contempla-dos en este artículo, no merezca la calificación de falta muy grave.
10. Incumplir, el promotor de ensayos clínicos, los plazos de comunicación alas autoridades sanitarias de las reacciones adversas graves e inesperadasocurridas en un ensayo clínico.
11. Facilitar, al Comité Ético de Investigación Clínica o a las autoridades sa-nitarias, información y/o documentación, relacionada con un ensayo clí-nico, no veraz o que dé lugar a conclusiones inexactas.
12. Incumplir, el promotor, la obligación de publicación de los resultados deun ensayo clínico según lo establecido en el artículo 62.
13. Actuar, los integrantes del Comité Ético de Investigación Clínica, sin ajus-tarse a los requisitos de funcionamiento establecidos legalmente o sin es-tar debidamente acreditados.
14. Incumplir, los laboratorios farmacéuticos, almacenes mayoristas o perso-nal sanitario, el deber de fármacovigilancia.
15. Negarse a dispensar medicamentos o productos sanitarios sin causa justi-ficada.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
131

16. Dispensar medicamentos o productos sanitarios sin receta, cuando éstaresulte obligada.
17. Suministrar, adquirir o vender medicamentos o productos sanitarios a en-tidades no autorizadas para la realización de tales actividades.
18. Actuar, los profesionales sanitarios implicados en el ciclo de prescripción,dispensación y administración y siempre que estén en ejercicio, en funcio-nes de delegados de visita médica, representantes, comisionistas o agentesinformadores de los laboratorios de medicamentos o productos sanitarios.
19. Incumplir, el personal sanitario, el deber de garantizar la confidencialidady la intimidad de los pacientes en la tramitación de las recetas y órdenesmédicas.
20. Funcionar, los servicios farmacéuticos y oficinas de farmacia, sin la presen-cia y actuación profesional del farmacéutico responsable.
21. Incumplir, las oficinas de farmacia, las exigencias que conlleva la facturaciónal Sistema Nacional de Salud de los productos contemplados en esta Ley.
22. Defraudar, las oficinas de farmacia, al Sistema Nacional de Salud o al bene-ficiario del mismo con motivo de la facturación y cobro de recetas oficiales.
23. Dispensar o suministrar medicamentos o productos sanitarios en estable-cimientos distintos a los autorizados.
24. No ajustar los precios de los medicamentos a lo determinado por la Admi-nistración.
25. Sustituir medicamentos en la dispensación, contraviniendo lo dispuesto enel artículo 86 de esta Ley.
26. Cualquier acto u omisión encaminado a coartar la libertad del usuario enla elección de la oficina de farmacia.
27. Ofrecer directa o indirectamente cualquier tipo de incentivo, bonificacio-nes, descuentos prohibidos, primas u obsequios, efectuados por quientenga intereses directos o indirectos en la producción, fabricación y comer-cialización de medicamentos, a los profesionales sanitarios, con motivode la prescripción, dispensación y administración de los mismos, o a susparientes y personas de su convivencia.
28. Ofrecer directa o indirectamente cualquier tipo de incentivo, bonificaciones,descuentos prohibidos, primas u obsequios, efectuados por quien tenga in-tereses directos o indirectos en la producción, fabricación y comercializaciónde productos sanitarios, a los profesionales sanitarios, con motivo de la pres-cripción de los mismos, o a sus parientes y personas de su convivencia.
132

29. Aceptar, los profesionales sanitarios, con motivo de la prescripción, dispen-sación y administración de medicamentos y/o productos sanitarios concargo al Sistema Nacional de Salud, o sus parientes y personas de su con-vivencia, cualquier tipo de incentivo, bonificaciones, descuentos prohibi-dos, primas u obsequios efectuados por quien tenga intereses directos oindirectos en la producción, fabricación y comercialización de medicamen-tos y productos sanitarios.
30. No comunicar los laboratorios farmacéuticos al Ministerio de Sanidad yConsumo las unidades de medicamentos vendidas para ser dispensadas enterritorio nacional.
31. No informar los almacenes mayoristas a las autoridades sanitarias de lasComunidades Autónomas en las que tengan su domicilio social y al Minis-terio de Sanidad y Consumo, de las unidades suministradas a oficinas defarmacia o servicios de farmacia que radiquen en territorio nacional, asícomo, en su caso, a otros almacenes mayoristas, con independencia de laComunidad Autónoma en que éstos últimos radiquen.
32. No comunicar las oficinas de farmacia la información sobre medicamen-tos dispensados a que se refiere esta Ley.
33. Cometer tres infracciones calificadas como leves en el plazo de un año.
c) Infracciones muy graves:
1. La puesta en el mercado de medicamentos o productos sanitarios de cual-quier naturaleza sin haber obtenido la preceptiva autorización sanitaria paraello.
2. La falsificación de medicamentos.
3. Incumplir, el titular de la autorización, la obligación de presentar los in-formes periódicos de seguridad.
4. Preparar remedios secretos.
5. Importar y exportar sangre, fluidos, glándulas y tejidos humanos y sus com-ponentes y derivados sin la previa autorización.
6. Realizar ensayos clínicos sin la previa autorización administrativa.
7. Realizar ensayos clínicos sin contar con el consentimiento del sujeto delensayo o, en su caso, de su representante legal, o el incumplimiento, porparte del investigador, del deber de información sobre el ensayo clínico aquien participa como sujeto del mismo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
133

8. No comunicar, el promotor de un ensayo clínico, a las autoridades sanitariaslas reacciones adversas ocurridas en el desarrollo del mismo o los informes pe-riódicos de seguridad.
9. Incumplir, el promotor o investigador de un ensayo clínico, las obligacionesestablecidas en la legislación vigente o en las normas de buena práctica clí-nica, así como realización de un ensayo clínico sin ajustarse al protocolo apro-bado cuando suponga perjuicio en los derechos, seguridad y bienestar de lossujetos o afecte a la credibilidad de los datos obtenidos.
10. Distribuir o conservar los medicamentos sin observar las condiciones exigi-das, así como poner a la venta medicamentos alterados, en malas condicio-nes o, cuando se haya señalado, pasado el plazo de validez.
11. Vender medicamentos o productos sanitarios a domicilio o a través de interneto de otros medios telemáticos o indirectos, en contra de lo previsto en esta Ley.
12. Incumplir los almacenes de distribución y las oficinas de farmacia sus obliga-ciones legales y, en particular, no disponer de las existencias de medicamen-tos adecuadas para la normal prestación de sus actividades o servicios.
13. Incumplir los almacenes de distribución y las oficinas de farmacia sus obliga-ciones legales y, en particular, no disponer de existencias mínimas de medi-camentos para supuestos de emergencia o catástrofes, en los casos queresulte obligado.
14. La elaboración, fabricación, importación, exportación, distribución, comercia-lización, prescripción y dispensación de productos, preparados, sustancias ocombinaciones de las mismas, que se presenten como medicamentos sin es-tar legalmente reconocidos como tales.
15. El incumplimiento de la obligación de suscribir un seguro, aval o garantía fi-nanciera equivalente en los supuestos exigidos por esta Ley.
16. Realizar promoción, información o publicidad de medicamentos no autoriza-dos o sin ajustarse a las condiciones establecidas en la autorización, en lo dis-puesto en esta Ley y en la legislación general sobre publicidad.
17. Efectuar promoción, publicidad o información destinada al público de pro-ductos o preparados, con fines medicinales, aún cuando el propio productono haga referencia explícita a dichos fines, incluidas las sustancias medici-nales y sus combinaciones, que no se encuentren autorizados como medica-mentos.
18. Ofrecer primas, obsequios, premios, concursos, bonificaciones, descuentos osimilares como métodos vinculados a la promoción o venta al público de losproductos regulados en esta Ley.
134

19. Incumplir las medidas cautelares y definitivas sobre medicamentos que las au-toridades sanitarias competentes acuerden por causa grave de salud pública.
20. No cumplir los requisitos y condiciones reglamentariamente exigidos en mate-ria de publicidad y promoción comercial de los productos, materiales, sustan-cias, energías o métodos a los que se atribuyan efectos beneficiosos sobre lasalud.
21. Cometer tres infracciones calificadas como graves en el plazo de dos años.
Artículo 102. Sanciones.
1. Las infracciones en materia de medicamentos serán sancionadas con multa, deconformidad con lo establecido en el artículo 101 aplicando una graduación de mínimo,medio y máximo a cada nivel de infracción, en función de la negligencia e intencionali-dad del sujeto infractor, fraude, connivencia, incumplimiento de las advertencias previas,cifra de negocios de la empresa, número de personas afectadas, perjuicio causado, bene-ficios obtenidos a causa de la infracción, permanencia o transitoriedad de los riesgos yreincidencia por comisión en el término de un año de más de una infracción de la mismanaturaleza cuando así haya sido declarado por resolución firme:
a) Infracciones leves:
1. Grado mínimo: Hasta 6.000 euros.
2. Grado medio: Desde 6.001 a 18.000 euros.
3. Grado máximo: Desde 18.001 a 30.000 euros.
a) Infracciones graves:
1. Grado mínimo: Desde 30.001 a 60.000 euros.
2. Grado medio: Desde 60.001 a 78.000 euros.
3. Grado máximo: Desde 78.001 a 90.000 euros.
a) Infracciones muy graves:
1. Grado mínimo: Desde 90.001 a 300.000 euros.
2. Grado medio: Desde 300.001 a 600.000 euros.
3. Grado máximo: Desde 600.001 a 1.000.000 de euros, pudiendo rebasardicha cantidad hasta alcanzar el quíntuplo del valor de los productos oservicios objeto de la infracción.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
135

No obstante, en el caso de infracciones en materia de medicamentos veterina-rios, la sanción sólo se impondrá en el grado máximo cuando la actuación infractora hayaproducido un daño directo o provocado un riesgo grave y directo en la salud pública o enla seguridad alimentaria.
2. Sin perjuicio de la multa que proceda imponer conforme a lo dispuesto en elapartado anterior, las infracciones en materia de medicamentos serán sancionadas con elcomiso, en favor del Tesoro Público, del beneficio ilícito obtenido como consecuencia dela perpetración de la infracción. La resolución de la Administración determinará a estosefectos la cuantía del beneficio ilícito obtenido.
3. Las sanciones por la comisión de infracciones graves y muy graves serán publi-cadas en el diario oficial correspondiente una vez que adquieran firmeza.
4. Corresponde el ejercicio de la potestad sancionadora a la Administración Ge-neral del Estado o a las Comunidades Autónomas que ostentan la función inspectora, deacuerdo con lo regulado en el artículo 98 de esta Ley.
5. Además, en los supuestos de infracciones muy graves podrá acordarse, por elConsejo de Ministros o por los órganos competentes de las Comunidades Autónomas a lasque corresponda la ejecución de la legislación sobre productos farmacéuticos, el cierretemporal del establecimiento, instalación o servicio por un plazo máximo de cinco años.En tal caso, será de aplicación lo previsto en el artículo 53 de la Ley 31/1995, de 8 denoviembre, de Prevención de Riesgos Laborales.
Artículo 103. Otras medidas.
1. No tendrán carácter de sanción la clausura y cierre de establecimientos, insta-laciones o servicios que no cuenten con las previas autorizaciones o registros sanitariospreceptivos, o la suspensión de su funcionamiento hasta tanto se subsanen los defectoso se cumplan los requisitos exigidos por razones de sanidad, higiene o seguridad.
2. La autoridad a que corresponda resolver el expediente podrá acordar el comisode productos y medicamentos deteriorados, caducados, no autorizados o que puedan en-trañar riesgo para la salud.
3. Los gastos de transporte, distribución o destrucción de los productos y medica-mentos, así como los derivados de la suspensión, clausura y cierre de establecimientos, ins-talaciones o servicios señalados en los apartados anteriores, serán por cuenta del infractor.
136

Artículo 104. Prescripción.
1. Las infracciones muy graves prescribirán a los cinco años, las graves a los dosaños y las leves al año; en los mismos plazos prescribirán las sanciones.
2. El plazo de prescripción de las infracciones comenzará a contarse desde el díaen que la infracción se hubiera cometido.
Interrumpirá la prescripción la iniciación, con conocimiento del interesado, delprocedimiento sancionador, reanudándose el plazo de prescripción si el expediente san-cionador estuviera paralizado durante más de un mes por causa no imputable al presuntoresponsable.
3. El plazo de prescripción de las sanciones comenzará a contarse desde el día si-guiente a aquél en que adquiera firmeza la resolución por la que se impone la sanción.
Interrumpirá la prescripción la iniciación, con conocimiento del interesado, delprocedimiento de ejecución, volviendo a transcurrir el plazo si aquél está paralizado du-rante más de un mes por causa no imputable al infractor.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
137


REAL DECRETO 223/2004, DE 6 DE FEBRERO, POR EL QUE SE REGULAN LOS ENSAYOS CLÍNICOS
CON MEDICAMENTOS
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
139


REAL DECRETO 223/2004, DE 6 DE FEBRERO, POR EL QUE SE REGULANLOS ENSAYOS CLÍNICOS CON MEDICAMENTOS
Los ensayos clínicos con medicamentos han sido objeto de regulación en el títuloIII de la Ley 25/1990, de 20 de diciembre, del Medicamento, y su desarrollo reglamen-tario en esta materia mediante el Real Decreto 561/1993, de 16 de abril, por el que seestablecen los requisitos para la realización de ensayos clínicos con medicamentos.
La Directiva 2001/20/CE del Parlamento Europeo y del Consejo, de 4 de abril de2001, relativa a la aproximación de las disposiciones legales, reglamentarias y adminis-trativas de los Estados miembros sobre la aplicación de buenas prácticas clínicas en la re-alización de ensayos clínicos de medicamentos de uso humano, ha venido a armonizarlas legislaciones de los Estados miembros de la Unión Europea sobre ensayos clínicos conmedicamentos en seres humanos, lo que ha hecho necesario la modificación de la legis-lación española vigente en esta materia. En este sentido, el artículo 125 de la Ley53/2002, de 30 de diciembre, de medidas fiscales, administrativas y del orden social, haintroducido diversas modificaciones en el título III de la Ley 25/1990, de 20 de diciem-bre, del Medicamento, con la finalidad de eliminar las discrepancias de la citada normacon la Directiva 2001/20/CE, dotando así de la necesaria cobertura legal a este regla-mento. Asimismo, se ha tenido en cuenta en la elaboración lo dispuesto en la Ley41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de de-rechos y obligaciones en materia de información y documentación clínica.
Este real decreto, por tanto, viene a incorporar en su totalidad al ordenamiento ju-rídico interno la Directiva 2001/20/CE, y sustituye al actual Real Decreto 561/1993, de16 de abril, por el que se establecen los requisitos para la realización de ensayos clínicoscon medicamentos, dotando de nuevo desarrollo reglamentario a la Ley 25/1990, de 20de diciembre, del Medicamento, en cuanto a ensayos clínicos se refiere.
En este real decreto se han tenido en cuenta los principios básicos para la reali-zación de ensayos clínicos con seres humanos fundamentados en la protección de los de-rechos humanos y la dignidad del ser humano respecto a la aplicación de la biología y la
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
141

medicina, reflejados en la Declaración de Helsinki y en el Convenio de Oviedo sobre losderechos humanos y la biomedicina, así como las normas para la adecuada protección delos datos personales.
Por otra parte, se habilitan nuevos procedimientos administrativos para la autori-zación de los ensayos clínicos por parte de la Administración General del Estado, lo quesupone agilizar y simplificar los trámites actualmente existentes, equiparando las distin-tas reglamentaciones en esta materia de los Estados miembros de la Unión Europea y per-mitiendo el mutuo reconocimiento entre las autoridades sanitarias de dichos Estadosrespecto a los resultados de los ensayos clínicos realizados.
En cuanto a la evaluación del ensayo clínico, en el ámbito que les concierne, porparte del Comité Ético de Investigación Clínica, además del establecimiento de plazosmáximos para dicha evaluación se recoge la exigencia del dictamen único. En este sen-tido, en los ensayos clínicos multicéntricos, en los que participen dos o más centros ubi-cados en España, se designará un comité de referencia entre los distintos comités éticosimplicados para la emisión del citado dictamen único, lo cual hace necesaria la creaciónde un organismo de coordinación denominado Centro Coordinador de Comités Éticos deInvestigación Clínica.
Especial mención merece la obligación de aplicar las normas de buena prácticaclínica a la planificación, realización, registro y comunicación de todos los ensayos clíni-cos que se realicen en España, como conjunto de requisitos éticos y científicos de cali-dad reconocidos a escala internacional y como garantía de la protección de los derechos,la seguridad y el bienestar de los sujetos del ensayo, así como la fiabilidad de sus resul-tados.
La verificación de la conformidad con las normas de buenas práctica clínica y lainspección de los datos, la información y los documentos para comprobar que han sido pro-ducidos, registrados y comunicados correctamente, así como el cumplimiento de las nor-mas de correcta fabricación en la elaboración, importación y etiquetado de losmedicamentos en investigación, la vigilancia de la seguridad de estos medicamentos ylas comunicaciones entre las autoridades competentes en la materia, han sido también re-cogidos y tenidos en cuenta como requisitos indispensables para justificar la participaciónde seres humanos en los ensayos clínicos.
Por último, complementan este real decreto las normas de buena práctica clínica ylas instrucciones para la realización de ensayos clínicos en España, o en su caso, las direc-trices de la Comisión Europea, que se publicarán por el Ministerio de Sanidad y Consumo.
142

Este real decreto mediante el que se incorpora al ordenamiento jurídico interno laDirectiva 2001/20/CE desarrolla el título III de la Ley 25/1990, de 20 de diciembre, delMedicamento, y se dicta al amparo de lo dispuesto en el artículo 149.1.16.ª de la Cons-titución Española, en concordancia con el artículo 2.1 de la citada ley.
En la elaboración de este real decreto han sido oídas las comunidades autónomasy los sectores afectados.
En su virtud, a propuesta de la Ministra de Sanidad y Consumo, con la aproba-ción previa de la Ministra de Administraciones Públicas, de acuerdo con el Consejo deEstado y previa deliberación del Consejo de Ministros en su reunión del día 6 de febrerode 2004,
D I S P O N G O :
CAPÍTULO I. Disposiciones generales
Artículo 1. Ámbito de aplicación.
1. Este real decreto se aplicará a los ensayos clínicos con medicamentos de usohumano que se realicen en España.
A estos efectos, no tendrá la consideración de ensayo clínico la administración deun medicamento en investigación a un solo paciente, en el ámbito de la práctica médicahabitual y con el único propósito de conseguir un beneficio terapéutico para el paciente,que se regirá por lo dispuesto sobre uso compasivo en el artículo 28.
La práctica médica y la libertad profesional de prescripción del médico no ampa-rarán, en ningún caso, la realización de ensayos clínicos no autorizados ni la utilizaciónde remedios secretos o no declarados a la autoridad sanitaria.
2. Se excluyen del ámbito de aplicación de este real decreto los estudios obser-vacionales, definidos en el artículo 2.c), que se regirán por su normativa específica.
3. Quedan prohibidos los ensayos clínicos con medicamentos de terapia génicaque produzcan modificaciones en la identidad génica de la línea germinal del sujeto.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
143

Artículo 2. Definiciones.
A los efectos de lo dispuesto en este real decreto, se aplicarán las siguientes defini-ciones:
a) Ensayo clínico: toda investigación efectuada en seres humanos para determinar oconfirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o de de-tectar las reacciones adversas, y/o de estudiar la absorción, distribución, metabolismo y excre-ción de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/osu eficacia.
A estos efectos, se aplicará la definición de medicamento en investigación prevista enel párrafo d).
b) Ensayo clínico multicéntrico: ensayo clínico realizado de acuerdo con un protocoloúnico pero en más de un centro y, por tanto, realizado por más de un investigador.
c) Estudio observacional: estudio en el que los medicamentos se prescriben de la ma-nera habitual, de acuerdo con las condiciones normales de la práctica clínica (aquellas esta-blecidas en la autorización de comercialización). La asignación de un paciente a una estrategiaterapéutica concreta no estará decidida de antemano por un protocolo de ensayo, sino que es-tará determinada por la práctica habitual de la medicina, y la decisión de prescribir un medi-camento determinado estará claramente disociada de la decisión de incluir al paciente en elestudio. No se aplicará a los pacientes ninguna intervención, ya sea diagnóstica o de segui-miento, que no sea la habitual de la práctica clínica, y se utilizarán métodos epidemiológicospara el análisis de los datos recogidos.
d) Medicamento en investigación: forma farmacéutica de una sustancia activa o pla-cebo que se investiga o se utiliza como referencia en un ensayo clínico, incluidos los produc-tos con autorización de comercialización cuando se utilicen o combinen (en la formulación oen el envase) de forma diferente a la autorizada, o cuando se utilicen para tratar una indica-ción no autorizada, o para obtener más información sobre un uso autorizado.
e) Promotor: individuo, empresa, institución u organización responsable del inicio,gestión y/o financiación de un ensayo clínico.
f) Monitor: profesional capacitado con la necesaria competencia clínica, elegido por elpromotor, que se encarga del seguimiento directo de la realización del ensayo. Sirve de vínculoentre el promotor y el investigador principal, cuando éstos no concurran en la misma persona.
144

g) Organización de investigación por contrato (CRO): persona física o jurídica contra-tada por el promotor para realizar funciones o deberes del promotor en relación con el ensayoclínico.
h) Investigador: médico o persona que ejerce una profesión reconocida para llevar acabo investigaciones en razón de su formación científica y de su experiencia en la atenciónsanitaria requerida. El investigador es responsable de la realización del ensayo clínico en uncentro. Si es un equipo el que realiza el ensayo en un centro, el investigador es el responsa-ble del equipo y puede denominarse investigador principal.
i) Investigador coordinador: investigador responsable de la coordinación de los inves-tigadores de todos los centros españoles que participan en un ensayo clínico multicéntrico.
j) Manual del investigador: conjunto de datos clínicos y no clínicos sobre el medica-mento en investigación pertinente para el estudio de dicho medicamento en seres humanos.
k) Protocolo: documento donde se describen los objetivos, el diseño, la metodología,las consideraciones estadísticas y la organización de un ensayo. El término protocolo se re-fiere al protocolo original, a sus sucesivas versiones y a sus modificaciones.
l) Sujeto del ensayo: individuo que participa en un ensayo clínico, bien recibiendoel medicamento en investigación, bien como control.
m) Consentimiento informado: decisión, que debe figurar por escrito y estar fechaday firmada, de participar en un ensayo clínico adoptada voluntariamente por una persona ca-paz de dar su consentimiento tras haber sido debidamente informada y documentada acercade su naturaleza, importancia, implicaciones y riesgos.
En el supuesto de que el sujeto tenga un impedimento para escribir, el consenti-miento podrá otorgarse en casos excepcionales de forma oral en presencia de al menos untestigo.
Cuando el sujeto del ensayo no sea una persona capaz para dar su consentimiento, ladecisión deberá adoptarse por su representante legal en los términos previstos en el artículo 7.
n) Comité Ético de Investigación Clínica (CEIC): Organismo independiente, consti-tuido por profesionales sanitarios y miembros no sanitarios, encargado de velar por la pro-tección de los derechos, seguridad y bienestar de los sujetos que participen en un ensayo yde ofrecer garantía pública al respecto, mediante un dictamen sobre el protocolo del ensayo,
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
145

la idoneidad de los investigadores y la adecuación de las instalaciones, así como los méto-dos y los documentos que vayan a utilizarse para informar a los sujetos del ensayo con el finde obtener su consentimiento informado. En el caso de ensayos clínicos multicéntricos, elComité Ético de Investigación Clínica encargado de emitir el dictamen se denomina ComitéÉtico de Investigación Clínica de Referencia.
ñ) Inspección: revisión oficial por una autoridad competente de los documentos,las instalaciones, los archivos, los sistemas de garantía de calidad y cualesquiera otros ele-mentos que la autoridad competente considere relacionados con el ensayo clínico y quepuedan encontrarse en el lugar del ensayo, en las instalaciones del promotor y/o de la or-ganización de investigación por contrato, o en cualquier otro establecimiento que la auto-ridad competente considere oportuno inspeccionar.
o) Acontecimiento adverso: cualquier incidencia perjudicial para la salud en un pa-ciente o sujeto de ensayo clínico tratado con un medicamento, aunque no tenga necesa-riamente relación causal con dicho tratamiento.
p) Reacción adversa: toda reacción nociva y no intencionada a un medicamentoen investigación, independientemente de la dosis administrada.
q) Acontecimiento adverso grave o reacción adversa grave: cualquier aconteci-miento adverso o reacción adversa que, a cualquier dosis, produzca la muerte, amenacela vida del sujeto, haga necesaria la hospitalización o la prolongación de ésta, produzcainvalidez o incapacidad permanente o importante, o dé lugar a una anomalía o malforma-ción congénita. A efectos de su notificación, se tratarán también como graves aquellas sos-pechas de acontecimiento adverso o reacción adversa que se consideren importantes desdeel punto de vista médico, aunque no cumplan los criterios anteriores.
r) Reacción adversa inesperada: reacción adversa cuya naturaleza o gravedad nose corresponde con la información referente al producto (por ejemplo, el manual del in-vestigador en el caso de un medicamento en investigación no autorizado para su comer-cialización, o la ficha técnica del producto en el caso de un medicamento autorizado).
CAPÍTULO II. Protección de los sujetos del ensayo
Artículo 3. Postulados éticos.
1. Sólo se podrá iniciar un ensayo clínico cuando el Comité Ético de InvestigaciónClínica que corresponda y la Agencia Española de Medicamentos y Productos Sanitarios
146

hayan considerado que los beneficios esperados para el sujeto del ensayo y para la socie-dad justifican los riesgos; asimismo, sólo podrá proseguir si se supervisa permanente-mente el cumplimiento de este criterio.
2. Los ensayos clínicos se realizarán en condiciones de respeto a los derechosdel sujeto y a los postulados éticos que afectan a la investigación biomédica con sereshumanos. En particular, se deberá salvaguardar la integridad física y mental del sujeto,así como su intimidad y la protección de sus datos, de acuerdo con la Ley Orgánica15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal. Se obten-drá y documentará el consentimiento informado de cada uno de los sujetos del ensayo,libremente expresado, antes de su inclusión en el ensayo en los términos previstos enel artículo 7 de este real decreto.
3. Sólo se podrán realizar ensayos clínicos cuando se cumplan todos los requi-sitos siguientes:
a) Disponer de suficientes datos científicos y, en particular, ensayos farmacoló-gicos y toxicológicos en animales, que garanticen que los riesgos que implica en la per-sona en que se realiza son admisibles.
b) Que el estudio se base en los conocimientos disponibles, la información bus-cada suponga, presumiblemente, un avance en el conocimiento científico sobre el serhumano o para mejorar su estado de salud y su diseño minimice los riesgos para los su-jetos participantes en él.
c) Que los riesgos e inconvenientes previsibles para los sujetos del ensayo se ha-yan ponderado con respecto a los beneficios previsibles para cada sujeto del ensayo yfuturos pacientes.
4. Con el fin de garantizar una protección óptima de la salud y los derechos delos sujetos, no se podrán llevar a cabo investigaciones obsoletas o repetitivas.
5. El ensayo clínico debe estar diseñado para reducir al mínimo posible el do-lor, la incomodidad, el miedo y cualquier otro riesgo previsible en relación con la enfer-medad y edad o grado de desarrollo del sujeto ; tanto el umbral de riesgo como el gradode incomodidad deben ser definidos de forma específica y monitorizados durante el en-sayo, especialmente cuando los sujetos del ensayo sean menores, adultos incapaces oconstituyan una población especialmente vulnerable en razón de su situación econó-mica, médica o social.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
147

6. El tratamiento, comunicación y cesión de los datos de carácter personal de lossujetos participantes en el ensayo se ajustará a lo dispuesto en la Ley Orgánica 15/1999,de 13 de diciembre, de Protección de Datos de Carácter Personal, y constará expresa-mente en el consentimiento informado.
7. La atención sanitaria que se dispense y las decisiones médicas que se adop-ten sobre los sujetos serán responsabilidad de un médico o de un odontólogo debidamentecualificados.
8. Los sujetos participantes en ensayos clínicos sin beneficio potencial directopara el sujeto en investigación recibirán del promotor la compensación pactada por las mo-lestias sufridas. La cuantía de la compensación económica estará en relación con las ca-racterísticas del ensayo, pero en ningún caso será tan elevada como para inducir a unsujeto a participar por motivos distintos del interés por el avance científico.
La contraprestación que se hubiera pactado por la participación voluntaria en elensayo se percibirá en todo caso, si bien se reducirá proporcionalmente según la partici-pación del sujeto en la experimentación, en el supuesto de que decida revocar su consen-timiento y abandonar el ensayo.
En los casos extraordinarios de investigaciones sin beneficio potencial directo parael sujeto en investigación en menores e incapaces, para evitar la posible explotación deestos sujetos, no se producirá ninguna compensación económica por parte del promotor,a excepción del reintegro de los gastos extraordinarios y pérdidas de productividad que sederiven de la participación del sujeto en el ensayo.
9. Los sujetos que participen en ensayos con un posible beneficio potencial directopara el sujeto de investigación o sus representantes legales únicamente podrán recibir delpromotor el reintegro de los gastos extraordinarios y pérdidas de productividad que se de-riven de su participación en el ensayo.
10. Los sujetos del ensayo dispondrán de un punto donde puedan obtener mayorinformación sobre el ensayo, que constará en la hoja de información para el sujeto.
Artículo 4. De los ensayos clínicos con menores.
Sin perjuicio de la aplicación de las disposiciones generales establecidas en el ar-tículo anterior, solo se podrán realizar ensayos clínicos en menores de edad cuando secumplan, además, las siguientes condiciones especiales:
148

a) Que los ensayos sean de interés específico para la población que se investiga,y solo cuando dicha investigación sea esencial para validar datos procedentes de ensayosclínicos efectuados en personas capaces de otorgar su consentimiento informado u obte-nidos por otros medios de investigación. Además, la investigación deberá guardar relacióndirecta con alguna enfermedad que padezca el menor o bien ser de naturaleza tal quesolo pueda ser realizada en menores.
b) Que el bienestar del sujeto prevalezca siempre sobre los intereses de la cien-cia y de la sociedad, y existan datos que permitan prever que los beneficios esperados su-peran los riesgos o que el riesgo que conlleva el ensayo es mínimo.
c) Que la obtención del consentimiento informado se ajuste a lo especificado enel artículo. 7.3.
d) Que el protocolo sea aprobado por un Comité Ético de Investigación Clínicaque cuente con expertos en pediatría o que haya recabado asesoramiento sobre las cues-tiones clínicas, éticas y psicosociales en el ámbito de la pediatría.
e) Que se sigan las directrices científicas correspondientes de la Agencia Europeapara la Evaluación de Medicamentos.
Artículo 5. De los ensayos clínicos con adultos incapacitados.
Sin perjuicio de la aplicación de las disposiciones generales establecidas en elartículo 3, solo se podrán realizar ensayos clínicos en adultos que no estén en condicio-nes de dar su consentimiento informado y que no lo hayan dado con anterioridad al co-mienzo de su incapacidad, cuando se cumplan, además, las siguientes condicionesespeciales:
a) Que los ensayos sean de interés específico para la población que se investiga,y dicha investigación sea esencial para validar datos procedentes de ensayos clínicos efec-tuados en personas capaces de otorgar su consentimiento informado u obtenidos por otrosmedios de investigación. Además, la investigación deberá guardar relación directa con al-guna enfermedad que padezca el adulto incapaz, y que ésta le debilite o ponga en peli-gro su vida.
b) Que el bienestar del sujeto prevalezca sobre los intereses de la ciencia y de lasociedad, y existan datos que permitan prever que reporta algún beneficio al paciente queprevalezca sobre los riesgos o no produzca ningún riesgo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
149

c) Que el consentimiento informado se ajuste a lo especificado en el artículo 7.3.En todo caso, los sujetos no deben haberse negado a dar su consentimiento informado conanterioridad al comienzo de su incapacidad.
d) Que el protocolo sea aprobado por un Comité Ético de Investigación Clínicaque cuente con expertos en la enfermedad en cuestión o que haya recabado asesoramientode este tipo de expertos sobre las cuestiones clínicas, éticas y psicosociales en el ámbitode la enfermedad y del grupo de pacientes afectado.
Artículo 6. De los ensayos clínicos sin beneficio directo para la salud de los sujetos.
1. En los ensayos clínicos sin beneficio potencial directo para la salud de los su-jetos participantes, el riesgo que estos sujetos asuman estará justificado en razón del be-neficio esperado para la colectividad.
2. En menores y en sujetos incapacitados podrán realizarse ensayos sin beneficiopotencial directo para el sujeto únicamente si, además de tenerse en cuenta lo dispuestoen los artículos 4 y 5, el Comité Ético de Investigación Clínica considera que se cumplenlos siguientes requisitos:
a) Que se adoptan las medidas necesarias para garantizar que el riesgo seamínimo.
b) Que las intervenciones a que van a ser sometidos los sujetos del ensayo sonequiparables a las que corresponden a la práctica médica habitual en función de su si-tuación médica, psicológica o social.
c) Que del ensayo se pueden obtener conocimientos relevantes sobre la enfer-medad o situación objeto de investigación, de vital importancia para entenderla, pa-liarla o curarla.
d) Que estos conocimientos no pueden ser obtenidos de otro modo.
e) Que existen garantías sobre la correcta obtención del consentimiento infor-mado, de acuerdo con lo contemplado en el artículo 7.
3. En mujeres gestantes o en período de lactancia, sólo se podrán realizar ensa-yos clínicos sin beneficio potencial directo para ellas cuando el Comité Ético de Investi-gación Clínica concluya que no suponen ningún riesgo previsible para su salud ni para la
150

del feto o niño, y que se obtendrán conocimientos útiles y relevantes sobre el embarazo ola lactancia.
Artículo 7. Del consentimiento informado.
1. La obtención del consentimiento informado debe tener en cuenta los aspectosindicados en las recomendaciones europeas al respecto y que se recogen en las instruc-ciones para la realización de ensayos clínicos en España o, en su caso, en las directricesde la Unión Europea.
2. El sujeto del ensayo deberá otorgar su consentimiento después de haber enten-dido, mediante una entrevista previa con el investigador o un miembro del equipo de in-vestigación, los objetivos del ensayo, sus riesgos e inconvenientes, así como lascondiciones en las que se llevará a cabo, y después de haber sido informado de su dere-cho a retirarse del ensayo en cualquier momento sin que ello le ocasione perjuicio alguno.
El consentimiento se documentará mediante una hoja de información para el su-jeto y el documento de consentimiento. La hoja de información contendrá únicamente in-formación relevante, expresada en términos claros y comprensibles para los sujetos, yestará redactada en la lengua propia del sujeto.
3. Cuando el sujeto del ensayo no sea una persona capaz para dar su consenti-miento o no esté en condiciones de hacerlo, la decisión deberá adoptarse, teniendo encuenta lo indicado en este artículo.
a) Si el sujeto del ensayo es menor de edad:
1.º Se obtendrá el consentimiento informado previo de los padres o del represen-tante legal del menor ; el consentimiento deberá reflejar la presunta voluntad del menor ypodrá retirarse en cualquier momento sin perjuicio alguno para él. Cuando el menor tenga12 o más años, deberá prestar además su consentimiento para participar en el ensayo.
2.º El menor recibirá, de personal que cuente con experiencia en el trato con me-nores, una información sobre el ensayo, los riesgos y los beneficios adecuada a su capa-cidad de entendimiento.
3.º El investigador aceptará el deseo explícito del menor de negarse a participaren el ensayo o de retirarse en cualquier momento, cuando éste sea capaz de formarse unaopinión en función de la información recibida.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
151

4.º El promotor pondrá en conocimiento del Ministerio Fiscal las autorizaciones delos ensayos clínicos cuya población incluya a menores.
b) Si el sujeto es un adulto sin capacidad para otorgar su consentimiento infor-mado:
1.º Deberá obtenerse el consentimiento informado de su representante legal, trashaber sido informado sobre los posibles riesgos, incomodidades y beneficios del ensayo.El consentimiento deberá reflejar la presunta voluntad del sujeto y podrá ser retirado encualquier momento sin perjuicio para éste.
2.º Cuando las condiciones del sujeto lo permitan, este deberá prestar además suconsentimiento para participar en el ensayo, después de haber recibido toda la informa-ción pertinente adaptada a su nivel de entendimiento. En este caso, el investigador de-berá tener en cuenta la voluntad de la persona incapaz de retirarse del ensayo.
4. Cuando el ensayo clínico tenga un interés específico para la población en la quese realiza la investigación y lo justifiquen razones de necesidad en la administración delmedicamento en investigación, podrá someterse a un sujeto a un ensayo clínico sin obte-ner el consentimiento previo en los siguientes casos:
a) Si existe un riesgo inmediato grave para la integridad física o psíquica del su-jeto, se carece de una alternativa terapéutica apropiada en la práctica clínica y no es po-sible obtener su consentimiento o el de su representante legal. En este caso, siempre quelas circunstancias lo permitan, se consultará previamente a las personas vinculadas a élpor razones familiares o de hecho.
b) Si el sujeto no es capaz para tomar decisiones debido a su estado físico o psí-quico y carece de representante legal. En este caso, el consentimiento lo prestarán las per-sonas vinculadas a él por razones familiares o de hecho.
En ambos casos, esta eventualidad y la forma en que se procederá debe hallarseprevista en la documentación del ensayo aprobada por el Comité Ético de Investigación Clí-nica, y el sujeto o su representante legal será informado en cuanto sea posible y deberáotorgar su consentimiento para continuar en el ensayo si procediera.
5. El sujeto participante en un ensayo clínico, o su representante legal, podrán re-vocar su consentimiento en cualquier momento, sin expresión de causa y sin que por ellose derive para el sujeto participante responsabilidad ni perjuicio alguno.
152

Artículo 8. Del seguro u otra garantía financiera de los sujetos del ensayo.
1. Sólo podrá realizarse un ensayo clínico con medicamentos en investigación si,previamente, se ha concertado un seguro u otra garantía financiera que cubra los daños yperjuicios que como consecuencia del ensayo puedan resultar para la persona en que hu-biera de realizarse, salvo que el ensayo se refiera únicamente a medicamentos autoriza-dos en España, su utilización en el ensayo se ajuste a las condiciones de uso autorizadasy el Comité Ético de Investigación Clínica considere que las intervenciones a las que se-rán sometidos los sujetos por su participación en el ensayo suponen un riesgo equivalenteo inferior al que correspondería a su atención en la práctica clínica habitual.
2. El promotor del ensayo es el responsable de la contratación de dicho seguro deresponsabilidad o de la garantía financiera y éstos cubrirán las responsabilidades del pro-motor, del investigador principal y sus colaboradores, y del hospital o centro donde selleve a cabo el ensayo clínico.
3. En el supuesto previsto al final del apartado 1 de este artículo, cuando no seconcierte seguro u otra garantía financiera o, por cualquier circunstancia, el seguro o lagarantía financiera concertados no cubran enteramente los daños, el promotor del ensayoclínico, el investigador principal y el hospital o centro donde se realice el ensayo serán res-ponsables solidariamente, sin necesidad de que medie culpa, del daño que en su saludsufra el sujeto sometido al ensayo clínico, así como de los perjuicios económicos que sederiven, incumbiéndoles la carga de la prueba de que no son consecuencia del ensayo clí-nico o de las medidas terapéuticas o diagnósticas que se adopten durante su realización.Ni la autorización administrativa, ni el dictamen favorable del Comité Ético de Investiga-ción Clínica eximirán de responsabilidad al promotor del ensayo clínico, al investigadorprincipal y sus colaboradores o al hospital o centro donde se realice el ensayo clínico enestas circunstancias.
4. Se presume, salvo prueba en contrario, que los daños que afecten a la saluddel sujeto del ensayo durante su realización y en el año siguiente a la terminación del tra-tamiento se han producido como consecuencia del ensayo. Sin embargo, una vez con-cluido el año, el sujeto del ensayo esta obligado a probar el nexo entre el ensayo y el dañoproducido.
5. A los efectos del régimen de responsabilidad previsto en este artículo, serán ob-jeto de resarcimiento todos los gastos derivados del menoscabo en la salud o estado físicodel sujeto sometido al ensayo clínico, así como los perjuicios económicos que se derivendirectamente de dicho menoscabo, siempre que éste no sea inherente a la patología ob-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
153

jeto de estudio, o se incluya dentro de las reacciones adversas propias de la medicaciónprescrita para dicha patología, así como la evolución propia de su enfermedad como con-secuencia de la ineficacia del tratamiento.
6. El importe mínimo que en concepto de responsabilidad estará garantizado seráde 250.000 euros por sujeto sometido a ensayo clínico, como indemnización a tanto al-zado. En caso de que la indemnización se fije como renta anual constante o creciente, ellímite de la cobertura del seguro o de la garantía financiera será de al menos 25.000 eu-ros anuales por cada sujeto sometido al ensayo clínico, pudiéndose establecer como ca-pital asegurado máximo o como importe máximo de la garantía financiera un sublímite porensayo clínico y año de 2.500.000 euros.
Se autoriza al Ministerio de Sanidad y Consumo para revisar los límites anterior-mente establecidos.
7. Cuando el promotor e investigador principal sean la misma persona y el ensayoclínico se realice en un centro sanitario dependiente de una Administración pública, éstapodrá adoptar las medidas que considere oportunas para facilitar la garantía de los ries-gos específicos derivados del ensayo en los términos señalados en los apartados anterio-res, con el objeto de fomentar la investigación.
CAPÍTULO III. De los Comités Éticos de Investigación Clínica
Artículo 9. Del Centro Coordinador de los Comités Éticos de Investigación Clínica.
1. Con el objeto de facilitar el dictamen único se creará el Centro Coordinador deComités Éticos de Investigación Clínica. Dicha organización se constituye como la unidadtécnica operativa que tiene como objetivo facilitar que los Comité Ético de InvestigaciónClínica acreditados por las comunidades autónomas puedan compartir estándares de ca-lidad y criterios de evaluación adecuados y homogéneos y favorecer la agilidad en el pro-ceso de obtención del dictamen único.
2. El Centro Coordinador de Comités Éticos de Investigación Clínica se adscribeal Ministerio de Sanidad y Consumo, a través de la Secretaría General de Sanidad.
3. El Centro Coordinador de Comités Éticos de Investigación Clínica colaborará conlas autoridades competentes de las comunidades autónomas y rendirá cuentas de su ac-tividad en el Consejo Interterritorial del Sistema Nacional de Salud.
154

4. El Centro Coordinador de Comités Éticos de Investigación Clínica desarrollarálas siguientes actividades:
a) Facilitar el dictamen único en los ensayos multicéntricos.
b) Coordinar con las comunidades autónomas el desarrollo de un sistema informá-tico de comunicación entre Comités Éticos de Investigación Clínica.
c) Gestionar la base de datos de ensayos clínicos de la red nacional de ComitésÉticos de Investigación Clínica.
d) Promover criterios de evaluación comunes en los Comités Éticos de Investiga-ción Clínica.
e) Promover la formación de los miembros de los Comités Éticos de InvestigaciónClínica.
f) Promover foros de debate entre Comités Éticos de Investigación Clínica.
g) Actuar como punto de contacto para proporcionar información sobre el funcio-namiento de la red nacional de Comités Éticos de Investigación Clínica.
h) Proporcionar asesoramiento a los Comités Éticos de Investigación Clínica encuestiones de procedimiento.
i) Elaborar la memoria anual de actividades.
Artículo 10. Funciones de los Comités Éticos de Investigación Clínica.
Los Comités Éticos de Investigación Clínica desempeñarán las siguientesfunciones:
a) Evaluar los aspectos metodológicos, éticos y legales de los ensayos clínicos queles sean remitidos, de conformidad con lo establecido en la sección 2.ª del capítulo IV.
b) Evaluar las modificaciones relevantes de los ensayos clínicos autorizados.
c) Realizar un seguimiento del ensayo, desde su inicio hasta la recepción del in-forme final.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
155

Artículo 11. Acreditación de los Comités Éticos de Investigación Clínica.
Los Comités Éticos de Investigación Clínica serán acreditados por la autoridad sani-taria competente en cada comunidad autónoma, quien determinará el ámbito geográfico einstitucional de cada comité. Dicha acreditación deberá ser renovada periódicamente por di-cha autoridad sanitaria según los procedimientos y plazos que ésta determine. Tanto la acre-ditación inicial como sus renovaciones deberán ser notificadas a la Agencia Española deMedicamentos y Productos Sanitarios y al Centro Coordinador de Comités Éticos de Investi-gación Clínica.
Artículo 12. Composición de los Comités Éticos de Investigación Clínica.
1. El Comité Ético de Investigación Clínica deberá estar constituido por al menosnueve miembros, de manera que se asegure la independencia de sus decisiones, así comosu competencia y experiencia en relación con los aspectos metodológicos, éticos y lega-les de la investigación, la farmacología y la práctica clínica asistencial en medicina hos-pitalaria y extrahospitalaria.
2. Entre los miembros del citado comité figurarán médicos, uno de los cuales será far-macólogo clínico ; un farmacéutico de hospital, y un Diplomado universitario en Enfermería.
Al menos un miembro deberá ser independiente de los centros en los que se lleven acabo proyectos de investigación que requieran la evaluación ética por parte del comité.
Al menos dos miembros deben ser ajenos a las profesiones sanitarias, uno de loscuales deberá ser Licenciado en Derecho.
3. Se garantizará un sistema de renovación de miembros que permita nuevas in-corporaciones de forma regular, a la vez que se mantiene la experiencia del comité.
4. Tal como establece el artículo 4 de la Ley 25/1990, de 20 de diciembre, delMedicamento, la pertenencia a un Comité Ético de Investigación Clínica será incompati-ble con cualquier clase de intereses derivados de la fabricación y venta de medicamentosy productos sanitarios.
Artículo 13. Requisitos mínimos respecto a los medios e infraestructura de los Comités Éti-cos de Investigación Clínica.
Las autoridades sanitarias de las comunidades autónomas correspondientes ase-
156

gurarán que cada Comité Ético de Investigación Clínica acreditado cuente al menos con lossiguientes medios:
a) Instalaciones específicas que permitan la realización de su trabajo, en condi-ciones que garanticen la confidencialidad. Deberán disponer de un espacio apropiado parala secretaría del comité, para la realización de las reuniones y para el manejo y archivo dedocumentos confidenciales.
b) Equipamiento informático con capacidad suficiente para manejar toda la infor-mación generada por el comité y disponibilidad de un sistema rápido de transmisión deinformación.
c) Personal administrativo y técnico que permita al comité poder ejercer de ma-nera apropiada sus funciones.
Artículo 14. Normas generales de funcionamiento de los Comités Éticos de InvestigaciónClínica.
1. Ni el Comité Ético de Investigación Clínica ni ninguno de sus miembros podránpercibir directa ni indirectamente remuneración alguna por parte del promotor del ensayo.
2. Los Comités Éticos de Investigación Clínica deberán elaborar y seguir parasu funcionamiento unos procedimientos normalizados de trabajo que como mínimo sereferirán a:
a) La composición y requisitos que deben cumplir sus miembros.
b) La periodicidad de las reuniones, que al menos deberá ser mensual.
c) El procedimiento para convocar a sus miembros.
d) Los aspectos administrativos, incluyendo la documentación que debe presentarse.
e) Los casos en que se pueda realizar una revisión rápida de la documentación co-rrespondiente a un ensayo clínico y el procedimiento que debe seguirse en estos casos.
f) La evaluación inicial de los protocolos y sistema de seguimiento de los ensayos.
g) Los mecanismos de toma de decisiones.
h) La preparación y aprobación de las actas de las reuniones.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
157

i) El archivo y conservación de la documentación del comité y de la relacionadacon los ensayos clínicos evaluados.
3. En los casos que exista Comisión de Investigación o Comité de Ética Asisten-cial, deberá formar parte del comité un miembro de cada una de ellas.
4. Cuando el Comité Ético de Investigación Clínica no reúna los conocimientos yexperiencia necesarios para evaluar un determinado ensayo clínico recabará el asesora-miento de alguna persona experta no perteneciente al comité, que respetará el principiode confidencialidad. De esta manera:
a) Cuando el comité evalúe protocolos de investigación clínica con procedimien-tos quirúrgicos, técnicas diagnósticas o productos sanitarios, contará con el asesoramientode al menos una persona experta en el procedimiento o tecnología que se vaya a evaluar.
b) Cuando el comité evalúe ensayos clínicos que se refieran a menores o a suje-tos incapacitados, contará con el asesoramiento de al menos una persona con experien-cia en el tratamiento de la población que se incluya en el ensayo.
5. El investigador principal o los colaboradores de un ensayo clínico no podrán par-ticipar en la evaluación, ni en el dictamen de su propio protocolo, aun cuando sean miem-bros del comité.
6. Cada reunión del comité quedará recogida en el acta correspondiente, en la quese detallarán como mínimo los miembros asistentes, que para cada estudio evaluado sehan ponderado los aspectos contemplados en este real decreto y la decisión adoptada so-bre cada ensayo.
CAPÍTULO IV. De la intervención sobre los ensayos clínicos con medicamentos
SECCIÓN 1.ª Disposiciones Comunes
Artículo 15. Requisitos para la realización de ensayos clínicos.
Para la realización de ensayos clínicos con medicamentos se precisará del previodictamen favorable del Comité Ético de Investigación Clínica correspondiente, de la con-formidad de la dirección de cada uno de los centros donde el ensayo vaya a realizarse yde la autorización de la Agencia Española de Medicamentos y Productos Sanitarios.
158

El dictamen y la autorización citados en el párrafo anterior podrán solicitarse deforma simultánea o no, según las preferencias del promotor.
SECCIÓN 2.ª Del dictamen de los Comités Éticos De Investigación Clínica
Artículo 16. Iniciación del procedimiento.
1. El promotor deberá solicitar por escrito el dictamen del Comité Ético de Inves-tigación Clínica, de acuerdo con las instrucciones para la realización de ensayos clínicosen España o, en su caso, las directrices de la Comisión Europea, que publicará el Minis-terio de Sanidad y Consumo.
2. La solicitud deberá acompañarse de la siguiente documentación:
a) El protocolo.
b) El manual del investigador.
c) Los documentos referentes al consentimiento informado, incluyendo la hoja deinformación para el sujeto de ensayo.
d) Los documentos sobre la idoneidad del investigador y sus colaboradores.
e) Los documentos sobre la idoneidad de las instalaciones.
f) Las cantidades y el modo en que los investigadores y sujetos puedan ser, en sucaso, remunerados o indemnizados por su participación en el ensayo clínico, así como loselementos pertinentes de todo contrato previsto entre el promotor y el centro.
g) Una copia de la póliza del seguro o del justificante de la garantía financiera delensayo clínico o un certificado de ésta, cuando proceda.
h) En los casos previstos en el artículo 8.3 de ausencia de seguro o de seguro concobertura parcial, deberá acompañarse documento firmado de asunción de responsabili-dad en caso de daños producidos como consecuencia del ensayo.
i) Los procedimientos y material utilizado para el reclutamiento de los sujetos delensayo.
j) El compromiso de los investigadores que está previsto que participen en el ensayo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
159

Artículo 17. Criterios de evaluación para la emisión del dictamen.
1. El Comité Ético de Investigación Clínica correspondiente evaluará el protocolo,el manual del investigador y el resto de la documentación que acompañe a la solicitud yemitirá su dictamen tomando en consideración, en particular, las siguientes cuestiones:
a) La pertinencia del ensayo clínico, teniendo en cuenta el conocimiento disponible.
b) La pertinencia de su diseño para obtener conclusiones fundamentadas con elnúmero adecuado de sujetos en relación con el objetivo del estudio.
c) Los criterios de selección y retirada de los sujetos del ensayo, así como la se-lección equitativa de la muestra.
d) La justificación de los riesgos e inconvenientes previsibles en relación con los bene-ficios esperables para los sujetos del ensayo, para otros pacientes y para la comunidad, teniendoen cuenta el principio de protección de los sujetos del ensayo desarrollado en el artículo 3.
e) La justificación del grupo control (ya sea placebo o un tratamiento activo).
f) Las previsiones para el seguimiento del ensayo.
g) La idoneidad del investigador y de sus colaboradores.
h) La idoneidad de las instalaciones.
i) La idoneidad de la información escrita para los sujetos del ensayo y el procedi-miento de obtención del consentimiento informado, y la justificación de la investigaciónen personas incapaces de dar su consentimiento informado.
j) El seguro o la garantía financiera previstos para el ensayo.
k) Las cantidades y, en su caso, previsiones de remuneración o compensaciónpara los investigadores y sujetos del ensayo y los aspectos relevantes de cualquier acuerdoentre el promotor y el centro, que han de constar en el contrato previsto en el artículo 30.
l) El plan previsto para el reclutamiento de los sujetos.
2. Las cuestiones indicadas en los párrafos g), h) y k) del apartado anterior debe-rán ser evaluadas para cada uno de los centros implicados en el ensayo clínico.
160

Artículo 18. Procedimiento para la emisión del dictamen en ensayos unicéntricos.
1. En el caso de los ensayos clínicos unicéntricos, la solicitud se presentaráante el Comité Ético de Investigación Clínica correspondiente. Éste, en el plazo de 10días naturales, verificará que la solicitud reúne los requisitos previstos en el artículo16 y, sin perjuicio de su subsanación cuando proceda, comunicará al promotor la ad-misión a trámite de la solicitud con indicación del calendario de evaluación o, en sucaso, su inadmisión a trámite.
2. El Comité Ético de Investigación Clínica dispondrá de un plazo máximo de60 días naturales, a contar desde la notificación de la admisión a trámite de la solici-tud, para comunicar su dictamen motivado al promotor y a la Agencia Española deMedicamentos y Productos Sanitarios.
3. Durante el periodo establecido en el apartado anterior, el comité podrá so-licitar una sola vez información complementaria al promotor ; en tal caso se suspen-derá el cómputo del plazo de evaluación hasta que se reciba la información solicitada.
4. En el caso de ensayos clínicos que se refieran a medicamentos de terapiagénica, de terapia celular somática o que contengan organismos modificados genéti-camente, el plazo establecido en el apartado 2 será de 90 días naturales. Dicho plazopodrá prorrogarse por otros 90 días cuando se recabe dictamen de un comité de ex-pertos.
5. En el caso de ensayos clínicos que se refieran a terapia celular xenogénica,no existirá ninguna limitación de plazo para la emisión del dictamen motivado.
Artículo 19. Dictamen único en ensayos clínicos multicéntricos.
1. En los ensayos clínicos en los que participen dos o más centros ubicadosen España, se emitirá un único dictamen con independencia del número de ComitésÉticos de Investigación Clínica implicados.
El dictamen único se adoptará de conformidad con el procedimiento previstoen los apartados siguientes.
2. El promotor presentará la solicitud de evaluación del ensayo ante el ComitéÉtico de Investigación Clínica que actuará como comité de referencia y que se respon-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
161

sabilizará de la emisión del dictamen único y al resto de los Comités Éticos de Inves-tigación Clínica implicados.
3. El Comité Ético de Investigación Clínica de referencia, en el plazo máximode 10 días naturales, verificará que la solicitud reúne los requisitos previstos en el ar-tículo 16 y, sin perjuicio de su subsanación cuando proceda, comunicará al promotory a los Comité Ético de Investigación Clínica implicados en el ensayo la admisión a trá-mite de la solicitud con indicación del calendario de evaluación o, en su caso, su in-admisión a trámite.
4. El Comité Ético de Investigación Clínica de referencia dispondrá de un plazomáximo de 60 días naturales, a contar desde la notificación de la admisión a trámiteal promotor para comunicar su dictamen motivado al promotor, a la Agencia Españolade Medicamentos y Productos Sanitarios y a los demás comités implicados en el ensayo.
Cada comité implicado remitirá con tiempo suficiente al comité de referenciaun informe sobre los aspectos locales del ensayo, así como sobre cualquier otro aspectodel ensayo que considere relevante.
5. Durante el periodo establecido en el apartado anterior, el Comité Ético deInvestigación Clínica de referencia podrá solicitar una sola vez información comple-mentaria al promotor ; en tal caso se suspenderá el cómputo del plazo de evaluaciónhasta que se reciba la información solicitada. Dicha información se presentará tambiéna los demás comités implicados.
6. Los informes de los demás comités implicados deberán ser tenidos encuenta por el Comité Ético de Investigación Clínica de referencia para la emisión deldictamen único, que habrá de ser motivado, especialmente, en caso de discrepar dela opinión de otro comité sobre cualquier aspecto del ensayo, pero sólo vincularán alcomité de referencia respecto a los aspectos locales.
7. En el caso de ensayos clínicos que se refieran a medicamentos de terapiagénica, de terapia celular somática o que contengan organismos modificados genéti-camente, el plazo establecido en el apartado 4 será de 90 días naturales. Dicho plazopodrá prorrogarse por otros 90 días cuando se recabe dictamen de un comité de ex-pertos.
8. En el caso de ensayos clínicos que se refieran a terapia celular xenogénica,no existirá ninguna limitación de plazo para la emisión del dictamen motivado.
162

SECCIÓN 3.ª DE LA AUTORIZACIÓN DE LA AGENCIA ESPAÑOLA DE MEDICAMENTOSY PRODUCTOS SANITARIOS
Artículo 20. Iniciación del procedimiento.
1. La autorización del ensayo clínico deberá solicitarse mediante escrito del pro-motor dirigido al Director de la Agencia Española de Medicamentos y Productos Sanita-rios, de acuerdo con las instrucciones para la realización de ensayos clínicos en Españao, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sa-nidad y Consumo.
2. La solicitud deberá acompañarse de la siguiente documentación:
a) Protocolo del ensayo.
b) Manual del investigador.
c) Hoja de información para los sujetos del ensayo.
d) Expediente del medicamento en investigación, cuando proceda.
e) Acreditación del pago de la tasa prevista en el artículo 117.1, grupo V, epígrafe5.2, de la Ley 25/1990, de 20 de diciembre, del Medicamento.
3. Además, será necesaria la calificación como producto en fase de investigaciónclínica para aquellos medicamentos en investigación que se definan en las instruccionespara la realización de ensayos clínicos en España o, en su caso, las directrices de la Co-misión Europea, que publicará el Ministerio de Sanidad y Consumo.
Para la calificación del medicamento como producto en fase de investigaciónclínica, a los efectos previstos en el artículo 24, se deberá aportar la siguiente docu-mentación:
a) Formulario de solicitud.
b) Expediente del medicamento en investigación.
c) Acreditación del pago de la tasa prevista en el artículo 117.1, grupo V, epígrafe5.1, de la Ley 25/1990, de 20 de diciembre, del Medicamento.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
163

Artículo 21. Validación de la solicitud.
1. La Agencia Española de Medicamentos y Productos Sanitarios, en el plazo de10 días naturales, verificará que la solicitud reúne los requisitos previstos en el artículoanterior, y notificará al solicitante la admisión a trámite de la solicitud con indicación delprocedimiento aplicable, así como del plazo para la notificación de la resolución.
2. En el caso de que la solicitud no reúna los requisitos establecidos en el apar-tado anterior, se requerirá al solicitante para que subsane las deficiencias en el plazo má-ximo de 10 días, con indicación de que, si así no lo hiciera, se archivará la solicitud previaresolución que se dictará en los términos establecidos en el artículo 42 de la Ley 30/1992,de 26 de noviembre, de Régimen Jurídico para las Administraciones Públicas y del Pro-cedimiento Administrativo Común.
3. En el plazo de 10 días naturales, a contar desde la presentación de la docu-mentación requerida, se notificará al solicitante la admisión a trámite de su solicitud enlos términos previstos en el apartado 1 de este artículo o, en su caso, su inadmisión a trá-mite.
Artículo 22. Procedimiento ordinario.
1. La autorización se entenderá otorgada si en el plazo de 60 días naturales, a con-tar desde la notificación de la admisión a trámite de la solicitud, la Agencia Española deMedicamentos y Productos Sanitarios no comunica objeciones motivadas al solicitantesiempre y cuando se haya notificado de forma previa a la Agencia Española de Medicamen-tos y Productos Sanitarios el dictamen favorable del Comité Ético de Investigación Clínicay la conformidad de la dirección de los centros participantes en el ensayo.
2. En el caso de que se comuniquen objeciones motivadas, el solicitante dispon-drá del plazo de 15 días naturales para modificar su solicitud de acuerdo con las objecio-nes planteadas o, en caso de discrepancia con dichas objeciones, efectuar las alegacionesy presentar los documentos que estime pertinentes en apoyo de su solicitud.
Transcurrido el plazo establecido en el párrafo anterior sin que el solicitante hayamodificado la solicitud o presentado alegaciones, se le entenderá desistido de su solicitud.
3. A la vista de la modificación propuesta por el solicitante o, en su caso, de susalegaciones, la Agencia Española de Medicamentos y Productos Sanitarios emitirá reso-lución expresa, autorizando o denegando el ensayo, que deberá ser notificada al solicitante
164

en el plazo de 15 días a contar desde la entrada en su registro general del escrito de mo-dificación o alegaciones.
4. La autorización del ensayo clínico se entenderá sin perjuicio de la aplicación,cuando proceda, de la legislación vigente sobre la utilización confinada de organismosmodificados genéticamente, y la liberación intencional en el medio ambiente de organis-mos modificados genéticamente.
Artículo 23. Procedimientos especiales.
1. Sin perjuicio de lo previsto en al artículo anterior, no podrá iniciarse un ensayoclínico sin la previa autorización por escrito de la Agencia Española de Medicamentos yProductos Sanitarios en los siguientes casos:
a) Ensayos clínicos en los que la Agencia Española de Medicamentos y ProductosSanitarios haya comunicado objeciones al promotor dentro del plazo establecido en elapartado 1 del artículo anterior.
b) Ensayos clínicos con medicamentos que requieren la calificación de productoen fase de investigación clínica.
c) Ensayos clínicos con medicamentos de terapia génica, terapia celular somática(incluidos los de terapia celular xenogénica), así como todos los medicamentos que con-tengan organismos modificados genéticamente.
2. En los casos previstos en el apartado anterior, la Agencia Española de Medica-mentos y Productos Sanitarios emitirá resolución expresa que autorice o deniegue el en-sayo clínico, de conformidad con el procedimiento y los plazos previstos en el artículo 22,con las particularidades que se establecen en los apartados 3 y 4 siguientes.
Una vez transcurrido el plazo correspondiente sin que se notifique al interesadola resolución, se podrá entender desestimada la solicitud.
3. En los ensayos clínicos con medicamentos de terapia génica, terapia celularsomática (excluidos los de terapia celular xenogénica), así como todos los medicamen-tos que contengan organismos modificados genéticamente, el plazo máximo para autori-zar expresamente el ensayo clínico será de 90 días naturales. Dicho plazo se ampliará en90 días naturales cuando resulte preceptivo recabar algún informe de acuerdo con lanormativa vigente.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
165

4. En los ensayos clínicos con medicamentos de terapia celular xenogénica, laAgencia Española de Medicamentos y Productos Sanitarios no tendrá limite temporal parala comunicación de objeciones ni para la autorización o denegación del ensayo.
Artículo 24. Productos en fase de investigación clínica.
1. La Agencia Española de Medicamentos y Productos Sanitarios, cuando autoriceun ensayo clínico con un medicamento en investigación, hará constar en la autorizacióndel ensayo la calificación de dicho medicamento como producto en fase de investigaciónclínica, en los casos que proceda.
2. La Agencia Española de Medicamentos y Productos Sanitarios mantendrá unregistro actualizado de los medicamentos en investigación calificados como productos enfase de investigación clínica, en el que se enumerarán las indicaciones concretas quepueden ser objeto de investigación clínica, así como las limitaciones, plazos, condicionesy garantías que, en su caso, se establezcan.
3. Para la autorización de posteriores ensayos clínicos con un medicamento en in-vestigación previamente calificado como producto en fase de investigación clínica deberáactualizarse, cuando resulte necesario, la documentación citada en el artículo 20.3 con-forme a las instrucciones para la realización de ensayos clínicos en España o, en su caso,las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
Las solicitudes que se acojan a lo dispuesto en el párrafo anterior harán referen-cia a dichas circunstancias.
Artículo 25. Modificación de las condiciones de autorización de ensayos clínicos.
1. Cualquier modificación en las condiciones autorizadas para un ensayo clínicoque se considere relevante no podrá llevarse a efecto sin el previo dictamen favorable delComité Ético de Investigación Clínica correspondiente y la autorización de la Agencia Es-pañola de Medicamentos y Productos Sanitarios.
Sin perjuicio de lo anterior, si la modificación se refiere exclusivamente a docu-mentos específicos que deben ser evaluados por el Comité Ético de Investigación Clínica,únicamente se requerirá el dictamen favorable de dicho comité para su aplicación. Por elcontrario, si la modificación se refiere a la documentación que deba ser evaluada única-mente por la Agencia Española de Medicamentos y Productos Sanitarios, solamente se re-querirá la autorización de ésta.
166

Si se dieran circunstancias que pudieran poner en peligro la seguridad de los su-jetos, el promotor y el investigador adoptarán las medidas urgentes oportunas para prote-ger a los sujetos de cualquier riesgo inmediato. El promotor informará lo antes posibletanto a la Agencia Española de Medicamentos y Productos Sanitarios como a los ComitésÉticos de Investigación Clínica implicados en el ensayo de dichas circunstancias y de lasmedidas adoptadas.
2. Se consideran modificaciones relevantes aquellas que se detallen en las instruc-ciones para la realización de ensayos clínicos en España o, en su caso, las directrices dela Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
3. La solicitud deberá presentarse por escrito, fechada y firmada por el promotore investigador, ante la Agencia Española de Medicamentos y Productos Sanitarios y antelos Comités Éticos de Investigación Clínica correspondientes. La solicitud se adecuará alo establecido en las instrucciones para la realización de ensayos clínicos en España o, ensu caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad yConsumo.
4. El dictamen previo se adoptará conforme al procedimiento establecido en losartículos 18 y 19. No obstante:
a) El Comité Ético de Investigación Clínica correspondiente dispondrá de un plazomáximo de 35 días naturales, a contar desde la notificación de la admisión a trámite dela solicitud, para comunicar su dictamen motivado al promotor, al Centro Coordinador deComités Éticos de Investigación Clínica, a la Agencia Española de Medicamentos y Pro-ductos Sanitarios y, en el caso de ensayos multicéntricos, a los Comités Éticos de Inves-tigación Clínica implicados.
b) En los ensayos multicéntricos, el informe de los Comités Éticos de Investiga-ción Clínica implicados distintos del comité de referencia sólo resultará preceptivo cuandola modificación suponga la incorporación de nuevos centros o investigadores principalesal ensayo, y sólo será vinculante en los aspectos locales del ensayo. Sólo será necesarioel informe del comité ético correspondiente al centro o investigador que se incorpore.
5. La autorización de la Agencia Española de Medicamentos y Productos Sanita-rios se adoptará conforme al procedimiento establecido en los artículos 21, 22 y 23. Noobstante, la autorización se entenderá otorgada si en el plazo de 35 días naturales, a con-tar desde la notificación de la admisión a trámite de la solicitud, la Agencia Española deMedicamentos y Productos Sanitarios no comunica objeciones motivadas al solicitante.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
167

6. En caso de modificaciones en ensayos clínicos con medicamentos de terapiagénica, de terapia celular somática (incluidos los de terapia celular xenogénica), asícomo todos los medicamentos que contengan organismos modificados genéticamente,los plazos para su autorización podrán ser ampliados, notificando al promotor el nuevoplazo.
Artículo 26. Suspensión y revocación de la autorización del ensayo clínico.
1. La autorización del ensayo clínico se suspenderá o revocará, de oficio o a peti-ción justificada del promotor, mediante resolución de la Agencia Española de Medicamen-tos y Productos Sanitarios en los siguientes supuestos:
a) Si se viola la ley.
b) Si se alteran las condiciones de su autorización.
c) Si no se cumplen los principios éticos recogidos en el artículo 60 de la Ley25/1990, de 20 de diciembre, del Medicamento.
d) Para proteger a los sujetos del ensayo.
e) En defensa de la salud pública.
2. La resolución por la que se suspenda o revoque la autorización del ensayo seadoptará previa instrucción del oportuno procedimiento, con audiencia al interesado quedeberá pronunciarse en el plazo de siete días a contar desde la notificación del inicio delprocedimiento.
Una vez adoptada la resolución citada en el párrafo anterior, la Agencia Españolade Medicamentos y Productos Sanitarios notificará la decisión adoptada, con expresa in-dicación de los motivos, a los Comités Éticos de Investigación Clínica participantes, a laComisión Europea, a la Agencia Europea para la Evaluación de Medicamentos, a las au-toridades sanitarias de las comunidades autónomas y a las autoridades sanitarias de losdemás Estados miembros.
3. Las autoridades sanitarias de las comunidades autónomas, por propia inicia-tiva o a propuesta del Comité Ético de Investigación Clínica correspondiente, podrán re-solver la suspensión cautelar del ensayo clínico en los casos previstos en el apartado 1,y lo notificarán de inmediato a la Agencia Española de Medicamentos y Productos Sa-
168

nitarios, la cual, conforme a lo establecido en el apartado 2, resolverá la suspensión orevocación de la autorización del ensayo o, en su caso, el levantamiento de la medidacautelar.
Artículo 27. Informe final del ensayo clínico.
1. Una vez finalizada la realización del ensayo clínico, en el plazo de 90 días,el promotor notificará a la Agencia Española de Medicamentos y Productos Sanitariosy a los comités éticos implicados el final del ensayo.
2. En caso de terminación anticipada, en el plazo de 15 días el promotor re-mitirá a la Agencia Española de Medicamentos y Productos Sanitarios y a los ComitésÉticos de Investigación Clínica implicados un informe que incluya los datos obtenidoshasta el momento de su conclusión anticipada, así como los motivos de ésta, y en sucaso las medidas adoptadas en relación con los sujetos participantes en el ensayo.
3. En el plazo de un año desde el final del ensayo, el promotor remitirá a laAgencia Española de Medicamentos y Productos Sanitarios y a los Comités Éticos deInvestigación Clínica implicados un resumen del informe final sobre los resultados delensayo.
4. Cuando la duración del ensayo sea superior a un año, será necesario ade-más que el promotor remita un informe anual sobre la marcha del ensayo.
5. En todos los casos se seguirán las instrucciones para la realización de en-sayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que pu-blicará el Ministerio de Sanidad y Consumo.
CAPÍTULO V. Del uso compasivo
Artículo 28. Uso compasivo de medicamentos.
1. Se entiende por uso compasivo de medicamentos la utilización en pacien-tes aislados y al margen de un ensayo clínico de medicamentos en investigación, in-cluidas especialidades farmacéuticas para indicaciones o condiciones de uso distintasde las autorizadas, cuando el médico bajo su exclusiva responsabilidad considere in-dispensable su utilización.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
169

2. Para utilizar un medicamento bajo las condiciones de uso compasivo se reque-rirá el consentimiento informado del paciente o de su representante legal, un informe clí-nico en el que el médico justifique la necesidad de dicho tratamiento, la conformidad deldirector del centro donde se vaya a aplicar el tratamiento y la autorización de la AgenciaEspañola de Medicamentos y Productos Sanitarios.
3. El médico responsable comunicará a la Agencia Española de Medicamentos yProductos Sanitarios los resultados del tratamiento, así como las sospechas de reaccionesadversas que puedan ser debidas a éste.
Artículo 29. Continuación del tratamiento tras la finalización del ensayo.
Una vez finalizado el ensayo, toda continuación en la administración del medica-mento en investigación, en tanto no se autorice el medicamento para esas condiciones deuso, se regirá por las normas establecidas para el uso compasivo en el artículo anterior.
CAPÍTULO VI. Aspectos económicos
Artículo 30. Aspectos económicos del ensayo clínico.
1. Todos los aspectos económicos relacionados con el ensayo clínico quedarán re-flejados en un contrato entre el promotor y cada uno de los centros donde se vaya a rea-lizar el ensayo. Esta documentación se pondrá a disposición del Comité Ético deInvestigación Clínica correspondiente.
2. Las Administraciones sanitarias competentes para cada servicio de salud esta-blecerán los requisitos comunes y condiciones de financiación, así como el modelo decontrato de conformidad con los principios generales de coordinación que acuerde el Con-sejo Interterritorial del Sistema Nacional de Salud.
3. En el contrato constará el presupuesto inicial del ensayo, que especificará loscostes indirectos que aplicará el centro, así como los costes directos extraordinarios, con-siderando como tales aquellos gastos ajenos a los que hubiera habido si el sujeto no hu-biera participado en el ensayo, como análisis y exploraciones complementarias añadidas,cambios en la duración de la atención a los enfermos, reembolso por gastos a los pacien-tes, compra de aparatos y compensación para los sujetos del ensayo e investigadores.También constarán los términos y plazos de los pagos, así como cualquier otra responsa-bilidad subsidiaria que contraigan las partes.
170

CAPÍTULO VII. Medicamentos en investigación
Artículo 31. Fabricación.
1. La fabricación de medicamentos no autorizados en España para su utilizaciónen el ámbito de un ensayo clínico únicamente podrá realizarse previa autorización de laAgencia Española de Medicamentos y Productos Sanitarios. Esta autorización estará vi-gente durante el tiempo de realización del ensayo clínico en el que se utilicen.
2. El fabricante de un medicamento en investigación ha de estar autorizado parael ejercicio de su actividad de acuerdo con lo establecido en el Real Decreto 1564/1992,de 18 de diciembre, por el que se desarrolla y regula el régimen de autorización de los la-boratorios farmacéuticos e importadores de medicamentos y la garantía de calidad de sufabricación industrial.
3. En el caso de que alguna de las fases de la fabricación, como el acondiciona-miento final, se realicen en un servicio de farmacia hospitalario, dicho servicio quedará ex-cluido de la autorización contemplada en el apartado 2.
4. En todas las fases de la fabricación de un medicamento en investigación se hande seguir las normas de correcta fabricación de medicamentos en la Unión Europea, in-cluido su anexo 13, publicadas por el Ministerio de Sanidad y Consumo.
Artículo 32. Importación.
1. La importación de medicamentos en investigación, para su utilización en elámbito de un ensayo clínico, únicamente podrá realizarse previa autorización de la Agen-cia Española de Medicamentos y Productos Sanitarios.
2. El laboratorio farmacéutico importador garantizará que el medicamento ha sidoelaborado por un fabricante debidamente autorizado en el país de origen y que cumple nor-mas de correcta fabricación, al menos equivalentes a las establecidas por la Unión Euro-pea, sin perjuicio de la responsabilidad del promotor establecida en el artículo 35.
3. La solicitud de fabricación o importación de medicamentos en investigación po-drá solicitarse en unidad de acto con la solicitud de realización del ensayo clínico al queestén destinados.
La fabricación o importación de medicamentos hemoderivados, estupefacientes opsicótropos se regirán por su normativa específica en la materia.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
171

Artículo 33. Etiquetado.
El etiquetado de los medicamentos en investigación deberá figurar al menos enlengua española oficial del Estado y adecuarse a lo establecido en el anexo 13 de las denormas de correcta fabricación de medicamentos en la Unión Europea.
CAPÍTULO VIII. Normas de Buena Práctica Clínica
Artículo 34. Normas de buena práctica clínica.
Todos los ensayos clínicos con medicamentos que se realicen en España deberán lle-varse a cabo de acuerdo con las normas de buena práctica clínica publicadas por el Ministe-rio de Sanidad y Consumo, siempre que no se opongan a lo dispuesto en este real decreto.
Artículo 35. Promotor.
1. El promotor o su representante legal habrá de estar establecido en uno de losEstados miembros de la Unión Europea.
2. Corresponde al promotor firmar las solicitudes de dictamen y autorización diri-gidas al Comité Ético de Investigación Clínica y a la Agencia Española de Medicamentosy Productos Sanitarios.
3. Son responsabilidades del promotor:
a) Establecer y mantener un sistema de garantías y control de calidad, con proce-dimientos normalizados de trabajo escritos, de forma que los ensayos sean realizados y losdatos generados, documentados y comunicados de acuerdo con el protocolo, las normasde buena práctica clínica y lo dispuesto en este real decreto.
b) Firmar, junto con el investigador que corresponda, el protocolo y cualquiera desus modificaciones.
c) Seleccionar al investigador más adecuado según su cualificación y medios dis-ponibles, y asegurarse de que éste llevará a cabo el estudio tal como está especificado enel protocolo.
d) Proporcionar la información básica y clínica disponible del producto en inves-tigación y actualizarla a lo largo del ensayo.
172

e) Solicitar el dictamen del Comité Ético de Investigación Clínica y la autorizaciónde la Agencia Española de Medicamentos y Productos Sanitarios, así como suministrarlesla información y recabar las autorizaciones que procedan, sin perjuicio de la comunica-ción a las comunidades autónomas, en caso de modificación o violación del protocolo ointerrupción del ensayo, y las razones para ello.
f) Suministrar de forma gratuita los medicamentos en investigación, garantizarque se han cumplido las normas de correcta fabricación y que las muestras están adecua-damente envasadas y etiquetadas. También es responsable de la conservación de mues-tras y sus protocolos de fabricación y control, del registro de las muestras entregadas y deasegurarse que en el centro donde se realiza el ensayo existirá un procedimiento correctode manejo, conservación y uso de dichas muestras.
Excepcionalmente, se podrán acordar con el centro otras vías de suministro.
g) Designar el monitor que vigilará la marcha del ensayo.
h) Comunicar a las autoridades sanitarias, a los investigadores y a los Comités Éti-cos de Investigación Clínica involucrados en el ensayo las sospechas de reacciones adver-sas graves e inesperadas de conformidad con lo establecido en los artículos 43 a 46.
i) Proporcionar al investigador y al Comité Ético de Investigación Clínica, de formainmediata, cualquier información de importancia a la que tenga acceso durante el ensayo.
j) Proporcionar compensación económica a los sujetos en caso de lesión o muerte re-lacionadas con el ensayo. Proporcionar al investigador cobertura legal y económica en estoscasos excepto cuando la lesión sea consecuencia de negligencia o mala práctica del inves-tigador.
k) Acordar con el investigador las obligaciones en cuanto al tratamiento de datos, ela-boración de informes y publicación de resultados. En cualquier caso, el promotor es responsa-ble de elaborar los informes finales o parciales del ensayo y comunicarlos a quien corresponda.
l) El promotor dispondrá de un punto de contacto, donde los sujetos del ensayopuedan obtener mayor información sobre éste, que podrá delegar en el investigador.
Artículo 36. Monitor.
Son responsabilidades del monitor:
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
173

a) Trabajar de acuerdo con los procedimientos normalizados de trabajo del promo-tor, visitar al investigador antes, durante y después del ensayo para comprobar el cumpli-miento del protocolo, garantizar que los datos son registrados de forma correcta ycompleta, así como asegurarse de que se ha obtenido el consentimiento informado de to-dos los sujetos antes de su inclusión en el ensayo.
b) Cerciorarse de que los investigadores y el centro donde se realizará la investi-gación son adecuados para este propósito durante el periodo de realización del ensayo.
c) Asegurarse de que tanto el investigador principal como sus colaboradores hansido informados adecuadamente y garantizar en todo momento una comunicación rápidaentre investigador y promotor.
d) Verificar que el investigador cumple el protocolo y todas sus modificacionesaprobadas.
e) Comprobar que el almacenamiento, distribución, devolución y documentaciónde los medicamentos en investigación es seguro y adecuado.
f) Remitir al promotor informes de las visitas de monitorización y de todos los con-tactos relevantes con el investigador.
Artículo 37. Investigador.
1. El investigador dirige y se responsabiliza de la realización práctica del ensayo clí-nico en un centro, y firma junto con el promotor la solicitud, corresponsabilizándose con él.
2. Solamente podrá actuar como investigador un médico o persona que ejerza unaprofesión reconocida en España para llevar a cabo las investigaciones en razón de su for-mación científica y de su experiencia en la atención sanitaria requerida.
3. Son responsabilidades del investigador:
a) Estar de acuerdo y firmar junto con el promotor el protocolo del ensayo.
b) Conocer a fondo las propiedades de los medicamentos en investigación.
c) Garantizar que el consentimiento informado se recoge de conformidad a lo es-tablecido en este real decreto.
174

d) Recoger, registrar y notificar los datos de forma correcta y garantizar su veracidad.
e) Notificar inmediatamente los acontecimientos adversos graves o inesperados alpromotor.
f) Garantizar que todas las personas implicadas respetarán la confidencialidad decualquier información acerca de los sujetos del ensayo, así como la protección de sus datosde carácter personal.
g) Informar regularmente al Comité Ético de Investigación Clínica de la marcha delensayo.
h) Corresponsabilizarse con el promotor de la elaboración del informe final del en-sayo, dando su acuerdo con su firma.
Artículo 38. Publicaciones.
1. El promotor está obligado a publicar los resultados, tanto positivos como negati-vos, de los ensayos clínicos autorizados en revistas científicas y con mención al Comité Éticode Investigación Clínica que aprobó el estudio.
2. Cuando se hagan públicos estudios y trabajos de investigación sobre medicamen-tos, dirigidos a la comunidad científica, se harán constar los fondos obtenidos por el autorpor o para su realización y la fuente de financiación.
3. Se mantendrá en todo momento el anonimato de los sujetos participantes en elensayo.
4. Los resultados o conclusiones de los ensayos clínicos se comunicarán preferen-temente en publicaciones científicas antes de ser divulgados al público no sanitario.
No se darán a conocer de modo prematuro o sensacionalista tratamientos de efica-cia todavía no determinada, ni se exagerará ésta.
5. La publicidad de medicamentos en investigación queda terminantemente prohi-bida, tal como se establece en la Ley 25/1990, de 20 de diciembre, del Medicamento, elReal Decreto 1416/1994, de 25 de junio, por el que se regula la publicidad de los medica-mentos, el Real Decreto 1907/1996, de 2 de agosto, sobre publicidad y promoción comer-cial de productos, actividades o servicios con pretendida finalidad sanitaria, y la Ley 34/1988,de 11 de noviembre, General de Publicidad.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
175

Artículo 39. Archivo de la documentación del ensayo clínico.
Los documentos que constituyen el archivo maestro de un ensayo clínico deberánconservarse durante el tiempo y conforme a las especificaciones establecidas en las Ins-trucciones para la realización de ensayos clínicos en España o, en su caso, las directricesde la Comisión Europea que publicará el Ministerio de sanidad y Consumo.
CAPÍTULO IX. Verificación del cumplimiento de las Normas de Buena Práctica Clínica
Artículo 40. Inspecciones.
1. La Agencia Española de Medicamentos y Productos Sanitarios y las autoridadessanitarias competentes de las comunidades autónomas, en el ámbito de sus respectivascompetencias, verificarán la aplicación de este real decreto, de las normas de buena prác-tica clínica y de las normas de correcta fabricación en los ensayos clínicos que se realicenen España, a través de las correspondientes inspecciones.
2. La Agencia Española de Medicamentos y Productos Sanitarios informará a laAgencia Europea de Evaluación de Medicamentos de las inspecciones efectuadas y de susresultados.
Asimismo, será responsable de la introducción de los datos relativos a inspeccionesen la base de datos europea de ensayos clínicos EUDRACT, de acuerdo con lo especificadoen las instrucciones para la realización de ensayos clínicos en España o, en su caso, las di-rectrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
3. Las inspecciones serán llevadas a cabo por inspectores debidamente cualifica-dos y designados para tal efecto en los lugares relacionados con la realización de los ensa-yos clínicos y, entre otros, en el centro o centros en los que se lleve a cabo el ensayo, el lugarde fabricación del medicamento en investigación, cualquier laboratorio de análisis utilizadoen el ensayo clínico y/o en las instalaciones del promotor.
4. Tras la inspección se elaborará un informe que se pondrá a disposición de los ins-peccionados. También se podrá poner a disposición del Comité Ético de Investigación Clí-nica implicado, de las autoridades competentes en España y demás Estados miembros dela Unión Europea y de la Agencia Europea de Evaluación de Medicamentos.
5. Corresponde a la Agencia Española de Medicamentos y Productos Sanitarios re-cabar la colaboración de las autoridades competentes de otros Estados miembros de la
176

Unión Europea y terceros Estados para la inspección del centro del ensayo, instalaciones delpromotor o instalaciones del fabricante del medicamento en investigación establecido fueradel territorio nacional.
CAPÍTULO X. Comunicaciones
Artículo 41. Bases de datos.
1. La Agencia Española de Medicamentos y Productos Sanitarios se responsabi-lizará de la inclusión en la base de datos europea de ensayos clínicos EUDRACT de los da-tos relativos a los ensayos clínicos que se lleven a cabo en el territorio nacional, de acuerdocon lo establecido en las instrucciones para la realización de ensayos clínicos en Españao, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sa-nidad y Consumo.
2. La Agencia Española de Medicamentos y Productos Sanitarios se responsabi-lizará de mantener actualizada su base de datos de los ensayos clínicos autorizados quese lleven a cabo en el territorio nacional.
Las autoridades competentes de las comunidades autónomas tendrán acceso alos datos relativos a los ensayos que se realicen en su ámbito territorial.
3. La Agencia Española de Medicamentos y Productos Sanitarios pondrá a dispo-sición de los ciudadanos a través de su página web información referente al título del en-sayo, promotor, centros implicados, patología y población en estudio de los ensayosclínicos autorizados.
Se considerará que no existe oposición, por parte del promotor del ensayo, a la pu-blicación de los datos antes indicados de los ensayos promovidos por él, siempre que nose haga indicación expresa en su contra en la solicitud de autorización del ensayo clínicodirigida a la Agencia Española de Medicamentos y Productos Sanitarios.
CAPÍTULO XI. De la vigilancia de la seguridad de los medicamentos en investigación
Artículo 42. Obligaciones de los investigadores en el registro y comunicación de aconte-cimientos adversos.
1. El investigador comunicará inmediatamente al promotor todos los aconte-cimientos adversos graves, salvo cuando se trate de los señalados en el protocolo o
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
177

en el manual del investigador como acontecimientos que no requieran comunica-ción inmediata. La comunicación inicial irá seguida de comunicaciones escritas por-menorizadas. En las comunicaciones iniciales y en las de seguimiento se identificaráa los sujetos del ensayo mediante un número de código específico para cada uno deellos.
2. Los acontecimientos adversos y/o los resultados de laboratorio anómalos ca-lificados en el protocolo como determinantes para las evaluaciones de seguridad se co-municarán al promotor con arreglo a los requisitos de comunicación y dentro de losperiodos especificados en el protocolo.
3. En caso de que se haya comunicado un fallecimiento de un sujeto partici-pante en un ensayo clínico, el investigador proporcionará al promotor y a los ComitésÉticos de Investigación Clínica implicados toda la información complementaria que lesoliciten.
Artículo 43. Obligaciones del promotor en el registro, evaluación y comunicación deacontecimientos adversos.
1. El promotor mantendrá unos registros detallados de todos los acontecimien-tos adversos que le sean comunicados por los investigadores. Estos registros se pre-sentarán a la Agencia Española de Medicamentos y Productos Sanitarios cuando éstaasí lo solicite.
2. El promotor tiene la obligación de evaluar de forma continua la seguridadde los medicamentos en investigación utilizando toda la información a su alcance.
Asimismo, debe comunicar sin tardanza a la Agencia Española de Medica-mentos y Productos Sanitarios, a los órganos competentes de las comunidades au-tónomas donde se realice el ensayo clínico y a los Comités Éticos de InvestigaciónClínica implicados cualquier información importante que afecte a la seguridad delmedicamento en investigación. Dicha comunicación se realizará según los criteriosque se especifican en los artículos siguientes y de acuerdo con los procedimientosestablecidos en las instrucciones para la realización de ensayos clínicos en Españao, en su caso, directrices de la Comisión Europea, que publicará el Ministerio de Sa-nidad y Consumo.
3. La comunicación de información de seguridad del promotor a los investiga-dores seguirá lo especificado en las normas de buena práctica clínica.
178

Artículo 44. Notificación expeditiva de casos individuales de sospecha de reacción ad-versa a la Agencia Española de Medicamentos y Productos Sanitarios.
1. El promotor notificará a la Agencia Española de Medicamentos y Productos Sa-nitarios todas las sospechas de reacciones adversas graves y a la vez inesperadas asocia-das a los medicamentos en investigación, tanto si ocurren en España como en otrosEstados, y tanto si han ocurrido en el ensayo clínico autorizado como en otros ensayos clí-nicos o en un contexto de uso diferente, siempre que dichos medicamentos no se encuen-tren comercializados en España. Para los productos comercializados, incluyendo elmedicamento utilizado como control o los medicamentos utilizados como concomitantes,la Agencia Española de Medicamentos y Productos Sanitarios emitirá unas directrices es-pecíficas que tendrán en cuenta los requerimientos de farmacovigilancia al objeto de evi-tar posibles duplicaciones en la notificación.
2. El plazo máximo de notificación será de 15 días naturales a partir del momentoen que el promotor haya tenido conocimiento de la sospecha de reacción adversa.
Cuando la sospecha de reacción adversa grave e inesperada haya ocasionado lamuerte del sujeto, o puesto en peligro su vida, el promotor informará a la Agencia Espa-ñola de Medicamentos y Productos Sanitarios en el plazo máximo de siete días naturalesa partir del momento en que el promotor tenga conocimiento del caso. Dicha informacióndeberá ser completada, en lo posible, en los ocho días siguientes.
3. Cuando las sospechas de reacciones adversas graves e inesperadas ocurran enun ensayo clínico doble ciego, se deberá desvelar el código de tratamiento de ese pacienteconcreto a efectos de notificación. Siempre que sea posible, se mantendrá el carácterciego para el investigador, y para las personas encargadas del análisis e interpretación delos resultados, así como de la elaboración de las conclusiones del estudio. En aquellos ca-sos en que se considere que este sistema de notificación pueda interferir con la validezdel estudio, podrá acordarse con la Agencia Española de Medicamentos y Productos Sa-nitarios un sistema de notificación específico.
4. Las sospechas de reacciones adversas atribuibles a placebo no estarán sujetasa este sistema de notificación individualizada.
5. Las notificaciones se realizarán preferiblemente utilizando el formato electró-nico estándar europeo.
Cuando esto no sea posible, debido a un motivo justificado, se utilizará el formu-lario de notificación en papel para las notificaciones de sospechas de reacción adversa que
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
179

ocurran en España. Para las notificaciones de sospechas de reacción adversa que ocurranfuera de España, podrá utilizarse un formulario estándar internacional. Las notificacionesque ocurran en España, con independencia del formato utilizado, tendrán que ser comu-nicadas en la lengua española oficial del Estado.
6. La Agencia Española de Medicamentos y Productos Sanitarios mantendrá unared de proceso de datos para registrar todas las sospechas de reacciones adversas gravesinesperadas de un medicamento en investigación de las cuales tenga conocimiento. Di-cha red permitirá, además de la recepción de comunicaciones electrónicas, el acceso entiempo real de las comunidades autónomas y la comunicación electrónica a la Agencia Eu-ropea de Evaluación de Medicamentos de las sospechas de reacciones adversas gravesinesperadas que hayan ocurrido en España.
Artículo 45. Notificación expeditiva de casos individuales de sospecha de reacción ad-versa a los órganos competentes de las comunidades autónomas.
1. El promotor notificará a los órganos competentes de las comunidades autóno-mas en cuyo territorio se esté realizando el ensayo, de forma individual y en el plazo má-ximo de 15 días, todas las sospechas de reacciones adversas que sean a la vez graves einesperadas asociadas al medicamento en investigación y que hayan ocurrido en pacien-tes seleccionados en sus respectivos ámbitos territoriales. Este plazo máximo será de sietedías cuando se trate de sospechas de reacciones adversas que produzcan la muerte o ame-nacen la vida. Para los medicamentos comercializados, incluyendo el utilizado como con-trol o los utilizados como concomitantes, la Agencia Española de Medicamentos yProductos Sanitarios emitirá unas directrices específicas que tendrán en cuenta los reque-rimientos de farmacovigilancia al objeto de evitar posibles duplicaciones en la notificación.
2. El promotor notificará cualquier otra información sobre reacciones adversasgraves e inesperadas asociadas al medicamento en investigación cuando así lo disponganlas normativas específicas de las comunidades autónomas y, en cualquier caso, si la in-formación supone un cambio importante en el perfil de seguridad del producto investigado.
3. Cuando el promotor realice la comunicación en formato electrónico no será pre-cisa la notificación a las comunidades autónomas, dado que dicha información les será ac-cesible en tiempo real a través de la red de proceso de datos.
Artículo 46. Notificación expeditiva de casos individuales de sospecha de reacción ad-versa a los Comités Éticos de Investigación Clínica.
1. El promotor notificará a los comités éticos implicados, de forma individual y
180

en el plazo máximo de 15 días, todas las sospechas de reacciones adversas que sean ala vez graves e inesperadas asociadas al medicamento en investigación y que hayan ocu-rrido en pacientes seleccionados en sus respectivos ámbitos. El plazo máximo será desiete días cuando se trate de sospechas de reacciones adversas que produzcan la muerteo amenacen la vida.
2. El promotor notificará cualquier otra información sobre reacciones adversasgraves e inesperadas asociadas al medicamento de investigación cuando así lo dispon-gan los comités éticos implicados en el momento del dictamen favorable del estudio y,en cualquier caso, si la información supone un cambio importante en el perfil de segu-ridad del producto investigado. Los comités éticos implicados podrán establecer queesta información adicional le sea suministrada periódicamente de forma resumida. LaAgencia Española de Medicamentos y Productos Sanitarios, en consonancia con las dis-posiciones europeas, publicará las correspondientes directrices para orientar a los pro-motores.
Artículo 47. Informes periódicos de seguridad.
1. Adicionalmente a la notificación expeditiva, los promotores de ensayos clíni-cos prepararán un informe periódico en el que se evalúe la seguridad del medicamentoen investigación teniendo en cuenta toda la información disponible.
2. El informe periódico de seguridad se presentará a la Agencia Española deMedicamentos y Productos Sanitarios, a los órganos competentes de las comunidadesautónomas correspondientes y a los Comités Éticos de Investigación Clínica implicados,anualmente hasta el final del ensayo y siempre que lo soliciten las autoridades sanita-rias o los comités éticos implicados.
3. La Agencia Española de Medicamentos y Productos Sanitarios determinará elformato del informe periódico de seguridad teniendo en cuenta la normativa europea alrespecto. El informe periódico de seguridad no sustituirá a la solicitud de modificacio-nes a los documentos del ensayo, que seguirá su procedimiento específico.
4. Sin perjuicio de la periodicidad señalada para los informes de seguridad, elpromotor preparará un informe de evaluación ad hoc siempre que exista un problema deseguridad relevante. Dicho informe se presentará sin tardanza a la Agencia Española deMedicamentos y Productos Sanitarios, a los órganos competentes de las comunidadesautónomas y a los Comités Éticos de Investigación Clínica concernidos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
181

5. El informe periódico de seguridad podrá ser una parte del informe anual y fi-nal correspondientes o bien ser preparado de forma independiente.
CAPÍTULO XII. Infracciones
Artículo 48. Infracciones administrativas en materia de ensayos clínicos.
Constituirán infracciones administrativas las previstas en el artículo 108 de la Ley25/1990, de 20 de diciembre, del Medicamento, y serán sancionadas de acuerdo con elartículo 109 de la misma ley.
DISPOSICIÓN ADICIONAL ÚNICA. Ensayos clínicos con productos sanitarios.
Los ensayos clínicos con productos sanitarios se regirán por los principios recogi-dos en este real decreto en lo que les resulte de aplicación.
DISPOSICIÓN TRANSITORIA PRIMERA. Régimen transitorio.
Los procedimientos iniciados con anterioridad a la entrada en vigor de estereal decreto se regirán por lo establecido en el Real Decreto 561/1993, de 16 de abril,por el que se establecen los requisitos para la realización de ensayos clínicos con me-dicamentos.
DISPOSICIÓN TRANSITORIA SEGUNDA. Aplicación de este real decreto al ámbito de losservicios sanitarios de las Fuerzas Armadas.
En tanto no se desarrollen las previsiones contenidas en la disposición adicionalsegunda de la Ley 25/1990, de 20 de diciembre, del Medicamento, en la aplicación deeste real decreto al ámbito de los servicios sanitarios de las Fuerzas Armadas se observa-rán las siguientes normas:
a) Corresponderá al Ministerio de Sanidad y Consumo la acreditación de los Co-mités Éticos de Investigación Clínica.
b) El Ministerio de Defensa, a través de la Inspección General de Sanidad dela Defensa, ejercerá las competencias en materia de inspección, recepción de comu-nicaciones y notificaciones y las demás que este real decreto atribuye a las comuni-dades autónomas.
182

DISPOSICIÓN DEROGATORIA ÚNICA. Derogación normativa.
Queda derogado el Real Decreto 561/1993, de 16 de abril, por el que se estable-cen los requisitos para la realización de ensayos clínicos con medicamentos, sin perjuiciode lo dispuesto en la disposición transitoria primera, así como cualquier otra disposiciónde igual o inferior rango que se oponga a lo dispuesto en este real decreto.
DISPOSICIÓN FINAL PRIMERA. Título habilitante.
Este real decreto tiene carácter de legislación sobre productos farmacéuticos alos efectos previstos en el artículo 149.1.16..ª de la Constitución Española, y se adoptaen desarrollo del título III de la Ley 25/1990, de 20 de diciembre, del Medicamento.
DISPOSICIÓN FINAL SEGUNDA. Facultad de desarrollo.
Se faculta al Ministro de Sanidad y Consumo para dictar las disposiciones nece-sarias para el desarrollo de este real decreto, conforme al avance de los conocimientoscientíficos y técnicos y de acuerdo con las orientaciones de la Unión Europea.
Asimismo, se faculta al Ministro de Sanidad y Consumo para la adopción de lasnormas de buena práctica clínica y las instrucciones para la realización de ensayos clíni-cos en España, de acuerdo con las directrices que adopte la Comisión Europea.
DISPOSICIÓN FINAL TERCERA. Entrada en vigor.
El presente real decreto entrará en vigor el día 1 de mayo de 2004.
Dado en Madrid, a 6 de febrero de 2004.
JUAN CARLOS R.
La Ministra de Sanidad y Consumo, ANA MARÍA PASTOR JULIÁN
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
183


LEY 41/2002, DE 14 DE NOVIEMBRE, BÁSICA REGULADORA DE LA AUTONOMÍA DEL PACIENTE Y DE DERECHOS Y OBLIGACIONES EN MATERIA DE INFORMACIÓN Y DOCUMENTACIÓN CLÍNICA
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
185


LEY 41/2002, DE 14 DE NOVIEMBRE, BÁSICA REGULADORA DE LA AUTONOMÍA DEL PACIENTE Y DE DERECHOS Y OBLIGACIONES EN MATERIA DE INFORMACIÓN Y DOCUMENTACIÓN CLÍNICA
Don Juan Carlos I,Rey de España
A todos los que la presente vieren y entendieren.
Sabed: Que las Cortes Generales han aprobado y Yo vengo en sancionar la si-guiente Ley.
EXPOSICIÓN DE MOTIVOS
La importancia que tienen los derechos de los pacientes como eje básico de las re-laciones clínico-asistenciales se pone de manifiesto al constatar el interés que han demos-trado por los mismos casi todas las organizaciones internacionales con competencia en lamateria. Ya desde el fin de la Segunda Guerra Mundial, organizaciones como Naciones Uni-das, UNESCO o la Organización Mundial de la Salud, o, más recientemente, la Unión Eu-ropea o el Consejo de Europa, entre muchas otras, han impulsado declaraciones o, en algúncaso, han promulgado normas jurídicas sobre aspectos genéricos o específicos relaciona-dos con esta cuestión. En este sentido, es necesario mencionar la trascendencia de la De-claración universal de derechos humanos, del año 1948, que ha sido el punto de referenciaobligado para todos los textos constitucionales promulgados posteriormente o, en el ámbitomás estrictamente sanitario, la Declaración sobre la promoción de los derechos de los pa-cientes en Europa, promovida el año 1994 por la Oficina Regional para Europa de la Orga-nización Mundial de la Salud, aparte de múltiples declaraciones internacionales de mayoro menor alcance e influencia que se han referido a dichas cuestiones.
Últimamente, cabe subrayar la relevancia especial del Convenio del Consejo deEuropa para la protección de los derechos humanos y la dignidad del ser humano res-pecto de las aplicaciones de la biología y la medicina (Convenio sobre los derechos del
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
187

hombre y la biomedicina), suscrito el día 4 de abril de 1997, el cual ha entrado en vigoren el Reino de España el 1 de enero de 2000. Dicho Convenio es una iniciativa capital:en efecto, a diferencia de las distintas declaraciones internacionales que lo han precedido,es el primer instrumento internacional con carácter jurídico vinculante para los países quelo suscriben. Su especial valía reside en el hecho de que establece un marco común parala protección de los derechos humanos y la dignidad humana en la aplicación de la bio-logía y la medicina. El Convenio trata explícitamente, con detenimiento y extensión, so-bre la necesidad de reconocer los derechos de los pacientes, entre los cuales resaltan elderecho a la información, el consentimiento informado y la intimidad de la informaciónrelativa a la salud de las personas, persiguiendo el alcance de una armonización de las le-gislaciones de los diversos países en estas materias, en este sentido, es absolutamenteconveniente tener en cuenta el Convenio en el momento de abordar el reto de regularcuestiones tan importantes.
Es preciso decir, sin embargo, que la regulación del derecho a la protección de lasalud, recogido por el artículo 43 de la Constitución de 1978, desde el punto de vista delas cuestiones más estrechamente vinculadas a la condición de sujetos de derechos de laspersonas usuarias de los servicios sanitarios, es decir, la plasmación de los derechos re-lativos a la información clínica y la autonomía individual de los pacientes en lo relativo asu salud, ha sido objeto de una regulación básica en el ámbito del Estado, a través de laLey 14/1986, de 25 de abril, General de Sanidad.
De otra parte, esta Ley, a pesar de que fija básicamente su atención en el esta-blecimiento y ordenación del sistema sanitario desde un punto de vista organizativo, de-dica a esta cuestión diversas previsiones, entre las que destaca la voluntad dehumanización de los servicios sanitarios. Así mantiene el máximo respeto a la dignidad dela persona y a la libertad individual, de un lado, y, del otro, declara que la organizaciónsanitaria debe permitir garantizar la salud como derecho inalienable de la población me-diante la estructura del Sistema Nacional de Salud, que debe asegurarse en condicionesde escrupuloso respeto a la intimidad personal y a la libertad individual del usuario, ga-rantizando la confidencialidad de la información relacionada con los servicios sanitariosque se prestan y sin ningún tipo de discriminación.
A partir de dichas premisas, la presente Ley completa las previsiones que la LeyGeneral de Sanidad enunció como principios generales. En este sentido, refuerza y de untrato especial al derecho a la autonomía del paciente. En particular, merece mención es-pecial la regulación sobre instrucciones previas que contempla, de acuerdo con el crite-rio establecido en el Convenio de Oviedo, los deseos del paciente expresados conanterioridad dentro del ámbito del consentimiento informado. Asimismo, la Ley trata con
188

profundidad todo lo referente a la documentación clínica generada en los centros asisten-ciales, subrayando especialmente la consideración y la concreción de los derechos de losusuarios en este aspecto.
En septiembre de 1997, en desarrollo de un convenio de colaboración entre elConsejo General del Poder Judicial y el Ministerio de Sanidad y Consumo, tuvo lugar unseminario conjunto sobre información y documentación clínica, en el que se debatieronlos principales aspectos normativos y judiciales en la materia. Al mismo tiempo, se cons-tituyó un grupo de expertos a quienes se encargó la elaboración de unas directrices parael desarrollo futuro de este tema. Este grupo suscribió un dictamen el 26 de noviembrede 1997, que ha sido tenido en cuenta en la elaboración de los principios fundamenta-les de esta Ley.
La atención que a estas materias otorgó en su día la Ley General de Sanidadsupuso un notable avance como reflejan, entre otros, sus artículos 9, 10 y 61. Sinembargo, el derecho a la información, como derecho del ciudadano cuando demandala atención sanitaria, ha sido objeto en los últimos años de diversas matizaciones y am-pliaciones por Leyes y disposiciones de distinto tipo y rango, que ponen de manifiestola necesidad de una reforma y actualización de la normativa contenida en la Ley Ge-neral de Sanidad. Así, la Ley Orgánica 15/1999, de 13 de diciembre, de Protecciónde Datos de Carácter Personal, califica a los datos relativos a la salud de los ciudada-nos como datos especialmente protegidos, estableciendo un régimen singularmente ri-guroso para su obtención, custodia y eventual cesión. Esta defensa de laconfidencialidad había sido ya defendida por la Directiva comunitaria 95/46, de 24 deoctubre, en la que, además de reafirmarse la defensa de los derechos y libertades delos ciudadanos europeos, en especial de su intimidad relativa a la información relacio-nada con su salud, se apunta la presencia de otros intereses generales como los estu-dios epidemiológicos, las situaciones de riesgo grave para la salud de la colectividad,la investigación y los ensayos clínicos que, cuando estén incluidos en normas de rangode Ley, pueden justificar una excepción motivada a los derechos del paciente. Se ma-nifiesta así una concepción comunitaria del derecho a la salud, en la que, junto al in-terés singular de cada individuo, como destinatario por excelencia de la informaciónrelativa a la salud, aparecen también otros agentes y bienes jurídicos referidos a la sa-lud pública, que deben ser considerados, con la relevancia necesaria, en una sociedaddemocrática avanzada. En esta línea, el Consejo de Europa, en su Recomendación de13 de febrero de 1997, relativa a la protección de los datos médicos, después de afir-mar que deben recogerse y procesarse con el consentimiento del afectado, indica quela información puede restringirse si así lo dispone una Ley y constituye una medida ne-cesaria por razones de interés general.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
189

Todas estas circunstancias aconsejan una adaptación de la Ley General de Sani-dad con el objetivo de aclarar la situación jurídica y los derechos y obligaciones de los pro-fesionales sanitarios, de los ciudadanos y de las instituciones sanitarias. Se trata de ofreceren el terreno de la información y la documentación clínicas las mismas garantías a todoslos ciudadanos del Estado, fortaleciendo con ello el derecho a la protección de la saludque reconoce la Constitución.
CAPÍTULO I. Principios Generales.
Artículo 1. Ámbito de aplicación.
La presente Ley tiene por objeto la regulación de los derechos y obligaciones delos pacientes, usuarios y profesionales, así como de los centros y servicios sanitarios, pú-blicos y privados, en materia de autonomía del paciente y de información y documenta-ción clínica.
Artículo 2. Principios básicos.
1. La dignidad de la persona humana, el respeto a la autonomía de su voluntad ya su intimidad orientarán toda la actividad encaminada a obtener, utilizar, archivar, cus-todiar y transmitir la información y la documentación clínica.
2. Toda actuación en el ámbito de la sanidad requiere, con carácter general, el pre-vio consentimiento de los pacientes o usuarios. El consentimiento, que debe obtenersedespués de que el paciente reciba una información adecuada, se hará por escrito en lossupuestos previstos en la Ley.
3. El paciente o usuario tiene derecho a decidir libremente, después de recibir lainformación adecuada, entre las opciones clínicas disponibles.
4. Todo paciente o usuario tiene derecho a negarse al tratamiento, excepto en loscasos determinados en la Ley. Su negativa al tratamiento constará por escrito.
5. Los pacientes o usuarios tienen el deber de facilitar los datos sobre su estadofísico o sobre su salud de manera leal y verdadera, así como el de colaborar en su obten-ción, especialmente cuando sean necesarios por razones de interés público o con motivode la asistencia sanitaria.
190

6. Todo profesional que interviene en la actividad asistencial está obligado no sóloa la correcta prestación de sus técnicas, sino al cumplimiento de los deberes de informa-ción y de documentación clínica, y al respeto de las decisiones adoptadas libre y volunta-riamente por el paciente.
7. La persona que elabore o tenga acceso a la información y la documentación clí-nica está obligada a guardar la reserva debida.
Artículo 3. Las definiciones legales.
A efectos de esta Ley se entiende por:
• Centro sanitario: el conjunto organizado de profesionales, instalaciones y mediostécnicos que realiza actividades y presta servicios para cuidar la salud de los pacientes yusuarios.
• Certificado médico: la declaración escrita de un médico que de fe del estado desalud de una persona en un determinado momento.
• Consentimiento informado: la conformidad libre, voluntaria y consciente de unpaciente, manifestada en el pleno uso de sus facultades después de recibir la informaciónadecuada, para que tenga lugar una actuación que afecta a su salud.
• Documentación clínica: el soporte de cualquier tipo o clase que contiene unconjunto de datos e informaciones de carácter asistencial.
• Historia clínica: el conjunto de documentos que contienen los datos, valoracio-nes e informaciones de cualquier índole sobre la situación y la evolución clínica de un pa-ciente a lo largo del proceso asistencial.
• Información clínica: todo dato, cualquiera que sea su forma, clase o tipo, quepermite adquirir o ampliar conocimientos sobre el estado físico y la salud de una persona,o la forma de preservarla, cuidarla, mejorarla o recuperarla.
• Informe de alta médica: el documento emitido por el médico responsable en uncentro sanitario al finalizar cada proceso asistencial de un paciente, que especifica losdatos de éste, un resumen de su historial clínico, la actividad asistencial prestada, el diag-nóstico y las recomendaciones terapéuticas.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
191

• Intervención en el ámbito de la sanidad: toda actuación realizada con fines pre-ventivos, diagnósticos, terapéuticos, rehabilitadores o de investigación.
• Libre elección: la facultad del paciente o usuario de optar, libre y voluntaria-mente, entre dos o más alternativas asistenciales, entre varios facultativos o entre centrosasistenciales, en los términos y condiciones que establezcan los servicios de salud com-petentes, en cada caso.
• Médico responsable: el profesional que tiene a su cargo coordinar la informacióny la asistencia sanitaria del paciente o del usuario, con el carácter de interlocutor princi-pal del mismo en todo lo referente a su atención e información durante el proceso asis-tencial, sin perjuicio de las obligaciones de otros profesionales que participan en lasactuaciones asistenciales.
• Paciente: la persona que requiere asistencia sanitaria y está sometida a cuida-dos profesionales para el mantenimiento o recuperación de su salud.
• Servicio sanitario: la unidad asistencial con organización propia, dotada de losrecursos técnicos y del personal cualificado para llevar a cabo actividades sanitarias.
• Usuario: la persona que utiliza los servicios sanitarios de educación y promociónde la salud, de prevención de enfermedades y de información sanitaria.
CAPÍTULO II. El derecho de información sanitaria.
Artículo 4. Derecho a la información asistencial.
1. Los pacientes tienen derecho a conocer, con motivo de cualquier actuaciónen el ámbito de su salud, toda la información disponible sobre la misma, salvando lossupuestos exceptuados por la Ley. Además, toda persona tiene derecho a que se res-pete su voluntad de no ser informada. La información, que como regla general se pro-porcionará verbalmente dejando constancia en la historia clínica, comprende, comomínimo, la finalidad y la naturaleza de cada intervención, sus riesgos y sus conse-cuencias.
2. La información clínica forma parte de todas las actuaciones asistenciales, seráverdadera, se comunicará al paciente de forma comprensible y adecuada a sus necesida-des y le ayudará a tomar decisiones de acuerdo con su propia y libre voluntad.
192

3. El médico responsable del paciente le garantiza el cumplimiento de su derechoa la información. Los profesionales que le atiendan durante el proceso asistencial o le apli-quen una técnica o un procedimiento concreto también serán responsables de informarle.
Artículo 5. Titular del derecho a la información asistencial.
1. El titular del derecho a la información es el paciente. También serán informa-das las personas vinculadas a él, por razones familiares o de hecho, en la medida que elpaciente lo permita de manera expresa o tácita.
2. El paciente será informado, incluso en caso de incapacidad, de modo adecuadoa sus posibilidades de comprensión, cumpliendo con el deber de informar también a su re-presentante legal.
3. Cuando el paciente, según el criterio del médico que le asiste, carezca de capa-cidad para entender la información a causa de su estado físico o psíquico, la informaciónse pondrá en conocimiento de las personas vinculadas a él por razones familiares o de he-cho.
4. El derecho a la información sanitaria de los pacientes puede limitarse por laexistencia acreditada de un estado de necesidad terapéutica. Se entenderá por necesidadterapéutica la facultad del médico para actuar profesionalmente sin informar antes al pa-ciente, cuando por razones objetivas el conocimiento de su propia situación pueda perju-dicar su salud de manera grave. Llegado este caso, el médico dejará constancia razonadade las circunstancias en la historia clínica y comunicará su decisión a las personas vincu-ladas al paciente por razones familiares o de hecho.
Artículo 6. Derecho a la información epidemiológica.
Los ciudadanos tienen derecho a conocer los problemas sanitarios de la colecti-vidad cuando impliquen un riesgo para la salud pública o para su salud individual, y el de-recho a que esta información se difunda en términos verdaderos, comprensibles yadecuados para la protección de la salud, de acuerdo con lo establecido por la Ley.
CAPÍTULO III. Derecho a la intimidad.
Artículo 7. El derecho a la intimidad.
1. Toda persona tiene derecho a que se respete el carácter confidencial de los da-tos referentes a su salud, y a que nadie pueda acceder a ellos sin previa autorización am-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
193

parada por la Ley.2. Los centros sanitarios adoptarán las medidas oportunas para garantizar los de-
rechos a que se refiere el apartado anterior, y elaborarán, cuando proceda, las normas y losprocedimientos protocolizados que garanticen el acceso legal a los datos de los pacientes.
CAPÍTULO IV. El respeto de la autonomía del paciente.
Artículo 8. Consentimiento informado.
1. Toda actuación en el ámbito de la salud de un paciente necesita el consenti-miento libre y voluntario del afectado, una vez que, recibida la información prevista en elartículo 4, haya valorado las opciones propias del caso.
2. El consentimiento será verbal por regla general. Sin embargo, se prestarápor escrito en los casos siguientes: intervención quirúrgica, procedimientos diagnós-ticos y terapéuticos invasores y, en general, aplicación de procedimientos que supo-nen riesgos o inconvenientes de notoria y previsible repercusión negativa sobre lasalud del paciente.
3. El consentimiento escrito del paciente será necesario para cada una de las ac-tuaciones especificadas en el punto anterior de este artículo, dejando a salvo la posibili-dad de incorporar anejos y otros datos de carácter general, y tendrá información suficientesobre el procedimiento de aplicación y sobre sus riesgos.
4. Todo paciente o usuario tiene derecho a ser advertido sobre la posibilidad deutilizar los procedimientos de pronóstico, diagnóstico y terapéuticos que se le apliquen enun proyecto docente o de investigación, que en ningún caso podrá comportar riesgo adi-cional para su salud.
5. El paciente puede revocar libremente por escrito su consentimiento en cual-quier momento.
Artículo 9. Límites del consentimiento informado y consentimiento por representación.
1. La renuncia del paciente a recibir información está limitada por el interés dela salud del propio paciente, de terceros, de la colectividad y por las exigencias terapéu-ticas del caso. Cuando el paciente manifieste expresamente su deseo de no ser informado,se respetará su voluntad haciendo constar su renuncia documentalmente, sin perjuicio de
194

la obtención de su consentimiento previo para la intervención.2. Los facultativos podrán llevar a cabo las intervenciones clínicas indispensables
en favor de la salud del paciente, sin necesidad de contar con su consentimiento, en lossiguientes casos:
Cuando existe riesgo para la salud pública a causa de razones sanitarias estableci-das por la Ley. En todo caso, una vez adoptadas las medidas pertinentes, de conformidadcon lo establecido en la Ley Orgánica 3/1986, se comunicarán a la autoridad judicial en elplazo máximo de 24 horas siempre que dispongan el internamiento obligatorio de personas.
Cuando existe riesgo inmediato grave para la integridad física o psíquica del en-fermo y no es posible conseguir su autorización, consultando, cuando las circunstanciaslo permitan, a sus familiares o a las personas vinculadas de hecho a él.
3. Se otorgará el consentimiento por representación en los siguientes supuestos:
Cuando el paciente no sea capaz de tomar decisiones, a criterio del médico res-ponsable de la asistencia, o su estado físico o psíquico no le permita hacerse cargo de susituación. Si el paciente carece de representante legal, el consentimiento lo prestarán laspersonas vinculadas a él por razones familiares o de hecho.
Cuando el paciente esté incapacitado legalmente.
Cuando el paciente menor de edad no sea capaz intelectual ni emocionalmentede comprender el alcance de la intervención. En este caso, el consentimiento lo dará elrepresentante legal del menor después de haber escuchado su opinión si tiene doce añoscumplidos. Cuando se trate de menores no incapaces ni incapacitados, pero emancipadoso con dieciséis años cumplidos, no cabe prestar el consentimiento por representación.Sin embargo, en caso de actuación de grave riesgo, según el criterio del facultativo, lospadres serán informados y su opinión será tenida en cuenta para la toma de la decisióncorrespondiente.
4. La interrupción voluntaria del embarazo, la práctica de ensayos clínicos y lapráctica de técnicas de reproducción humana asistida se rigen por lo establecido con ca-rácter general sobre la mayoría de edad y por las disposiciones especiales de aplicación.
5. La prestación del consentimiento por representación será adecuada a las cir-cunstancias y proporcionada a las necesidades que haya que atender, siempre en favor delpaciente y con respeto a su dignidad personal. El paciente participará en la medida de lo
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
195

posible en la toma de decisiones a lo largo del proceso sanitario.Artículo 10. Condiciones de la información y consentimiento por escrito.
1. El facultativo proporcionará al paciente, antes de recabar su consentimiento es-crito, la información básica siguiente:
a. Las consecuencias relevantes o de importancia que la intervención origina conseguridad.
b. Los riesgos relacionados con las circunstancias personales o profesionales delpaciente.
c. Los riesgos probables en condiciones normales, conforme a la experiencia y alestado de la ciencia o directamente relacionados con el tipo de intervención.
d. Las contraindicaciones.
2. El médico responsable deberá ponderar en cada caso que cuanto más dudososea el resultado de una intervención más necesario resulta el previo consentimiento porescrito del paciente.
Artículo 11. Instrucciones previas.
1. Por el documento de instrucciones previas, una persona mayor de edad, capazy libre, manifiesta anticipadamente su voluntad, con objeto de que ésta se cumpla en elmomento en que llegue a situaciones en cuyas circunstancias no sea capaz de expresar-los personalmente, sobre los cuidados y el tratamiento de su salud o, una vez llegado elfallecimiento, sobre el destino de su cuerpo o de los órganos del mismo. El otorgante deldocumento puede designar, además, un representante para que, llegado el caso, sirvacomo interlocutor suyo con el médico o el equipo sanitario para procurar el cumplimientode las instrucciones previas.
2. Cada servicio de salud regulará el procedimiento adecuado para que, llegadoel caso, se garantice el cumplimiento de las instrucciones previas de cada persona, quedeberán constar siempre por escrito.
3. No serán aplicadas las instrucciones previas contrarias al ordenamiento jurídico,a la lex artis, ni las que no se correspondan con el supuesto de hecho que el interesadohaya previsto en el momento de manifestarlas. En la historia clínica del paciente quedará
196

constancia razonada de las anotaciones relacionadas con estas previsiones.4. Las instrucciones previas podrán revocarse libremente en cualquier momento
dejando constancia por escrito.
5. Con el fin de asegurar la eficacia en todo el territorio nacional de las instruc-ciones previas manifestadas por los pacientes y formalizadas de acuerdo con lo dispuestoen la legislación de las respectivas Comunidades Autónomas, se creará en el Ministerio deSanidad y Consumo el Registro Nacional de instrucciones previas que se regirá por las nor-mas que reglamentariamente se determinen, previo acuerdo del Consejo Interterritorialdel Sistema Nacional de Salud.
Artículo 12. Información en el Sistema Nacional de Salud.
1. Además de los derechos reconocidos en los artículos anteriores, los pacientes ylos usuarios del Sistema Nacional de Salud tendrán derecho a recibir información sobre losservicios y unidades asistenciales disponibles, su calidad y los requisitos de acceso a ellos.
2. Los servicios de salud dispondrán en los centros y servicios sanitarios de unaguía o carta de los servicios en la que se especifiquen los derechos y obligaciones de losusuarios, las prestaciones disponibles, las características asistenciales del centro o del ser-vicio, y sus dotaciones de personal, instalaciones y medios técnicos. Se facilitará a todos losusuarios información sobre las guías de participación y sobre sugerencias y reclamaciones.
3. Cada servicio de salud regulará los procedimientos y los sistemas para garan-tizar el efectivo cumplimiento de las previsiones de este artículo.
Artículo 13. Derecho a la información para la elección de médico y de centro.
Los usuarios y pacientes del Sistema Nacional de Salud, tanto en la atención pri-maria como en la especializada, tendrán derecho a la información previa correspondientepara elegir médico, e igualmente centro, con arreglo a los términos y condiciones que es-tablezcan los servicios de salud competentes.
CAPÍTULO V. La Historia Clínica.
Artículo 14. Definición y archivo de la historia clínica.
1. La historia clínica comprende el conjunto de los documentos relativos a los pro-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
197

cesos asistenciales de cada paciente, con la identificación de los médicos y de los demásprofesionales que han intervenido en ellos, con objeto de obtener la máxima integración po-sible de la documentación clínica de cada paciente, al menos, en el ámbito de cada centro.
2. Cada centro archivará las historias clínicas de sus pacientes, cualquiera que seael soporte papel, audiovisual, informático o de otro tipo en el que consten, de manera quequeden garantizadas su seguridad, su correcta conservación y la recuperación de la infor-mación.
3. Las Administraciones sanitarias establecerán los mecanismos que garanticenla autenticidad del contenido de la historia clínica y de los cambios operados en ella, asícomo la posibilidad de su reproducción futura.
4. Las Comunidades Autónomas aprobarán las disposiciones necesarias para quelos centros sanitarios puedan adoptar las medidas técnicas y organizativas adecuadas paraarchivar y proteger las historias clínicas y evitar su destrucción o su pérdida accidental.
Artículo 15. Contenido de la historia clínica de cada paciente.
1. La historia clínica incorporará la información que se considere trascendentalpara el conocimiento veraz y actualizado del estado de salud del paciente. Todo pacienteo usuario tiene derecho a que quede constancia, por escrito o en el soporte técnico másadecuado, de la información obtenida en todos sus procesos asistenciales, realizados porel servicio de salud tanto en el ámbito de atención primaria como de atención especiali-zada.
2. La historia clínica tendrá como fin principal facilitar la asistencia sanitaria, de-jando constancia de todos aquellos datos que, bajo criterio médico, permitan el conoci-miento veraz y actualizado del estado de salud. El contenido mínimo de la historia clínicaserá el siguiente:
a. La documentación relativa a la hoja clínico estadística.
b. La autorización de ingreso.
c. El informe de urgencia.
d. La anamnesis y la exploración física.
e. La evolución.
f. Las órdenes médicas.
198

g. La hoja de interconsulta.
h. Los informes de exploraciones complementarias.
i. El consentimiento informado.
j. El informe de anestesia.
k. El informe de quirófano o de registro del parto.
l. El informe de anatomía patológica.
m. La evolución y planificación de cuidados de enfermería.
n. La aplicación terapéutica de enfermería.
ñ. El gráfico de constantes.
o. El informe clínico de alta.
Los párrafos b, c, i, j, k, l, ñ y o sólo serán exigibles en la cumplimentación de lahistoria clínica cuando se trate de procesos de hospitalización o así se disponga.
3. La cumplimentación de la historia clínica, en los aspectos relacionados con laasistencia directa al paciente, será responsabilidad de los profesionales que intervenganen ella.
4. La historia clínica se llevará con criterios de unidad y de integración, en cadainstitución asistencial como mínimo, para facilitar el mejor y más oportuno conocimientopor los facultativos de los datos de un determinado paciente en cada proceso asistencial.
Artículo 16. Usos de la historia clínica.
1. La historia clínica es un instrumento destinado fundamentalmente a garanti-zar una asistencia adecuada al paciente. Los profesionales asistenciales del centro que re-alizan el diagnóstico o el tratamiento del paciente tienen acceso a la historia clínica deéste como instrumento fundamental para su adecuada asistencia.
2. Cada centro establecerá los métodos que posibiliten en todo momento el ac-ceso a la historia clínica de cada paciente por los profesionales que le asisten.
3. El acceso a la historia clínica con fines judiciales, epidemiológicos, de saludpública, de investigación o de docencia, se rige por lo dispuesto en la Ley Orgánica15/1999, de Protección de Datos de Carácter Personal, y en la Ley 14/1986, General deSanidad, y demás normas de aplicación en cada caso. El acceso a la historia clínica con
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
199

estos fines obliga a preservar los datos de identificación personal del paciente, separadosde los de carácter clínico-asistencial, de manera que como regla general quede aseguradoel anonimato, salvo que el propio paciente haya dado su consentimiento para no separar-los. Se exceptúan los supuestos de investigación de la autoridad judicial en los que se con-sidere imprescindible la unificación de los datos identificativos con losclínico-asistenciales, en los cuales se estará a lo que dispongan los jueces y tribunales enel proceso correspondiente. El acceso a los datos y documentos de la historia clínica quedalimitado estrictamente a los fines específicos de cada caso.
4. El personal de administración y gestión de los centros sanitarios sólo puedeacceder a los datos de la historia clínica relacionados con sus propias funciones.
5. El personal sanitario debidamente acreditado que ejerza funciones de inspec-ción, evaluación, acreditación y planificación, tiene acceso a las historias clínicas en elcumplimiento de sus funciones de comprobación de la calidad de la asistencia, el respetode los derechos del paciente o cualquier otra obligación del centro en relación con los pa-cientes y usuarios o la propia Administración sanitaria.
6. El personal que accede a los datos de la historia clínica en el ejercicio de susfunciones queda sujeto al deber de secreto.
7. Las Comunidades Autónomas regularán el procedimiento para que quede cons-tancia del acceso a la historia clínica y de su uso.
Artículo 17. La conservación de la documentación clínica.
1. Los centros sanitarios tienen la obligación de conservar la documentación clí-nica en condiciones que garanticen su correcto mantenimiento y seguridad, aunque no ne-cesariamente en el soporte original, para la debida asistencia al paciente durante el tiempoadecuado a cada caso y, como mínimo, cinco años contados desde la fecha del alta de cadaproceso asistencial.
2. La documentación clínica también se conservará a efectos judiciales de confor-midad con la legislación vigente. Se conservará, asimismo, cuando existan razones epide-miológicas, de investigación o de organización y funcionamiento del Sistema Nacional deSalud. Su tratamiento se hará de forma que se evite en lo posible la identificación de las per-sonas afectadas.
3. Los profesionales sanitarios tienen el deber de cooperar en la creación y elmantenimiento de una documentación clínica ordenada y secuencial del proceso asisten-
200

cial de los pacientes.4. La gestión de la historia clínica por los centros con pacientes hospitalizados, o
por los que atiendan a un número suficiente de pacientes bajo cualquier otra modalidadasistencial, según el criterio de los servicios de salud, se realizará a través de la unidadde admisión y documentación clínica, encargada de integrar en un solo archivo las histo-rias clínicas. La custodia de dichas historias clínicas estará bajo la responsabilidad de ladirección del centro sanitario.
5. Los profesionales sanitarios que desarrollen su actividad de manera individualson responsables de la gestión y de la custodia de la documentación asistencial que ge-neren.
6. Son de aplicación a la documentación clínica las medidas técnicas de seguri-dad establecidas por la legislación reguladora de la conservación de los ficheros que con-tienen datos de carácter personal y, en general, por la Ley Orgánica 15/1999, deProtección de Datos de Carácter Personal.
Artículo 18. Derechos de acceso a la historia clínica.
1. El paciente tiene el derecho de acceso, con las reservas señaladas en el apar-tado 3 de este artículo, a la documentación de la historia clínica y a obtener copia de losdatos que figuran en ella. Los centros sanitarios regularán el procedimiento que garanticela observancia de estos derechos.
2. El derecho de acceso del paciente a la historia clínica puede ejercerse tambiénpor representación debidamente acreditada.
3. El derecho al acceso del paciente a la documentación de la historia clínica nopuede ejercitarse en perjuicio del derecho de terceras personas a la confidencialidad delos datos que constan en ella recogidos en interés terapéutico del paciente, ni en perjui-cio del derecho de los profesionales participantes en su elaboración, los cuales puedenoponer al derecho de acceso la reserva de sus anotaciones subjetivas.
4. Los centros sanitarios y los facultativos de ejercicio individual sólo facilitaránel acceso a la historia clínica de los pacientes fallecidos a las personas vinculadas a él,por razones familiares o de hecho, salvo que el fallecido lo hubiese prohibido expresa-mente y así se acredite. En cualquier caso el acceso de un tercero a la historia clínica mo-tivado por un riesgo para su salud se limitará a los datos pertinentes. No se facilitaráinformación que afecte a la intimidad del fallecido ni a las anotaciones subjetivas de los
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
201

profesionales, ni que perjudique a terceros.Artículo 19. Derechos relacionados con la custodia de la historia clínica.
El paciente tiene derecho a que los centros sanitarios establezcan un meca-nismo de custodia activa y diligente de las historias clínicas. Dicha custodia permitirála recogida, la integración, la recuperación y la comunicación de la información some-tida al principio de confidencialidad con arreglo a lo establecido por el artículo 16 dela presente Ley.
CAPÍTULO VI. Informe de alta y otra documentación clínica.
Artículo 20. Informe de alta.
Todo paciente, familiar o persona vinculada a él, en su caso, tendrá el derecho arecibir del centro o servicio sanitario, una vez finalizado el proceso asistencial, un informede alta con los contenidos mínimos que determina el artículo 3. Las características, re-quisitos y condiciones de los informes de alta se determinarán reglamentariamente por lasAdministraciones sanitarias autonómicas.
Artículo 21. El alta del paciente.
1. En caso de no aceptar el tratamiento prescrito, se propondrá al paciente o usua-rio la firma del alta voluntaria. Si no la firmara, la dirección del centro sanitario, a pro-puesta del médico responsable, podrá disponer el alta forzosa en las condiciones reguladaspor la Ley. El hecho de no aceptar el tratamiento prescrito no dará lugar al alta forzosacuando existan tratamientos alternativos, aunque tengan carácter paliativo, siempre quelos preste el centro sanitario y el paciente acepte recibirlos. Estas circunstancias queda-rán debidamente documentadas.
2. En el caso de que el paciente no acepte el alta, la dirección del centro, pre-via comprobación del informe clínico correspondiente, oirá al paciente y, si persisteen su negativa, lo pondrá en conocimiento del juez para que confirme o revoque ladecisión.
Artículo 22. Emisión de certificados médicos.
Todo paciente o usuario tiene derecho a que se le faciliten los certificados acre-ditativos de su estado de salud. Estos serán gratuitos cuando así lo establezca una dispo-
202

sición legal o reglamentaria.Artículo 23. Obligaciones profesionales de información técnica, estadística y administrativa.
Los profesionales sanitarios, además de las obligaciones señaladas en materia deinformación clínica, tienen el deber de cumplimentar los protocolos, registros, informes,estadísticas y demás documentación asistencial o administrativa, que guarden relación conlos procesos clínicos en los que intervienen, y los que requieran los centros o servicios desalud competentes y las autoridades sanitarias, comprendidos los relacionados con la in-vestigación médica y la información epidemiológica.
DISPOSICIÓN ADICIONAL PRIMERA. Carácter de legislación básica.
Esta Ley tiene la condición de básica, de conformidad con lo establecido en el ar-tículo 149.1.1 y 16 de la Constitución.
El Estado y las Comunidades Autónomas adoptarán, en el ámbito de sus respec-tivas competencias, las medidas necesarias para la efectividad de esta Ley.
DISPOSICIÓN ADICIONAL SEGUNDA. Aplicación supletoria.
Las normas de esta Ley relativas a la información asistencial, la información para elejercicio de la libertad de elección de médico y de centro, el consentimiento informado delpaciente y la documentación clínica, serán de aplicación supletoria en los proyectos de inves-tigación médica, en los procesos de extracción y trasplante de órganos, en los de aplicaciónde técnicas de reproducción humana asistida y en los que carezcan de regulación especial.
DISPOSICIÓN ADICIONAL TERCERA. Coordinación de las historias clínicas.
El Ministerio de Sanidad y Consumo, en coordinación y con la colaboración de lasComunidades Autónomas competentes en la materia, promoverá, con la participación detodos los interesados, la implantación de un sistema de compatibilidad que, atendida laevolución y disponibilidad de los recursos técnicos, y la diversidad de sistemas y tipos dehistorias clínicas, posibilite su uso por los centros asistenciales de España que atiendana un mismo paciente, en evitación de que los atendidos en diversos centros se sometan aexploraciones y procedimientos de innecesaria repetición.
DISPOSICIÓN ADICIONAL CUARTA. Necesidades asociadas a la discapacidad.
El Estado y las Comunidades Autónomas, dentro del ámbito de sus respectivascompetencias, dictarán las disposiciones precisas para garantizar a los pacientes o usua-rios con necesidades especiales, asociadas a la discapacidad, los derechos en materia de
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
203

autonomía, información y documentación clínica regulados en esta Ley.DISPOSICIÓN ADICIONAL QUINTA. Información y documentación sobre medicamentos yproductos sanitarios.
La información, la documentación y la publicidad relativas a los medicamentos yproductos sanitarios, así como el régimen de las recetas y de las órdenes de prescripcióncorrespondientes, se regularán por su normativa específica, sin perjuicio de la aplicaciónde las reglas establecidas en esta Ley en cuanto a la prescripción y uso de medicamentoso productos sanitarios durante los procesos asistenciales.
DISPOSICIÓN ADICIONAL SEXTA. Régimen sancionador.
Las infracciones de lo dispuesto por la presente Ley quedan sometidas al régi-men sancionador previsto en el capítulo VI del Título I de la Ley 14/1986, General de Sa-nidad, sin perjuicio de la responsabilidad civil o penal y de la responsabilidad profesionalo estatutaria procedentes en derecho.
DISPOSICIÓN TRANSITORIA ÚNICA. Informe de alta.
El informe de alta se regirá por lo dispuesto en la Orden del Ministerio de Sani-dad, de 6 de septiembre de 1984, mientras no se desarrolle legalmente lo dispuesto enel artículo 20 de esta Ley.
DISPOSICIÓN DEROGATORIA ÚNICA. Derogación general y de preceptos concretos.
Quedan derogadas las disposiciones de igual o inferior rango que se opongan a lodispuesto en la presente Ley y, concretamente, los apartados 5, 6, 8, 9 y 11 del artículo10, el apartado 4 del artículo 11 y el artículo 61 de la Ley 14/1986, General de Sanidad.
DISPOSICIÓN FINAL ÚNICA. Entrada en vigor.
La presente Ley entrará en vigor en el plazo de seis meses a partir del día siguienteal de su publicación en el Boletín Oficial del Estado.
Por tanto, Mando a todos los españoles, particulares y autoridades, que guardeny hagan guardar esta Ley.
Madrid, 14 de noviembre de 2002.- Juan Carlos R. -
El Presidente del Gobierno, José María Aznar López.
204

LEY ORGÁNICA 15/1999, DE 13 DE DICIEMBRE, DE PROTECCIÓN DE DATOS DE CARÁCTER PERSONAL
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
205

206

LEY ORGÁNICA 15/1999, DE 13 DE DICIEMBRE, DE PROTECCIÓN DE DA-TOS DE CARÁCTER PERSONAL
TÍTULO I Disposiciones generales
Artículo 1. Objeto.
La presente Ley Orgánica tiene por objeto garantizar y proteger, en lo que concierneal tratamiento de los datos personales, las libertades públicas y los derechos fundamenta-les de las personas físicas, y especialmente de su honor e intimidad personal y familiar.
Artículo 2. Ámbito de aplicación.
1. La presente Ley Orgánica será de aplicación a los datos de carácter personalregistrados en soporte físico, que los haga susceptibles de tratamiento, y a toda modali-dad de uso posterior de estos datos por los sectores público y privado.
Se regirá por la presente Ley Orgánica todo tratamiento de datos de carácter personal:
a) Cuando el tratamiento sea efectuado en territorio español en el marco de las ac-tividades de un establecimiento del responsable del tratamiento.
b) Cuando al responsable del tratamiento no establecido en territorio español, lesea de aplicación la legislación española en aplicación de normas de Derecho Internacio-nal público.
c) Cuando el responsable del tratamiento no esté establecido en territorio de laUnión Europea y utilice en el tratamiento de datos medios situados en territorio español,salvo que tales medios se utilicen únicamente con fines de tránsito.
2. El régimen de protección de los datos de carácter personal que se establece enla presente Ley Orgánica no será de aplicación:
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
207

a) A los ficheros mantenidos por personas físicas en el ejercicio de actividades ex-clusivamente personales o domésticas.
b) A los ficheros sometidos a la normativa sobre protección de materias clasificadas.
c) A los ficheros establecidos para la investigación del terrorismo y de formas gra-ves de delincuencia organizada.
No obstante, en estos supuestos el responsable del fichero comunicará previa-mente la existencia del mismo, sus características generales y su finalidad a la Agenciade Protección de Datos.
3. Se regirán por sus disposiciones específicas, y por lo especialmente previsto,en su caso, por esta Ley Orgánica los siguientes tratamientos de datos personales:
a) Los ficheros regulados por la legislación de régimen electoral.
b) Los que sirvan a fines exclusivamente estadísticos, y estén amparados por la le-gislación estatal o autonómica sobre la función estadística pública.
c) Los que tengan por objeto el almacenamiento de los datos contenidos en los in-formes personales de calificación a que se refiere la legislación del régimen del personalde las Fuerzas Armadas.
d) Los derivados del Registro Civil y del Registro Central de penados y rebeldes.
e) Los procedentes de imágenes y sonidos obtenidos mediante la utilización de vi-deocámaras por las Fuerzas y Cuerpos de Seguridad, de conformidad con la legislación so-bre la materia.
Artículo 3. Definiciones.
A los efectos de la presente Ley Orgánica se entenderá por:
a) Datos de carácter personal: cualquier información concerniente a personas fí-sicas identificadas o identificables.
b) Fichero: todo conjunto organizado de datos de carácter personal, cualquieraque fuere la forma o modalidad de su creación, almacenamiento, organización y acceso.
208

c) Tratamiento de datos: operaciones y procedimientos técnicos de carácter auto-matizado o no, que permitan la recogida, grabación, conservación, elaboración, modifica-ción, bloqueo y cancelación, así como las cesiones de datos que resulten decomunicaciones, consultas, interconexiones y transferencias.
d) Responsable del fichero o tratamiento: persona física o jurídica, de naturalezapública o privada, u órgano administrativo, que decida sobre la finalidad, contenido y usodel tratamiento.
e) Afectado o interesado: persona física titular de los datos que sean objeto del tra-tamiento a que se refiere el apartado c) del presente artículo.
f) Procedimiento de disociación: todo tratamiento de datos personales de modoque la información que se obtenga no pueda asociarse a persona identificada o identifi-cable.
g) Encargado del tratamiento: la persona física o jurídica, autoridad pública, ser-vicio o cualquier otro organismo que, sólo o conjuntamente con otros, trate datos perso-nales por cuenta del responsable del tratamiento.
h) Consentimiento del interesado: toda manifestación de voluntad, libre, inequí-voca, específica e informada, mediante la que el interesado consienta el tratamiento dedatos personales que le conciernen.
i) Cesión o comunicación de datos: toda revelación de datos realizada a una per-sona distinta del interesado.
j) Fuentes accesibles al público: aquellos ficheros cuya consulta puede ser reali-zada, por cualquier persona, no impedida por una norma limitativa o sin más exigenciaque, en su caso, el abono de una contraprestación.
Tienen la consideración de fuentes de acceso público, exclusivamente, el censopromocional, los repertorios telefónicos en los términos previstos por su normativa espe-cífica y las listas de personas pertenecientes a grupos de profesionales que contenganúnicamente los datos de nombre, título, profesión, actividad, grado académico, direccióne indicación de su pertenencia al grupo.
Asimismo, tienen el carácter de fuentes de acceso público los diarios y boletinesoficiales y los medios de comunicación.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
209

TÍTULO II Principios de la protección de datos
Artículo 4. Calidad de los datos.
1. Los datos de carácter personal sólo se podrán recoger para su tratamiento, asícomo someterlos a dicho tratamiento, cuando sean adecuados, pertinentes y no excesivosen relación con el ámbito y las finalidades determinadas, explícitas y legítimas para las quese hayan obtenido.
2. Los datos de carácter personal objeto de tratamiento no podrán usarse para fi-nalidades incompatibles con aquellas para las que los datos hubieran sido recogidos.
No se considerará incompatible el tratamiento posterior de éstos con fines histó-ricos, estadísticos o científicos.
3. Los datos de carácter personal serán exactos y puestos al día de forma que res-pondan con veracidad a la situación actual del afectado.
4. Si los datos de carácter personal registrados resultaran ser inexactos, en todoo en parte, o incompletos, serán cancelados y sustituidos de oficio por los correspondien-tes datos rectificados o completados, sin perjuicio de las facultades que a los afectadosreconoce el artículo 16.
5. Los datos de carácter personal serán cancelados cuando hayan dejado de sernecesarios o pertinentes para la finalidad para la cual hubieran sido recabados o re-gistrados.
No serán conservados en forma que permita la identificación del interesado du-rante un período superior al necesario para los fines en base a los cuales hubieran sido re-cabados o registrados.
Reglamentariamente se determinará el procedimiento por el que, por excepción,atendidos los valores históricos, estadísticos o científicos de acuerdo con la legislaciónespecífica, se decida el mantenimiento integro de determinados datos.
6. Los datos de carácter personal serán almacenados de forma que permitan elejercicio del derecho de acceso, salvo que sean legalmente cancelados.
7. Se prohíbe la recogida de datos por medios fraudulentos, desleales o ilícitos.
210

Artículo 5. Derecho de información en la recogida de datos.
1. Los interesados a los que se soliciten datos personales deberán ser previa-mente informados de modo expreso, preciso e inequívoco:
a) De la existencia de un fichero o tratamiento de datos de carácter personal, dela finalidad de la recogida de éstos y de los destinatarios de la información.
b) Del carácter obligatorio o facultativo de su respuesta a las preguntas que lessean planteadas.
c) De las consecuencias de la obtención de los datos o de la negativa a sumi-nistrarlos.
d) De la posibilidad de ejercitar los derechos de acceso, rectificación, cancela-ción y oposición.
e) De la identidad y dirección del responsable del tratamiento o, en su caso, desu representante.
Cuando el responsable del tratamiento no esté establecido en el territorio de laUnión Europea y utilice en el tratamiento de datos medios situados en territorio espa-ñol, deberá designar, salvo que tales medios se utilicen con fines de trámite, un repre-sentante en España, sin perjuicio de las acciones que pudieran emprenderse contra elpropio responsable del tratamiento.
2. Cuando se utilicen cuestionarios u otros impresos para la recogida, figuraránen los mismos, en forma claramente legible, las advertencias a que se refiere el apar-tado anterior.
3. No será necesaria la información a que se refieren las letras b), c) y d) delapartado 1 si el contenido de ella se deduce claramente de la naturaleza de los datospersonales que se solicitan o de las circunstancias en que se recaban.
4. Cuando los datos de carácter personal no hayan sido recabados del intere-sado, éste deberá ser informado de forma expresa, precisa e inequívoca, por el respon-sable del fichero o su representante, dentro de los tres meses siguientes al momento delregistro de los datos, salvo que ya hubiera sido informado con anterioridad, del conte-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
211

nido del tratamiento, de la procedencia de los datos, así como de lo previsto en las le-tras a), d) y e) del apartado 1 del presente artículo.
5. No será de aplicación lo dispuesto en el apartado anterior, cuando expresa-mente una ley lo prevea, cuando el tratamiento tenga fines históricos, estadísticos o cien-tíficos, o cuando la información al interesado resulte imposible o exija esfuerzosdesproporcionados, a criterio de la Agencia de Protección de Datos o del organismo auto-nómico equivalente, en consideración al número de interesados, a la antigüedad de los da-tos y a las posibles medidas compensatorias.
Asimismo, tampoco regirá lo dispuesto en el apartado anterior cuando los datosprocedan de fuentes accesibles al público y se destinen a la actividad de publicidad o pros-pección comercial, en cuyo caso, en cada comunicación que se dirija al interesado se leinformará del origen de los datos y de la identidad del responsable del tratamiento asícomo de los derechos que le asisten.
Artículo 6. Consentimiento del afectado.
1. El tratamiento de los datos de carácter personal requerirá el consentimiento in-equívoco del afectado, salvo que la ley disponga otra cosa.
2. No será preciso el consentimiento cuando los datos de carácter personal se re-cojan para el ejercicio de las funciones propias de las Administraciones públicas en elámbito de sus competencias; cuando se refieran a las partes de un contrato o precontratode una relación negocial, laboral o administrativa y sean necesarios para su mantenimientoo cumplimiento; cuando el tratamiento de los datos tenga por finalidad proteger un inte-rés vital del interesado en los términos del artículo 7, apartado 6, de la presente Ley, ocuando los datos figuren en fuentes accesibles al público y su tratamiento sea necesariopara la satisfacción del interés legítimo perseguido por el responsable del fichero o por eldel tercero a quien se comuniquen los datos, siempre que no se vulneren los derechos ylibertades fundamentales del interesado.
3. El consentimiento a que se refiere el artículo podrá ser revocado cuando existacausa justificada para ello y no se le atribuyan efectos retroactivos.
4. En los casos en los que no sea necesario el consentimiento del afectado parael tratamiento de los datos de carácter personal, y siempre que una ley no disponga lo con-trario, éste podrá oponerse a su tratamiento cuando existan motivos fundados y legítimosrelativos a una concreta situación personal. En tal supuesto, el responsable del fichero ex-cluirá del tratamiento los datos relativos al afectado.
212

Artículo 7. Datos especialmente protegidos.
1. De acuerdo con lo establecido en el apartado 2 del artículo 16 de la Constitución,nadie podrá ser obligado a declarar sobre su ideología, religión o creencias.
Cuando en relación con estos datos se proceda a recabar el consentimiento a que serefiere el apartado siguiente, se advertirá al interesado acerca de su derecho a no prestarlo.
2. Sólo con el consentimiento expreso y por escrito del afectado podrán ser objeto detratamiento los datos de carácter personal que revelen la ideología, afiliación sindical, religióny creencias. Se exceptúan los ficheros mantenidos por los partidos políticos, sindicatos, igle-sias, confesiones o comunidades religiosas y asociaciones, fundaciones y otras entidades sinánimo de lucro, cuya finalidad sea política, filosófica, religiosa o sindical, en cuanto a los da-tos relativos a sus asociados o miembros, sin perjuicio de que la cesión de dichos datos preci-sará siempre el previo consentimiento del afectado.
3. Los datos de carácter personal que hagan referencia al origen racial, a la salud y ala vida sexual sólo podrán ser recabados, tratados y cedidos cuando, por razones de interés ge-neral, así lo disponga una ley o el afectado consienta expresamente.
4. Quedan prohibidos los ficheros creados con la finalidad exclusiva de almacenar da-tos de carácter personal que revelen la ideología, afiliación sindical, religión, creencias, origenracial o étnico, o vida sexual.
5. Los datos de carácter personal relativos a la comisión de infracciones penales o ad-ministrativas sólo podrán ser incluidos en ficheros de las Administraciones públicas competen-tes en los supuestos previstos en las respectivas normas reguladoras.
6. No obstante lo dispuesto en los apartados anteriores, podrán ser objeto de trata-miento los datos de carácter personal a que se refieren los apartados 2 y 3 de este artículo,cuando dicho tratamiento resulte necesario para la prevención o para el diagnóstico médicos,la prestación de asistencia sanitaria o tratamientos médicos o la gestión de servicios sanitarios,siempre que dicho tratamiento de datos se realice por un profesional sanitario sujeto al secretoprofesional o por otra persona sujeta asimismo a una obligación equivalente de secreto.
También podrán ser objeto de tratamiento los datos a que se refiere el párrafo anteriorcuando el tratamiento sea necesario para salvaguardar el interés vital del afectado o de otra per-sona, en el supuesto de que el afectado esté física o jurídicamente incapacitado para dar suconsentimiento.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
213

Artículo 8. Datos relativos a la salud.
Sin perjuicio de lo que se dispone en el artículo 11 respecto de la cesión, las ins-tituciones y los centros sanitarios públicos y privados y los profesionales correspondien-tes podrán proceder al tratamiento de los datos de carácter personal relativos a la saludde las personas que a ellos acudan o hayan de ser tratados en los mismos, de acuerdo conlo dispuesto en la legislación estatal o autonómica sobre sanidad.
Artículo 9. Seguridad de los datos.
1. El responsable del fichero, y, en su caso, el encargado del tratamiento deberánadoptar las medidas de índole técnica y organizativas necesarias que garanticen la segu-ridad de los datos de carácter personal y eviten su alteración, pérdida, tratamiento o ac-ceso no autorizado, habida cuenta del estado de la tecnología, la naturaleza de los datosalmacenados y los riesgos a que están expuestos, ya provengan de la acción humana o delmedio físico o natural.
2. No se registrarán datos de carácter personal en ficheros que no reúnan las con-diciones que se determinen por vía reglamentaria con respecto a su integridad y seguri-dad ya las de los centros de tratamiento, locales, equipos, sistemas y programas.
3. Reglamentariamente se establecerán los requisitos y condiciones que debanreunir los ficheros y las personas que intervengan en el tratamiento de los datos a que serefiere el artículo 7 de esta Ley.
Artículo 10. Deber de secreto.
El responsable del fichero y quienes intervengan en cualquier fase del tratamientode los datos de carácter personal están obligados al secreto profesional respecto de losmismos y al deber de guardarlos, obligaciones que subsistirán aun después de finalizar susrelaciones con el titular del fichero o, en su caso, con el responsable del mismo.
Artículo 11. Comunicación de datos.
1. Los datos de carácter personal objeto del tratamiento sólo podrán ser comuni-cados a un tercero para el cumplimiento de fines directamente relacionados con las fun-ciones legítimas del cedente y del cesionario con el previo consentimiento del interesado.
2. El consentimiento exigido en el apartado anterior no será preciso:
214

a) Cuando la cesión está autorizada en una ley.
b) Cuando se trate de datos recogidos de fuentes accesibles al público.
c) Cuando el tratamiento responda a la libre y legítima aceptación de una relación ju-rídica cuyo desarrollo, cumplimiento y control implique necesariamente la conexión de dichotratamiento con ficheros de terceros.
En este caso la comunicación sólo será legítima en cuanto se limite a la finalidad quela justifique.
d) Cuando la comunicación que deba efectuarse tenga por destinatario al Defensordel Pueblo, el Ministerio Fiscal o los Jueces o Tribunales o el Tribunal de Cuentas, en el ejer-cicio de las funciones que tiene atribuidas.
Tampoco será preciso el consentimiento cuando la comunicación tenga como destina-tario a instituciones autonómicas con funciones análogas al Defensor del Pueblo o al Tribunalde Cuentas.
e) Cuando la cesión se produzca entre Administraciones públicas y tenga por objetoel tratamiento posterior de los datos con fines históricos, estadísticos o científicos.
f) Cuando la cesión de datos de carácter personal relativos a la salud sea necesaria parasolucionar una urgencia que requiera acceder a un fichero o para realizar los estudios epide-miológicos en los términos establecidos en la legislación sobre sanidad estatal o autonómica.
3. Será nulo el consentimiento para la comunicación de los datos de carácter perso-nal a un tercero, cuando la información que se facilite al interesado no le permita conocer lafinalidad a que destinarán los datos cuya comunicación se autoriza o el tipo de actividad deaquel a quien se pretenden comunicar.
4. El consentimiento para la comunicación de los datos de carácter personal tienetambién un carácter de revocable.
5. Aquel a quien se comuniquen los datos de carácter personal se obliga, por el solohecho de la comunicación, a la observancia de las disposiciones de la presente Ley.
6. Si la comunicación se efectúa previo procedimiento de disociación, no será aplica-ble lo establecido en los apartados anteriores.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
215

Artículo 12. Acceso a los datos por cuenta de terceros.
1. No se considerará comunicación de datos el acceso de un tercero a los datoscuando dicho acceso sea necesario para la prestación de un servicio al responsable del tra-tamiento.
2. La realización de tratamientos por cuenta de terceros deberá estar regulada enun contrato que deberá constar por escrito o en alguna otra forma que permita acreditarsu celebración y contenido, estableciéndose expresamente que el encargado del trata-miento únicamente tratará los datos conforme a las instrucciones del responsable del tra-tamiento, que no los aplicará o utilizará con fin distinto al que figure en dicho contrato,ni los comunicará, ni siquiera para su conservación, a otras personas.
En el contrato se estipularán, asimismo, las medidas de seguridad a que se refiereel artículo 9 de esta Ley que el encargado del tratamiento está obligado a implementar.
3. Una vez cumplida la prestación contractual, los datos de carácter personal de-berán ser destruidos o devueltos al responsable del tratamiento, al igual que cualquier so-porte o documentos en que conste algún dato de carácter personal objeto del tratamiento.
4. En el caso de que el encargado del tratamiento destine los datos a otra finali-dad, los comunique o los utilice incumpliendo las estipulaciones del contrato, será con-siderado también responsable del tratamiento, respondiendo de las infracciones en quehubiera incurrido personalmente.
TÍTULO III Derechos de las personas
Artículo 13. Impugnación de valoraciones.
1. Los ciudadanos tienen derecho a no verse sometidos a una decisión con efectosjurídicos, sobre ellos o que les afecte de manera significativa, que se base únicamente enun tratamiento de datos destinados a evaluar determinados aspectos de su personalidad.
2. El afectado podrá impugnar los actos administrativos o decisiones privadas queimpliquen una valoración de su comportamiento, cuyo único fundamento sea un trata-
216

miento de datos de carácter personal que ofrezca una definición de sus características opersonalidad.
3. En este caso, el afectado tendrá derecho a obtener información del responsa-ble del fichero sobre los criterios de valoración y el programa utilizados en el tratamientoque sirvió para adoptar la decisión en que consistió el acto.
4. La valoración sobre el comportamiento de los ciudadanos, basada en un trata-miento de datos, únicamente podrá tener valor probatorio a petición del afectado.
Artículo 14. Derecho de consulta al Registro General de Protección de Datos.
Cualquier persona podrá conocer, recabando a tal fin la información oportuna delRegistro General de Protección de Datos, la existencia de tratamientos de datos de carác-ter personal, sus finalidades y la identidad del responsable del tratamiento. El Registro Ge-neral será de consulta pública y gratuita.
Artículo 15. Derecho de acceso.
1. El interesado tendrá derecho a solicitar y obtener gratuitamente información desus datos de carácter personal sometidos a tratamiento, el origen de dichos datos, asícomo las comunicaciones realizadas o que se prevén hacer de los mismos.
2. La información podrá obtenerse mediante la mera consulta de los datos pormedio de su visualización, o la indicación de los datos que son objeto de tratamiento me-diante escrito, copia, telecopia o fotocopia, certificada o no, en forma legible e inteligible,sin utilizar claves o códigos que requieran el uso de dispositivos mecánicos específicos.
3. El derecho de acceso a que se refiere este artículo sólo podrá ser ejercitado aintervalos no inferiores a doce meses, salvo que el interesado acredite un interés legítimoal efecto, en cuyo caso podrán ejercitarlo antes.
Artículo 16. Derecho de rectificación y cancelación.
1. El responsable del tratamiento tendrá la obligación de hacer efectivo el dere-cho de rectificación o cancelación del interesado en el plazo de diez días.
2. Serán rectificados o cancelados, en su caso, los datos de carácter personal cuyotratamiento no se ajuste a lo dispuesto en la presente Ley y, en particular, cuando talesdatos resulten inexactos o incompletos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
217

3. La cancelación dará lugar al bloqueo de los datos, conservándose únicamente a dis-posición de las Administraciones públicas, Jueces y Tribunales, para la atención de las posi-bles responsabilidades nacidas del tratamiento, durante el plazo de prescripción de éstas.
Cumplido el citado plazo deberá procederse a la supresión.
4. Si los datos rectificados o cancelados hubieran sido comunicados previamente,el responsable del tratamiento deberá notificar la rectificación o cancelación efectuada aquien se hayan comunicado, en el caso de que se mantenga el tratamiento por este último,que deberá también proceder a la cancelación.
5. Los datos de carácter personal deberán ser conservados durante los plazos pre-vistos en las disposiciones aplicables o, en su caso, en las relaciones contractuales entrela persona o entidad responsable del tratamiento y el interesado.
Artículo 17. Procedimiento de oposición, acceso, rectificación o cancelación.
1. Los procedimientos para ejercitar el derecho de oposición, acceso, así como losde rectificación y cancelación serán establecidos reglamentariamente.
2. No se exigirá contraprestación alguna por el ejercicio de los derechos de opo-sición, acceso, rectificación o cancelación.
Artículo 18. Tutela de los derechos.
1. Las actuaciones contrarias a lo dispuesto en la presente Ley pueden ser objetode reclamación por los interesados ante la Agencia de Protección de Datos, en la formaque reglamentariamente se determine.
2. El interesado al que se deniegue, total o parcialmente, el ejercicio de los derechosde oposición, acceso, rectificación o cancelación, podrá ponerlo en conocimiento de la Agen-cia de Protección de Datos o, en su caso, del organismo competente de cada Comunidad Au-tónoma, que deberá asegurarse de la procedencia o improcedencia de la denegación.
3. El plazo máximo en que debe dictarse la resolución expresa de tutela de dere-chos será de seis meses.
4. Contra las resoluciones de la Agencia de Protección de Datos procederá recursocontencioso-administrativo.
218

Artículo 19. Derecho a indemnización.
1. Los interesados que, como consecuencia del incumplimiento de lo dispuestoen la presente Ley por el responsable o el encargado del tratamiento, sufran daño o lesiónen sus bienes o derechos tendrán derecho a ser indemnizados.
2. Cuando se trate de ficheros de titularidad pública, la responsabilidad se exigiráde acuerdo con la legislación reguladora del régimen de responsabilidad de las Adminis-traciones públicas.
3. En el caso de los ficheros de titularidad privada, la acción se ejercitará ante losórganos de la jurisdicción ordinaria.
TÍTULO IV Disposiciones sectoriales
CAPÍTULO I Ficheros de titularidad pública
Artículo 20. Creación, modificación o supresión.
1. La creación, modificación o supresión de los ficheros de las Administracionespúblicas sólo podrán hacerse por medio de disposición general publicada en el «BoletínOficial del Estado» o Diario oficial correspondiente.
2. Las disposiciones de creación o de modificación de ficheros deberán indicar:
a) La finalidad del fichero y los usos previstos para el mismo.
b) Las personas o colectivos sobre los que se pretenda obtener datos de carácterpersonal o que resulten obligados a suministrarlos.
c) El procedimiento de recogida de los datos de carácter personal.
d) La estructura básica del fichero y la descripción de los tipos de datos de carác-ter personal incluidos en el mismo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
219

e) Las cesiones de datos de carácter personal y, en su caso, las transferencias dedatos que se prevean a países terceros.
f) Los órganos de las Administraciones responsables del fichero.
g) Los servicios o unidades ante los que pudiesen ejercitarse los derechos de ac-ceso, rectificación, cancelación y oposición.
h) Las medidas de seguridad con indicación del nivel básico, medio o alto exigible.
3. En las disposiciones que se dicten para la supresión de los ficheros, se estableceráel destino de los mismos o, en su caso, las previsiones que se adopten para su destrucción.
Artículo 21. Comunicación de datos entre Administraciones públicas.
1. Los datos de carácter personal recogidos o elaborados por las Administracionespúblicas para el desempeño de sus atribuciones no serán comunicados a otras Adminis-traciones públicas para el ejercicio de competencias diferentes o de competencias que ver-sen sobre materias distintas, salvo cuando la comunicación hubiere sido prevista por lasdisposiciones de creación del fichero o por disposición de superior rango que regule su uso,o cuando la comunicación tenga por objeto el tratamiento posterior de los datos con fineshistóricos, estadísticos o científicos.
2. Podrán, en todo caso, ser objeto de comunicación los datos de carácter perso-nal que una Administración pública obtenga o elabore con destino a otra.
3. No obstante lo establecido en el artículo 11.2.b).
La comunicación de datos recogidos de fuentes accesibles al público no podráefectuarse a ficheros de titularidad privada, sino con el consentimiento del interesado ocuando una ley prevea otra cosa.
4. En los supuestos previstos en los apartados 1 y 2 del presente artículo no seránecesario el consentimiento del afectado a que se refiere el artículo 11 de la presente Ley.
Artículo 22. Ficheros de las Fuerzas y Cuerpos de Seguridad.
Los ficheros creados por las Fuerzas y Cuerpos de Seguridad que contengan da-tos de carácter personal que, por haberse recogido para fines administrativos, deban serobjeto de registro permanente, estarán sujetos al régimen general de la presente Ley.
220

2. La recogida y tratamiento para fines policiales de datos de carácter personal porlas Fuerzas y Cuerpos de Seguridad sin consentimiento de las personas afectadas estánlimitados a aquellos supuestos y categorías de datos que resulten necesarios para la pre-vención de un peligro real para la seguridad pública o para la represión de infracciones pe-nales, debiendo ser almacenados en ficheros específicos establecidos al efecto, quedeberán clasificarse por categorías en función de su grado de fiabilidad.
3. La recogida y tratamiento por las Fuerzas y Cuerpos de Seguridad de los datos,a que hacen referencia los apartados 2 y 3 del artículo 7, podrán realizarse exclusiva-mente en los supuestos en que sea absolutamente necesario para los fines de una inves-tigación concreta, sin perjuicio del control de legalidad de la actuación administrativa ode la obligación de resolver las pretensiones formuladas en su caso por los interesados quecorresponden a los órganos jurisdiccionales.
4. Los datos personales registrados con fines policiales se cancelarán cuando nosean necesarios para las averiguaciones que motivaron su almacenamiento.
A estos efectos, se considerará especialmente la edad del afectado y el carácterde los datos almacenados, la necesidad de mantener los datos hasta la conclusión de unainvestigación o procedimiento concreto, la resolución judicial firme, en especial la abso-lutoria, el indulto, la rehabilitación y la prescripción de responsabilidad.
Artículo 23. Excepciones a los derechos de acceso, rectificación y cancelación.
1. Los responsables de los ficheros que contengan los datos a que se refieren losapartados 2, 3 y 4 del artículo anterior podrán denegar el acceso, la rectificación o can-celación en función de los peligros que pudieran derivarse para la defensa del Estado o laseguridad pública, la protección de los derechos y libertades de terceros o las necesida-des de las investigaciones que se estén realizando.
2. Los responsables de los ficheros de la Hacienda Pública podrán, igualmente,denegar el ejercicio de los derechos a que se refiere el apartado anterior cuando el mismoobstaculice las actuaciones administrativas tendentes a asegurar el cumplimiento de lasobligaciones tributarias y, en todo caso, cuando el afectado esté siendo objeto de actua-ciones inspectoras.
3. El afectado al que se deniegue, total o parcialmente, el ejercicio de los dere-chos mencionados en los apartados anteriores podrá ponerlo en conocimiento del Direc-tor de la Agencia de Protección de Datos o del organismo competente de cada Comunidad
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
221

Autónoma en el caso de ficheros mantenidos por Cuerpos de Policía propios de éstas, opor las Administraciones tributarias autonómicas, quienes deberán asegurarse de la pro-cedencia o improcedencia de la denegación.
Artículo 24. Otras excepciones a los derechos de los afectados.
1. Lo dispuesto en los apartados 1 y 2 del artículo 5 no será aplicable a la reco-gida de datos cuando la información al afectado impida o dificulte gravemente el cumpli-miento de las funciones de control y verificación de las Administraciones públicas ocuando afecte a la Defensa Nacional, a la seguridad pública o a la persecución de infrac-ciones penales o administrativas.
2. Lo dispuesto en el artículo 15 y en el apartado 1 del artículo 16 no será de apli-cación si, ponderados los intereses en presencia, resultase que los derechos que dichospreceptos conceden al afectado hubieran de ceder ante razones de interés público o anteintereses de terceros más dignos de protección. Si el órgano administrativo responsabledel fichero invocase lo dispuesto en este apartado, dictará resolución motivada e instruiráal afectado del derecho que le asiste a poner la negativa en conocimiento del Director dela Agencia de Protección de Datos o, en su caso, del órgano equivalente de las Comuni-dades Autónomas.
CAPÍTULO II Ficheros de titularidad privada
Artículo 25. Creación.
Podrán crearse ficheros de titularidad privada que contengan datos de carácterpersonal cuando resulte necesario para el logro de la actividad u objeto legítimos de la per-sona, empresa o entidad titular y se respeten las garantías que esta Ley establece para laprotección de las personas.
Artículo 26. Notificación e inscripción registral.
1. Toda persona o entidad que proceda a la creación de ficheros de datos de ca-rácter personal lo notificará previamente a la Agencia de Protección de Datos.
2. Por vía reglamentaria se procederá a la regulación detallada de los distintosextremos que debe contener la notificación, entre los cuales figurarán necesariamente elresponsable del fichero, la finalidad del mismo, su ubicación, el tipo de datos de carácter
222

personal que contiene, las medidas de seguridad, con indicación del nivel básico, medioo alto exigible y las cesiones de datos de carácter personal que se prevean realizar y, ensu caso, las transferencias de datos que se prevean a países terceros.
3. Deberán comunicarse a la Agencia de Protección de Datos los cambios que seproduzcan en la finalidad del fichero automatizado, en su responsable y en la dirección desu ubicación.
4. El Registro General de Protección de Datos inscribirá el fichero si la notificaciónse ajusta a los requisitos exigibles.
En caso contrario podrá pedir que se completen los datos que falten o se procedaa su subsanación.
5. Transcurrido un mes desde la presentación de la solicitud de inscripción sin quela Agencia de Protección de Datos hubiera resuelto sobre la misma, se entenderá inscrito elfichero automatizado a todos los efectos.
Artículo 27. Comunicación de la cesión de datos.
1. El responsable del fichero, en el momento en que se efectúe la primera cesión dedatos, deberá informar de ello a los afectados, indicando, asimismo, la finalidad del fichero,la naturaleza de los datos que han sido cedidos y el nombre y dirección del cesionario.
2. La obligación establecida en el apartado anterior no existirá en el supuesto pre-visto en los apartados 2, letras c), d), e) y 6 del artículo 11, ni cuando la cesión venga im-puesta por ley.
Artículo 28. Datos incluidos en las fuentes de acceso público.
1. Los datos personales que figuren en el censo promocional, o las listas de perso-nas pertenecientes a grupos de profesionales a que se refiere el artículo 3, j) de esta Ley de-berán limitarse a los que sean estrictamente necesarios para cumplir la finalidad a que sedestina cada listado. La inclusión de datos adicionales por las entidades responsables delmantenimiento de dichas fuentes requerirá el consentimiento del interesado, que podrá serrevocado en cualquier momento.
2. Los interesados tendrán derecho a que la entidad responsable del mantenimientode los listados de los Colegios profesionales indique gratuitamente que sus datos persona-les no pueden utilizarse para fines de publicidad o prospección comercial.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
223

Los interesados tendrán derecho a exigir gratuitamente la exclusión de la to-talidad de sus datos personales que consten en el censo promocional por las entida-des encargadas del mantenimiento de dichas fuentes.
La atención a la solicitud de exclusión de la información innecesaria o de in-clusión de la objeción al uso de los datos para fines de publicidad o venta a distanciadeberá realizarse en el plazo de diez días respecto de las informaciones que se reali-cen mediante consulta o comunicación telemática y en la siguiente edición del lis-tado cualquiera que sea el soporte en que se edite.
3. Las fuentes de acceso público que se editen en forma de libro o algún otrosoporte físico, perderán el carácter de fuente accesible con la nueva edición que se pu-blique.
En el caso de que se obtenga telemáticamente una copia de la lista en formatoelectrónico, ésta perderá el carácter de fuente de acceso público en el plazo de un año,contado desde el momento de su obtención.
4. Los datos que figuren en las guías de servicios de telecomunicaciones dis-ponibles al público se regirán por su normativa específica.
Artículo 29. Prestación de servicios de información sobre solvencia patrimonial ycrédito.
1. Quienes se dediquen a la prestación de servicios de información sobre la sol-vencia patrimonial y el crédito sólo podrán tratar datos de carácter personal obtenidosde los registros y las fuentes accesibles al público establecidos al efecto o proceden-tes de informaciones facilitadas por el interesado o con su consentimiento.
2. Podrán tratarse también datos de carácter personal relativos al cumpli-miento o incumplimiento de obligaciones dinerarias facilitados por el acreedor o porquien actúe por su cuenta o interés. En estos casos se notificará a los interesados res-pecto de los que hayan registrado datos de carácter personal en ficheros, en el plazode treinta días desde dicho registro, una referencia de los que hubiesen sido inclui-dos y se les informará de su derecho a recabar información de la totalidad de ellos, enlos términos establecidos por la presente Ley.
3. En los supuestos a que se refieren los dos apartados anteriores, cuando elinteresado lo solicite, el responsable del tratamiento le comunicará los datos, así como
224

las evaluaciones y apreciaciones que sobre el mismo hayan sido comunicadas durantelos últimos seis meses y el nombre y dirección de la persona o entidad a quien se ha-yan revelado los datos.
4. Sólo se podrán registrar y ceder los datos de carácter personal que sean de-terminantes para enjuiciar la solvencia económica de los interesados y que no se re-fieran, cuando sean adversos, a más de seis años, siempre que respondan converacidad a la situación actual de aquéllos.
Artículo 30. Tratamientos con fines de publicidad y de prospección comercial.
1. Quienes se dediquen a la recopilación de direcciones, reparto de documen-tos, publicidad, venta a distancia, prospección comercial y otras actividades análo-gas, utilizarán nombres y direcciones u otros datos de carácter personal cuando losmismos figuren en fuentes accesibles al público o cuando hayan sido facilitados porlos propios interesados u obtenidos con su consentimiento.
2. Cuando los datos procedan de fuentes accesibles al público de conformidadcon lo establecido en el párrafo segundo del artículo 5.5 de esta Ley, en cada comu-nicación que se dirija al interesado se informará del origen de los datos y de la iden-tidad del responsable del tratamiento, así como de los derechos que le asisten.
3. En el ejercicio del derecho de acceso los interesados tendrán derecho a co-nocer el origen de sus datos de carácter personal, así como del resto de informacióna que se refiere el artículo 15.
4. Los interesados tendrán derecho a oponerse, previa petición y sin gastos, altratamiento de los datos que les conciernan, en cuyo caso serán dados de baja del tra-tamiento, cancelándose las informaciones que sobre ellos figuren en aquél, a su sim-ple solicitud.
Artículo 31. Censo promocional.
1. Quienes pretendan realizar permanente o esporádicamente la actividad derecopilación de direcciones, reparto de documentos, publicidad, venta a distancia,prospección comercial u otras actividades análogas, podrán solicitar del Instituto Na-cional de Estadística o de los órganos equivalentes de las Comunidades Autónomas unacopia del censo promocional, formado con los datos de nombre, apellidos y domicilioque constan en el censo electoral.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
225

2. El uso de cada lista de censo promociona tendrá un plazo de vigencia de unaño. Transcurrido el plazo citado, la lista perderá su carácter de fuente de acceso pú-blico.
3. Los procedimientos mediante los que los interesados podrán solicitar no apa-recer en el censo promocional se regularán reglamentariamente. Entre estos procedimien-tos, que serán gratuitos para los interesados, se incluirá el documento deempadronamiento. Trimestralmente se editará una lista actualizada del censo promocio-nal, excluyendo los nombres y domicilios de los que así lo hayan solicitado.
4. Se podrá exigir una contraprestación por la facilitación de la citada lista en so-porte informático.
Artículo 32. Códigos tipo.
1. Mediante acuerdos sectoriales, convenios administrativos o decisiones de em-presa, los responsables de tratamientos de titularidad pública y privada, así como las or-ganizaciones en que se agrupen, podrán formular códigos tipo que establezcan lascondiciones de organización, régimen de funcionamiento, procedimientos aplicables, nor-mas de seguridad del entorno, programas o equipos, obligaciones de los implicados en eltratamiento y uso de la información personal, así como las garantías, en su ámbito, parael ejercicio de los derechos de las personas con pleno respeto a los principios y disposi-ciones de la presente Ley y sus normas de desarrollo.
2. Los citados códigos podrán contener o no reglas operacionales detalladas decada sistema particular y estándares técnicos de aplicación.
En el supuesto de que tales reglas o estándares no se incorporen directamente alcódigo, las instrucciones u órdenes que los establecieran deberán respetar los principiosfijados en aquél.
3. Los códigos tipo tendrán el carácter de códigos deontológicos o de buenapráctica profesional, debiendo ser depositados o inscritos en el Registro General deProtección de Datos y, cuando corresponda, en los creados a estos efectos por las Co-munidades Autónomas, de acuerdo con el artículo 41. El Registro General de Protec-ción de Datos podrá denegar la inscripción cuando considere que no se ajusta a lasdisposiciones legales y reglamentarias sobre la materia, debiendo, en este caso, el Di-rector de la Agencia de Protección de Datos requerir a los solicitantes para que efec-túen las correcciones oportunas.
226

TÍTULO V Movimiento internacional de datos
Artículo 33. Norma general.
1. No podrán realizarse transferencias temporales ni definitivas de datos de carác-ter personal que hayan sido objeto de tratamiento o hayan sido recogidos para someterlosa dicho tratamiento con destino a países que no proporcionen un nivel de protección equi-parable al que presta la presente Ley, salvo que, además de haberse observado lo dis-puesto en ésta, se obtenga autorización previa del Director de la Agencia de Protección deDatos, que sólo podrá otorgarla si se obtienen garantías adecuadas.
2. El carácter adecuado del nivel de protección que ofrece el país de destino seevaluará por la Agencia de Protección de Datos atendiendo a todas las circunstancias queconcurran en la transferencia o categoría de transferencia de datos. En particular, se to-mará en consideración la naturaleza de los datos, la finalidad y la duración del tratamientoo de los tratamientos previstos, el país de origen y el país de destino final, las normas dederecho, generales o sectoriales, vigentes en el país tercero de que se trate, el contenidode los informes de la Comisión de la Unión Europea, así como las normas profesionales ylas medidas de seguridad en vigor en dichos países.
Artículo 34. Excepciones.
Lo dispuesto en el artículo anterior no será de aplicación:
a) Cuando la transferencia internacional de datos de carácter personal resulte dela aplicación de tratados o convenios en los que sea parte España.
b) Cuando la transferencia se haga a efectos de prestar o solicitar auxilio judicialinternacional.
c) Cuando la transferencia sea necesaria para la prevención o para el diagnósticomédico, la prestación de asistencia sanitaria o tratamiento médico o la gestión de servi-cios sanitarios.
d) Cuando se refiera a transferencias dinerarias conforme a su legislación específica.
e) Cuando el afectado haya dado su consentimiento inequívoco a la transferenciaprevista.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
227

f) Cuando la transferencia sea necesaria para la ejecución de un contrato entre elafectado y el responsable del fichero o para la adopción de medidas precontractualesadoptadas a petición del afectado.
g) Cuando la transferencia sea necesaria para la celebración o ejecución de uncontrato celebrado o por celebrar, en interés del afectado, por el responsable del ficheroy un tercero.
h) Cuando la transferencia sea necesaria o legalmente exigida para la salvaguardade un interés público.
Tendrá esta consideración la transferencia solicitada por una Administración fis-cal o aduanera para el cumplimiento de sus competencias.
i) Cuando la transferencia sea precisa para el reconocimiento, ejercicio o defensade un derecho en un proceso judicial.
j) Cuando la transferencia se efectúe, a petición de persona con interés legítimo,desde un Registro público y aquélla sea acorde con la finalidad del mismo.
k) Cuando la transferencia tenga como destino un Estado miembro de la Unión Eu-ropea, o un Estado respecto del cual la Comisión de las Comunidades Europeas, en el ejer-cicio de sus competencias, haya declarado que garantiza un nivel de protección adecuado.
TÍTULO VI Agencia de Protección de Datos
Artículo 35. Naturaleza y régimen jurídico.
1. La Agencia de Protección de Datos es un ente de derecho público, con perso-nalidad jurídica propia y plena capacidad pública y privada, que actúa con plena indepen-dencia de las Administraciones públicas en el ejercicio de sus funciones. Se regirá por lodispuesto en la presente Ley y en un Estatuto propio, que será aprobado por el Gobierno.
2. En el ejercicio de sus funciones públicas, y en defecto de lo que disponga lapresente Ley y sus disposiciones de desarrollo, la Agencia de Protección de Datos actuará
228

de conformidad con la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Ad-ministraciones Públicas y del Procedimiento Administrativo Común. En sus adquisicionespatrimoniales y contratación estará sujeta al derecho privado.
3. Los puestos de trabajo de los órganos y servicios que integren la Agencia de Pro-tección de Datos serán desempeñados por funcionarios de las Administraciones públicasy por personal contratado al efecto, según la naturaleza de las funciones asignadas a cadapuesto de trabajo. Este personal está obligado a guardar secreto de los datos de carácterpersonal de que conozca en el desarrollo de su función.
4. La Agencia de Protección de Datos contará, para el cumplimiento de sus fines,con los siguientes bienes y medios económicos:
a) Las asignaciones que se establezcan anualmente con cargo a los PresupuestosGenerales del Estado.
b) Los bienes y valores que constituyan su patrimonio, así como los productos yrentas del mismo.
c) Cualesquiera otros que legalmente puedan serle atribuidos.
5. La Agencia de Protección de Datos elaborará y aprobará con carácter anual elcorrespondiente anteproyecto de presupuesto y lo remitirá al Gobierno para que sea inte-grado, con la debida independencia, en los Presupuestos Generales del Estado.
Artículo 36. El Director.
1. El Director de la Agencia de Protección de Datos dirige la Agencia y ostenta surepresentación. Será nombrado, de entre quienes componen el Consejo Consultivo, me-diante Real Decreto, por un período de cuatro años.
2. Ejercerá sus funciones con plena independencia y objetividad y no estará su-jeto a instrucción alguna en el desempeño de aquéllas.
En todo caso, el Director deberá oír al Consejo Consultivo en aquellas propuestasque éste le realice en el ejercicio de sus funciones.
3. El Director de la Agencia de Protección de Datos sólo cesará antes de la expi-ración del período a que se refiere el apartado 1, a petición propia o por separación acor-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
229

dada por el Gobierno, previa instrucción de expediente, en el que necesariamente seránoídos los restantes miembros del Consejo Consultivo, por incumplimiento grave de susobligaciones, incapacidad sobrevenida para el ejercicio de su función, incompatibilidad ocondena por delito doloso.
4. El Director de la Agencia de Protección de Datos tendrá la consideración de altocargo y quedará en la situación de servicios especiales si con anterioridad estuviera des-empeñando una función pública. En el supuesto de que sea nombrado para el cargo al-gún miembro de la carrera judicial o fiscal, pasará asimismo a la situación administrativade servicios especiales.
Artículo 37. Funciones.
Son funciones de la Agencia de Protección de Datos:
a) Velar por el cumplimiento de la legislación sobre protección de datos y contro-lar su aplicación, en especial en lo relativo a los derechos de información, acceso, recti-ficación, oposición y cancelación de datos.
b) Emitir las autorizaciones previstas en la Ley o en sus disposiciones reglamentarias.
c) Dictar, en su caso, y sin perjuicio de las competencias de otros órganos, las ins-trucciones precisas para adecuar los tratamientos a los principios de la presente Ley.
d) Atender las peticiones y reclamaciones formuladas por las personas afectadas.
e) Proporcionar información a las personas acerca de sus derechos en materia detratamiento de los datos de carácter personal.
f) Requerir a los responsables y los encargados de los tratamientos, previa audien-cia de éstos, la adopción de las medidas necesarias para la adecuación del tratamiento dedatos a las disposiciones de esta Ley y, en su caso, ordenar la cesación de los tratamien-tos y la cancelación de los ficheros, cuando no se ajuste a sus disposiciones.
g) Ejercer la potestad sancionadora en los términos previstos por el Título VII dela presente Ley.
h) Informar, con carácter preceptivo, los proyectos de disposiciones generales quedesarrollen esta Ley.
230

i) Recabar de los responsables de los ficheros cuanta ayuda e información estimenecesaria para el desempeño de sus funciones.
j) Velar por la publicidad de la existencia de los ficheros de datos con carácter per-sonal, a cuyo efecto publicará periódicamente una relación de dichos ficheros con la in-formación adicional que el Director de la Agencia determine.
k) Redactar una memoria anual y remitirla al Ministerio de Justicia.
l) Ejercer el control y adoptar las autorizaciones que procedan en relación con losmovimientos internacionales de datos, así como desempeñar las funciones de cooperacióninternacional en materia de protección de datos personales.
m) Velar por el cumplimiento de las disposiciones que la Ley de la Función Esta-dística Pública establece respecto a la recogida de datos estadísticos y al secreto estadís-tico, así como dictar las instrucciones precisas, dictaminar sobre las condiciones deseguridad de los ficheros constituidos con fines exclusivamente estadísticos y ejercer la po-testad a la que se refiere el artículo 46.
n) Cuantas otras le sean atribuidas por normas legales o reglamentarias.
Artículo 38. Consejo Consultivo.
El Director de la Agencia de Protección de Datos estará asesorado por un ConsejoConsultivo compuesto por los siguientes miembros:
Un Diputado, propuesto por el Congreso de los Diputados.
Un Senador, propuesto por el Senado.
Un representante de la Administración Central, designado por el Gobierno.
Un representante de la Administración Local, propuesto por la Federación Espa-ñola de Municipios y Provincias.
Un miembro de la Real Academia de la Historia, propuesto por la misma.
Un experto en la materia, propuesto por el Consejo Superior de Universidades.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
231

Un representante de los usuarios y consumidores, seleccionado del modo que seprevea reglamentariamente.
Un representante de cada Comunidad Autónoma que haya creado una agencia deprotección de datos en su ámbito territorial, propuesto de acuerdo con el procedimientoque establezca la respectiva Comunidad Autónoma.
Un representante del sector de ficheros privados, para cuya propuesta se seguiráel procedimiento que se regule reglamentariamente.
El funcionamiento del Consejo Consultivo se regirá por las normas reglamentariasque al efecto se establezcan.
Artículo 39. El Registro General de Protección de Datos.
1. El Registro General de Protección de Datos es un órgano integrado en la Agen-cia de Protección de Datos.
2. Serán objeto de inscripción en el Registro General de Protección de Datos:
a) Los ficheros de que sean titulares las Administraciones públicas.
b) Los ficheros de titularidad privada.
c) Las autorizaciones a que se refiere la presente Ley.
d) Los códigos tipo a que se refiere el artículo 32 de la presente Ley.
e) Los datos relativos a los ficheros que sean necesarios para el ejercicio de los de-rechos de información, acceso, rectificación, cancelación y oposición.
3. Por vía reglamentaria se regulará el procedimiento de inscripción de los ficheros,tanto de titularidad pública como de titularidad privada, en el Registro General de Protec-ción de Datos, el contenido de la inscripción, su modificación, cancelación, reclamacionesy recursos contra las resoluciones correspondientes y demás extremos pertinentes.
Artículo 40. Potestad de inspección.
1. Las autoridades de control podrán inspeccionar los ficheros a que hace referen-
232

cia la presente Ley, recabando cuantas informaciones precisen para el cumplimiento desus cometidos.
A tal efecto, podrán solicitar la exhibición o el envío de documentos y datos y exa-minarlos en el lugar en que se encuentren depositados, así como inspeccionar los equiposfísicos y lógicos utilizados para el tratamiento de los datos, accediendo a los locales dondese hallen instalados.
2. Los funcionarios que ejerzan la inspección a que se refiere el apartado anteriortendrán la consideración de autoridad pública en el desempeño de sus cometidos.
Estarán obligados a guardar secreto sobre las informaciones que conozcan en elejercicio de las mencionadas funciones, incluso después de haber cesado en las mismas.
Artículo 41. Órganos correspondientes de las Comunidades Autónomas.
1. Las funciones de la Agencia de Protección de Datos reguladas en el artículo 37,a excepción de las mencionadas en los apartados j), k) y 1), y en los apartados f) y g) en loque se refiere a las transferencias internacionales de datos, así como en los artículos 46 y49, en relación con sus específicas competencias serán ejercidas, cuando afecten a fiche-ros de datos de carácter personal creados o gestionados por las Comunidades Autónomas ypor la Administración Local de su ámbito territorial, por los órganos correspondientes decada Comunidad, que tendrán la consideración de autoridades de control, a los que garan-tizarán plena independencia y objetividad en el ejercicio de su cometido.
2. Las Comunidades Autónomas podrán crear y mantener sus propios registros deficheros para el ejercicio de las competencias que se les reconoce sobre los mismos.
3. El Director de la Agencia de Protección de Datos podrá convocar regularmente alos órganos correspondientes de las Comunidades Autónomas a efectos de cooperación insti-tucional y coordinación de criterios o procedimientos de actuación. El Director de la Agenciade Protección de Datos y los órganos correspondientes de las Comunidades Autónomas podránsolicitarse mutuamente la información necesaria para el cumplimiento de sus funciones.
Artículo 42. Ficheros de las Comunidades Autónomas en materia de su exclusiva competencia.
1. Cuando el Director de la Agencia de Protección de Datos constate que el man-tenimiento o uso de un determinado fichero de las Comunidades Autónomas contravienealgún precepto de esta Ley en materia de su exclusiva competencia podrá requerir a la Ad-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
233

ministración correspondiente que se adopten las medidas correctoras que determine enel plazo que expresamente se fije en el requerimiento.
2. Si la Administración pública correspondiente no cumpliera el requerimientoformulado, el Director de la Agencia de Protección de Datos podrá impugnar la resoluciónadoptada por aquella Administración.
TÍTULO VII Infracciones y sanciones
Artículo 43. Responsables.
1. Los responsables de los ficheros y los encargados de los tratamientos estaránsujetos al régimen sancionador establecido en la presente Ley.
2. Cuando se trate de ficheros de los que sean responsables las Administracionespúblicas se estará, en cuanto al procedimiento ya las sanciones, a lo dispuesto en el artí-culo 46, apartado 2.
Artículo 44. Tipos de infracciones.
1. Las infracciones se calificarán como leves, graves o muy graves.
2. Son infracciones leves:
a) No atender, por motivos formales, la solicitud del interesado de rectificación ocancelación de los datos personales objeto de tratamiento cuando legalmente proceda.
b) No proporcionar la información que solicite la Agencia de Protección de Datosen el ejercicio de las competencias que tiene legalmente atribuidas, en relación con as-pectos no sustantivos de la protección de datos.
c) No solicitar la inscripción del fichero de datos de carácter personal en el Re-gistro General de Protección de Datos, cuando no sea constitutivo de infracción grave.
d) Proceder a la recogida de datos de carácter personal de los propios afectadossin proporcionarles la información que señala el artículo 5 de la presente Ley.
234

e) Incumplir el deber de secreto establecido en el artículo 10 de esta Ley, salvo queconstituya infracción grave.
3. Son infracciones graves:
a) Proceder a la creación de ficheros de titularidad pública o iniciar la recogida dedatos de carácter personal para los mismos, sin autorización de disposición general, publi-cada en el «Boletín Oficial del Estado» o Diario oficial correspondiente.
b) Proceder a la creación de ficheros de titularidad privada o iniciar la recogida dedatos de carácter personal para los mismos con finalidades distintas de las que constituyenel objeto legítimo de la empresa o entidad.
c) Proceder a la recogida de datos de carácter personal sin recabar el consenti-miento expreso de las personas afectadas, en los casos en que éste sea exigible.
d) Tratar los datos de carácter personal o usarlos posteriormente con conculcaciónde los principios y garantías establecidos en la presente Ley o con incumplimiento de los pre-ceptos de protección que impongan las disposiciones reglamentarias de desarrollo, cuandono constituya infracción muy grave.
e) El impedimento o la obstaculización del ejercicio de los derechos de acceso y opo-sición y la negativa a facilitar la información que sea solicitada.
f) Mantener datos de carácter personal inexactos o no efectuar las rectificaciones ocancelaciones de los mismos que legalmente procedan cuando resulten afectados los dere-chos de las personas que la presente Ley ampara.
g) La vulneración del deber de guardar secreto sobre los datos de carácter personal in-corporados a ficheros que contengan datos relativos a la comisión de infracciones administra-tivas o penales, Hacienda Pública, servicios financieros, prestación de servicios de solvenciapatrimonial y crédito, así como aquellos otros ficheros que contengan un conjunto de datos decarácter personal suficientes para obtener una evaluación de la personalidad del individuo.
h) Mantener los ficheros, locales, programas o equipos que contengan datos de carác-ter personal sin las debidas condiciones de seguridad que por vía reglamentaria se determinen.
i) No remitir a la Agencia de Protección de Datos las notificaciones previstas enesta Ley o en sus disposiciones de desarrollo, así como no proporcionar en plazo a la
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
235

misma cuantos documentos e informaciones deba recibir o sean requeridos por aquéla tales efectos.
j) La obstrucción al ejercicio de la función inspectora.
k) No inscribir el fichero de datos de carácter personal en el Registro Generalde Protección Datos, cuando haya sido requerido para ello por el Director de la Agen-cia de Protección de Datos.
l) Incumplir el deber de información que se establece en los artículos 5,28 y 29 de esta Ley, cuando los datos hayan sido recabados de persona distinta delafectado.
4. Son infracciones muy graves:
a) La recogida de datos en forma engañosa y fraudulenta.
b) La comunicación o cesión de los datos de carácter personal, fuera de los ca-sos en que estén permitidas.
c) Recabar y tratar los datos de carácter personal a los que se refiere el apar-tado 2 del artículo 7 cuando no medie el consentimiento expreso del afectado; reca-bar y tratar los datos referidos en el apartado 3 del artículo 7 cuando no lo dispongauna ley o el afectado no haya consentido expresamente, o violentar la prohibición con-tenida en el apartado 4 del artículo 7.
d) No cesar en el uso ilegítimo de los tratamientos de datos de carácter perso-nal cuando sea requerido para ello por el Director de la Agencia de Protección de Da-tos o por las personas titulares del derecho de acceso.
e) La transferencia temporal o definitiva de datos de carácter personal que ha-yan sido objeto de tratamiento o hayan sido recogidos para someterlos a dicho trata-miento, con destino a países que no proporcionen un nivel de protección equiparablesin autorización del Director de la Agencia de Protección de Datos.
f) Tratar los datos de carácter personal de forma ilegítima o con menospreciode los principios y garantías que les sean de aplicación, cuando con ello se impida ose atente contra el ejercicio de los derechos fundamentales.
236

g) La vulneración del deber de guardar secreto sobre los datos de carácter perso-nal a que hacen referencia los apartados 2 y 3 del artículo 7, así como los que hayan sidorecabados para fines policiales sin consentimiento de las personas afectadas.
h) No atender, u obstaculizar de forma sistemática el ejercicio de los derechos deacceso, rectificación, cancelación u oposición.
i) No atender de forma sistemática el deber legal de notificación de la inclusiónde datos de carácter personal en un fichero.
Artículo 45. Tipo de sanciones.
1. Las infracciones leves serán sancionadas con multa de 100.000 a 10.000.000de pesetas.
2. Las infracciones graves serán sancionadas con multa de 10.000.000 a50.000.000 de pesetas.
3. Las infracciones muy graves serán sancionadas con multa de 50.000.000 a100.000.000 de pesetas.
4. La cuantía de las sanciones se graduará atendiendo a la naturaleza de los de-rechos personales afectados, al volumen de los tratamientos efectuados, a los beneficiosobtenidos, al grado de intencionalidad, a la reincidencia, a los daños y perjuicios causa-dos a las personas interesadas y a terceras personas, y a cualquier otra circunstancia quesea relevante para determinar el grado de antijuridicidad y de culpabilidad presentes enla concreta actuación infractora.
5. Si, en razón de las circunstancias concurrentes, se apreciara una cualificadadisminución de la culpabilidad del imputado o de la antijuridicidad del hecho, el órganosancionador establecerá la cuantía de la sanción aplicando la escala relativa a la clase deinfracciones que preceda inmediatamente en gravedad a aquella en que se integra la con-siderada en el caso de que se trate.
6. En ningún caso podrá imponerse una sanción más grave que la fijada en la Leypara la clase de infracción en la que se integre la que se pretenda sancionar.
7. El Gobierno actualizará periódicamente la cuantía de las sanciones de acuerdocon las variaciones que experimenten los índices de precios.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
237

Artículo 46. Infracciones de las Administraciones públicas.
1. Cuando las infracciones a que se refiere el artículo 4 fuesen cometidas en fi-cheros de los que sean responsables las Administraciones públicas, el Director de la Agen-cia de Protección de Datos dictará una resolución estableciendo las medidas que procedeadoptar para que cesen o se corrijan los efectos de la infracción.
Esta resolución se notificará al responsable del fichero, al órgano del que dependajerárquicamente y a los afectados si los hubiera.
2. El Director de la Agencia podrá proponer también la iniciación de actuacionesdisciplinarias, si procedieran.
El procedimiento y las sanciones a aplicar serán las establecidas en la legislaciónsobre régimen disciplinario de las Administraciones públicas.
3. Se deberán comunicar a la Agencia las resoluciones que recaigan en relacióncon las medidas y actuaciones a que se refieren los apartados anteriores.
4. El Director de la Agencia comunicará al Defensor del Pueblo las actuacionesque efectúe y las resoluciones que dicte al amparo de los apartados anteriores.
Artículo 47. Prescripción.
1. Las infracciones muy graves prescribirán a los tres años, las graves a los dosaños y las leves al año.
2. El plazo de prescripción comenzará a contarse desde el día en que la infrac-ción se hubiera cometido.
3. Interrumpirá la prescripción la iniciación, con conocimiento del interesado, delprocedimiento sancionador, reanudándose el plazo de prescripción si el expediente san-cionador estuviere paralizado durante más de seis meses por causas no imputables al pre-sunto infractor.
4. Las sanciones impuestas por faltas muy graves prescribirán a los tres años, lasimpuestas por faltas graves a los dos años y las impuestas por faltas leves al año.
5. El plazo de prescripción de las sanciones comenzará a contarse desde el día si-guiente a aquel en que adquiera firmeza la resolución por la que se impone la sanción.
238

6. La prescripción se interrumpirá por la iniciación, con conocimiento del intere-sado, del procedimiento de ejecución, volviendo a transcurrir el plazo si el mismo está pa-ralizado durante más de seis meses por causa no imputable al infractor.
Artículo 48. Procedimiento sancionador.
1. Por vía reglamentaria se establecerá el procedimiento a seguir para la determi-nación de las infracciones y la imposición de las sanciones a que hace referencia el pre-sente Título.
2. Las resoluciones de la Agencia de Protección de Datos u órgano correspon-diente de la Comunidad Autónoma agotan la vía administrativa.
Artículo 49. Potestad de inmovilización de ficheros.
En los supuestos, constitutivos de infracción muy grave, de utilización o cesión ilí-cita de los datos de carácter personal en que se impida gravemente o se atente de igual modocontra el ejercicio de los derechos de los ciudadanos y el libre desarrollo de la personalidadque la Constitución y las leyes garantizan, el Director de la Agencia de Protección de Datospodrá, además de ejercer la potestad sancionadora, requerir a los responsables de ficherosde datos de carácter personal, tanto de titularidad pública como privada, la cesación en lautilización o cesión ilícita de los datos. Si el requerimiento fuera desatendido, la Agencia deProtección de Datos podrá, mediante resolución motivada, inmovilizar tales ficheros a los so-los efectos de restaurar los derechos de las personas afectadas.
Disposición adicional primera. Ficheros preexistentes.
Los ficheros y tratamientos automatizados inscritos o no en el Registro General deProtección de Datos deberán adecuarse a la presente Ley Orgánica dentro del plazo de tresaños, a contar desde su entrada en vigor.
En dicho plazo, los ficheros de titularidad privada deberán ser comunicados a laAgencia de Protección de Datos y las Administraciones públicas, responsables de fiche-ros de titularidad pública, deberán aprobar la pertinente disposición de regulación del fi-chero o adaptar la existente.
En el supuesto de ficheros y tratamientos no automatizados, su adecuación a lapresente Ley Orgánica, y la obligación prevista en el párrafo anterior deberán cumplimen-tarse en el plazo de doce años a contar desde el 24 de octubre de 1995, sin perjuicio delejercicio de los derechos de acceso, rectificación y cancelación por parte de los afectados.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
239

Disposición adicional segunda. Ficheros y Registro de Población de las Administracionespúblicas.
1. La Administración General del Estado y las Administraciones de las ComunidadesAutónomas podrán solicitar al Instituto Nacional de Estadística, sin consentimiento del inte-resado, una copia actualizada del fichero formado con los datos del nombre, apellidos, domi-cilio, sexo y fecha de nacimiento que constan en los padrones municipales de habitantes y enel censo electoral correspondientes a los territorios donde ejerzan sus competencias, para la cre-ación de ficheros o registros de población.
2. Los ficheros o registros de población tendrán como finalidad la comunicación de losdistintos órganos de cada Administración pública con los interesados residentes en los respec-tivos territorios, respecto a las relaciones jurídico administrativas derivadas de las competen-cias respectivas de las Administraciones públicas.
Disposición adicional tercera. Tratamiento de los expedientes de las derogadas Leyes de Vagosy Maleantes y de Peligrosidad y Rehabilitación Social.
Los expedientes específicamente instruidos al amparo de las derogadas Leyes de Va-gos y Maleantes, y de Peligrosidad y Rehabilitación Social, que contengan datos de cualquieríndole susceptibles de afectar a la seguridad, al honor, a la intimidad o a la imagen de las per-sonas, no podrán ser consultados sin que medie consentimiento expreso de los afectados, o ha-yan transcurrido cincuenta años desde la fecha de aquéllos.
En este último supuesto, la Administración General del Estado, salvo que haya cons-tancia expresa del fallecimiento de los afectados, pondrá a disposición del solicitante la docu-mentación, suprimiendo de la misma los datos aludidos en el párrafo anterior, mediante lautilización de los procedimientos técnicos pertinentes en cada caso.
Disposición adicional cuarta. Modificación del artículo 112.4 de la Ley General Tributaria.
El apartado cuarto del artículo 112 de la Ley General Tributaria pasa a tener la si-guiente redacción:
«4. La cesión de aquellos datos de carácter personal, objeto de tratamiento, que sedebe efectuar a la Administración tributaria conforme a lo dispuesto en el artículo 111, en losapartados anteriores de este artículo o en otra norma de rango legal, no requerirá el consenti-miento del afectado.
En este ámbito tampoco será de aplicación lo que respecto a las Administraciones pú-
240

blicas establece el apartado 1 del artículo 21 de la Ley Orgánica de Protección de Datos de ca-rácter personal.»
Disposición adicional quinta. Competencias del Defensor del Pueblo y órganos auto-nómicos semejantes.
Lo dispuesto en la presente Ley Orgánica se entiende sin perjuicio de las competen-cias del Defensor del Pueblo y de los órganos análogos de las Comunidades Autónomas.
Disposición adicional sexta. Modificación del artículo 24.3 de la Ley de Ordenación ySupervisión de los Seguros Privados.
Se modifica el artículo 24.3, párrafo 2.º de la Ley 30/1995, de 8 de noviembre, deOrdenación y Supervisión de los Seguros Privados, con la siguiente redacción:
«Las entidades aseguradoras podrán establecer ficheros comunes que contengan da-tos de carácter personal para la liquidación de siniestros y la colaboración estadístico actua-rial con la finalidad de permitir la tarificación y selección de riesgos y la elaboración deestudios de técnica aseguradora. La cesión de datos a los citados ficheros no requerirá el con-sentimiento previo del afectado, pero sí la comunicación al mismo de la posible cesión de susdatos personales a ficheros comunes para los fines señalados con expresa indicación del res-ponsable para que se puedan ejercitar los derechos de acceso, rectificación y cancelaciónprevistos en la ley.
También podrán establecerse ficheros comunes cuya finalidad sea prevenir el fraudeen el seguro sin que sea necesario el consentimiento del afectado. No obstante, será necesa-ria en estos casos la comunicación al afectado, en la primera introducción de sus datos, dequién sea el responsable del fichero y de las formas de ejercicio de los derechos de acceso, rec-tificación y cancelación.
En todo caso, los datos relativos a la salud sólo podrán ser objeto de tratamiento conel consentimiento expreso del afectado.»
Disposición transitoria primera. Tratamientos creados por Convenios internacionales.
La Agencia de Protección de Datos será el organismo competente para la protecciónde las personas físicas en lo que respecta al tratamiento de datos de carácter personal res-pecto de los tratamientos establecidos en cualquier Convenio Internacional del que sea parteEspaña que atribuya a una autoridad nacional de control esta competencia, mientras no secree una autoridad diferente para este cometido en desarrollo del Convenio.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
241

Disposición transitoria segunda. Utilización del censo promocional.
Reglamentariamente se desarrollarán los procedimientos de formación del censopromocional, de oposición a aparecer en el mismo, de puesta a disposición de sus solici-tantes, y de control de las listas difundidas.
El Reglamento establecerá los plazos para la puesta en operación del censo pro-mocional.
Disposición transitoria tercera. Subsistencia de normas preexistentes.
Hasta tanto se lleven a efectos las previsiones de la disposición final primera deesta Ley, continuarán en vigor, con su propio rango, las normas reglamentarias existentesy, en especial, los Reales Decretos 428/1993, de 26 de marzo; 1332/1994, de 20 de ju-nio, y 994/1999, de 11 de junio, en cuanto no se opongan a la presente Ley.
Disposición derogatoria única. Derogación normativa.
Queda derogada la Ley Orgánica 5/1992, de 29 de octubre, de Regulación del tra-tamiento automatizado de los datos de carácter personal.
Disposición final primera. Habilitación para el desarrollo reglamentario.
El Gobierno aprobará, o modificará, las disposiciones reglamentarias necesariaspara la aplicación y desarrollo de la presente Ley.
Disposición final segunda. Preceptos con carácter de Ley ordinaria.
Los Títulos IV, VI excepto el último inciso del párrafo 4 del artículo 36 y VII de lapresente Ley, la disposición adicional cuarta, la disposición transitoria primera y la finalprimera tienen el carácter de Ley ordinaria.
Disposición final tercera. Entrada en vigor.
La presente Ley entrará en vigor en el plazo de un mes, contado desde su publi-cación en el «Boletín Oficial del Estado».
Por tanto, Mando a todos los españoles, particulares y autoridades, que guarden y hagan
guardar esta Ley Orgánica. Madrid, 13 de diciembre de 1999.
JUAN CARLOS R
242

ORDEN SCO/256/2007, DE 5 DE FEBRERO, POR LA QUE SE ESTABLECEN LOS PRINCIPIOS Y LAS DIRECTRICES DETALLADAS DE BUENA PRÁCTICA CLÍNICA Y LOS REQUISITOS PARA
AUTORIZAR LA FABRICACIÓN O IMPORTACIÓN DE MEDICAMENTOS EN INVESTIGACIÓN
DE USO HUMANO
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
243


ORDEN SCO/256/2007, DE 5 DE FEBRERO, POR LA QUE SE ESTABLECENLOS PRINCIPIOS Y LAS DIRECTRICES DETALLADAS DE BUENA PRÁCTICACLÍNICA Y LOS REQUISITOS PARA AUTORIZAR LA FABRICACIÓN O IMPOR-TACIÓN DE MEDICAMENTOS EN INVESTIGACIÓN DE USO HUMANO
Los ensayos clínicos con medicamentos de uso humano están regulados en el Tí-tulo III de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamen-tos y productos sanitarios, que recoge en su articulado parte muy importante de losprincipios garantistas que ya contaban con previsiones normativas precedentes. En parti-cular, el vigente Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensa-yos clínicos con medicamentos, que incorporó al ordenamiento jurídico español la Directiva2001/20/CE del Parlamento Europeo y del Consejo, de 4 de abril de 2001, relativa a laaproximación de las disposiciones legales, reglamentarias y administrativas de los Esta-dos miembros sobre la aplicación de buenas prácticas clínicas en la realización de ensa-yos clínicos de medicamentos de uso humano. Esta directiva estableció la obligación deadoptar los principios y directrices de buena práctica clínica y de las normas de correctafabricación de medicamentos en investigación, asi como las directrices detalladas para laautorización de la fabricación o importación de medicamentos en investigación.
Posteriormente, el Real Decreto 2183/2004, de 12 de noviembre, por el que semodifica el Real Decreto 1564/1992, de 18 de diciembre, por el que se desarrolla y re-gula el régimen de autorización de los laboratorios farmacéuticos e importadores de me-dicamentos y la garantía de calidad en su fabricación industrial, transpuso la Directiva2003/94/CE de la Comisión, de 8 de octubre de 2003, por la que se establecen los prin-cipios y directrices de las prácticas correctas de fabricación de los medicamentos de usohumano y de los medicamentos en investigación de uso humano.
Mas recientemente, la Directiva 2005/28/CE de la Comisión, de 8 de abril de2005, por la que se establecen los principios y las directrices detalladas de las buenasprácticas clínicas respecto a los medicamentos en investigación de uso humano, así comolos requisitos para autorizar la fabricación o importación de dichos productos, incluye asi-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
245

mismo las directrices detalladas sobre la documentación relativa a los ensayos clínicos,el archivo, la cualificación de los inspectores y los procedimientos de inspección.
Mediante esta disposición se incorpora al ordenamiento jurídico interno la Direc-tiva 2005/28/CE de la Comisión, de 8 de abril de 2005.
En la aplicación de los requisitos y las directrices contempladas en esta Orden sedeberán tener en cuenta las directrices comunitarias publicadas por la Comisión Europeaen los distintos volúmenes de las normas sobre medicamentos de la Unión Europea.
En la tramitación de esta Orden han sido oídos los sectores afectados.
Esta Orden se dicta al amparo de lo establecido en la disposición final segundadel Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicoscon medicamentos.
En su virtud, con la aprobación previa del Ministro de Administraciones Públicasy de acuerdo con el Consejo de Estado, dispongo:
ARTÍCULO 1. Objeto.
Esta Orden regula los principios y las directrices de buena práctica clínica en losensayos clínicos con medicamentos en investigación de uso humano, los requisitos paraautorizar la fabricación o importación de estos medicamentos, las directrices detalladassobre la documentación relativa a dichos ensayos, así como su archivo, y la cualificaciónde los inspectores y los procedimientos de inspección.
ARTÍCULO 2. Principios y directrices de buena práctica clínica.
En la realización de ensayos clínicos con medicamentos en investigación de usohumano se deberá observar lo siguiente:
a. Los derechos, la seguridad y el bienestar de los sujetos del ensayo prevalece-rán por encima de los intereses de la ciencia y de la sociedad.
b. Cada persona que participe en la realización de un ensayo estará capacitada porsu titulación, formación y experiencia para ejecutar sus tareas.
c. Los ensayos clínicos, en todos sus aspectos, deberán tener una sólida basecientífica y deberán regirse por principios éticos.
246

d. Deberán adoptarse los procedimientos necesarios que aseguren la calidad decada uno de los aspectos del ensayo clínico.
e. La información disponible sobre un medicamento en investigación, tanto clí-nica como no clínica, deberá ser adecuada para avalar el ensayo clínico propuesto.
f. Los ensayos clínicos deberán realizarse de acuerdo con la Declaración de Hel-sinki sobre los principios éticos para las investigaciones médicas en seres humanos,aprobada por la Asamblea General de la Asociación Médica Mundial.
g. El protocolo deberá establecer los criterios de inclusión, exclusión y retiradade los sujetos que participen en un ensayo, el plan de monitorización y el plan de pu-blicación.
h. El investigador y el promotor tendrán en cuenta todas las instrucciones parala realización de ensayos clínicos en España o, en su caso, las directrices de la Comi-sión Europea relativas al inicio, realización y finalización del ensayo clínico publicadaspor la Agencia Española de Medicamentos y Productos Sanitarios.
i. Toda la información sobre el ensayo clínico deberá registrarse, tratarse y con-servarse de forma que pueda ser comunicada, evaluada y verificada de manera precisa,protegiendo al mismo tiempo el carácter confidencial de los registros de los sujetos delensayo.
j. Los Comités Éticos de Investigación Clínica adoptarán los procedimientos nor-malizados de trabajo necesarios para la realización de sus funciones, de acuerdo con loestablecido en el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los en-sayos clínicos con medicamentos.
Los Comités conservarán todos los documentos esenciales, relacionados concada ensayo clínico evaluado, durante al menos tres años tras la finalización del mismo,o durante un periodo más largo si así lo establece la Agencia Española de Medicamen-tos y Productos Sanitarios o la autoridad competente de la comunidad autónoma co-rrespondiente. Esta documentación debe archivarse preferentemente agrupada porprotocolos, en un lugar que permita garantizar la confidencialidad de la información du-rante el tiempo de archivo requerido. En caso de cese de la actividad del Comité, la ins-titución en la que esté constituido el Comité debe mantener el archivo de ladocumentación durante el plazo establecido. Para cada ensayo clínico debe incluir, comomínimo, lo siguiente:
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
247

El protocolo, las modificaciones y toda la documentación presentada por el pro-motor o su representante legal.
Los dictámenes o informes emitidos por el Comité (Comité Ético de InvestigaciónClínica de Referencia si el ensayo clínico es multicéntrico), especificando la versión de losdocumentos revisados.
Copia de la correspondencia con el investigador y/o el promotor o su representante.
La documentación relativa a las actividades de seguimiento del ensayo clínico.
El informe anual sobre la marcha del ensayo clínico.
Copia de cualquier correspondencia con la Agencia Española de Medicamentos yProductos Sanitarios y/o con las autoridades competentes de la comunidad autónomadonde este localizado el comité.
Copia de las notificaciones y correspondencia relevantes con los Comités implica-dos y/o el Comité de referencia, de un ensayo multicéntrico.
Las sospechas de reacciones adversas recibidas y los informes de seguridad pre-sentados por el promotor.
Notificación de la finalización del ensayo clínico, ya sea prematura o programada.
Resumen del informe final del ensayo clínico presentado por el promotor.
Cualquier otra documentación relevante.
Los Comités deben mantener archivada la documentación relacionada con su fun-cionamiento y actividad. En caso de cese de la misma, esta documentación debe conser-varse en la institución durante al menos tres años, transcurridos desde la finalización delúltimo estudio evaluado. Debe incluir como mínimo, lo siguiente:
Resoluciones de acreditación y de posteriores modificaciones.
Currículum vitae de los miembros actuales o que hayan pertenecido al Comité.
Convocatoria y actas de las reuniones del Comité.
248

Procedimientos normalizados de trabajo del Comité, versión actual y archivo histórico.
k. La Agencia Española de Medicamentos y Productos Sanitarios establecerá losprocedimientos que garanticen la transmisión de información entre la Agencia y los Co-mités Éticos de Investigación Clínica.
l. El promotor de un ensayo clínico podrá delegar la totalidad o una parte de susfunciones en un particular, empresa, institución u organismo, que deberá poner en mar-cha un sistema de garantía y control de calidad. Las obligaciones del promotor estable-cidas en las normas de buena práctica clínica que se hayan delegado serán deaplicación al particular, empresa, institución u organismo contratado. No obstante, enestos casos, el promotor seguirá siendo el responsable de garantizar que la realizacióndel ensayo clínico y los datos finales generados en dicho estudio se ajustan a lo dis-puesto en el Real Decreto 223/2004, de 6 de febrero, y en esta Orden. Cualquier trans-ferencia de funciones del promotor en relación con un ensayo clínico debe quedarespecíficamente documentada.
m. El investigador y el promotor podrán ser la misma persona física.
n. La información del manual del investigador y sus actualizaciones se presenta-rán de forma concisa, sencilla, objetiva, equilibrada y no promocional, comprensible paralos posibles investigadores y que les permita realizar una evaluación no sesgada de los ries-gos y beneficios y de la pertinencia del ensayo clínico propuesto. Este documento servirácomo referencia para la evaluación del carácter esperado, o no, de las reacciones adver-sas graves que pudieran ocurrir durante la realización del ensayo.
ñ. En caso de medicamentos autorizados en algún Estado miembro de la UniónEuropea, cuando el medicamento se utilice en las condiciones de uso autorizadas, el ma-nual del investigador podrá reemplazarse por la ficha técnica autorizada.
o. El manual del investigador debe validarse y actualizarse de manera regular porel promotor, al menos una vez al año.
ARTÍCULO 3. Requisitos de la solicitud para la autorización de fabricación o importación.
1. Será necesaria la autorización previa otorgada por la Agencia Española de Me-dicamentos y Productos Sanitarios para la fabricación total o parcial, incluyendo las di-versas operaciones de división, acondicionamiento o presentación, de medicamentos eninvestigación, incluso en los casos en que los medicamentos fabricados estén destinados
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
249

a la exportación. La autorización será necesaria también para las importaciones proce-dentes de terceros países.
2. No se exigirá la citada autorización para el acondicionamiento final, en caso deque se realice en un Servicio de Farmacia autorizado, siempre que los medicamentos eninvestigación estén destinados a ser utilizados únicamente en un centro sanitario depen-diente de dicho servicio.
Cuando en el contexto de un ensayo clínico específico cuyo promotor sea un in-vestigador o un grupo de investigadores, un Servicio de Farmacia autorizado desee reali-zar una operación de fabricación distinta de las contempladas anteriormente, deberásolicitar una autorización previa a la Agencia Española de Medicamentos y Productos Sa-nitarios y únicamente se podrá utilizar el medicamento en el ensayo clínico concreto. LaAgencia Española de Medicamentos y Productos Sanitarios acordará con las comunidadesautónomas los procedimientos de verificación de las normas de correcta fabricación en es-tos casos.
3. El solicitante deberá disponer de manera permanente y continua de una per-sona cualificada o de un director técnico, conforme a lo establecido en el Real Decreto1564/1992, de 18 de diciembre, por el que se desarrolla y regula el régimen de autori-zación de los laboratorios farmacéuticos e importadores de medicamentos y la garantía ensu fabricación industrial.
4. La solicitud para la autorización de fabricación o importación deberá incorpo-rar los datos y documentos siguientes:
Los tipos de medicamentos y formas farmacéuticas que se pretenden fabricar oimportar.
Las operaciones de fabricación o importación de que se trate.
El proceso de fabricación, cuando proceda, como en el caso de inactivación deagentes virales o no convencionales.
La identificación del lugar en el que se fabricarán los productos, que deberá con-tar, para la fabricación o importación de los medicamentos y formas farmacéuticas, de loslocales, el equipo técnico y los medios de control adecuados y suficientes que se ajustena los requisitos que establece el Real Decreto 1564/1992, de 18 de diciembre, en lo querespecta a la fabricación, el control y el almacenamiento de los productos.
250

5. Los tipos de medicamentos referidos en el apartado 4.a de este artículo in-cluyen medicamentos hemoderivados, inmunológicos, de terapia celular, de terapiagénica, biotecnológicos, de origen humano o animal, plantas medicinales, homeopá-ticos, radiofármacos y de origen químico.
6. La autorización establecida en el apartado 1 de este artículo se notificaráen el plazo de 90 días desde la presentación de una solicitud válida, por la AgenciaEspañola de Medicamentos y Productos Sanitarios, una vez comprobada la exactitudde los datos facilitados por el solicitante. En caso de que la Agencia precise datoscomplementarios, el plazo para resolver quedará en suspenso hasta que se faciliten losmismos.
La citada autorización podrá estar condicionada al cumplimiento de determi-nadas obligaciones impuestas en el momento de la concesión de la autorización o enuna fecha posterior, con el fin de garantizar el cumplimiento de los requisitos estable-cidos en la misma.
La autorización únicamente será válida para las instalaciones, los tipos de me-dicamentos y las formas farmacéuticas solicitadas.
ARTÍCULO 4. Obligaciones del titular de la autorización de fabricación o importación.
1. El titular de la autorización deberá cumplir, como mínimo, los requisitos si-guientes:
Disponer de los medios personales, materiales y técnicos necesarios, que res-pondan a las exigencias legales para la fabricación y control establecidos en el RealDecreto 1564/1992, de 18 de diciembre.
Disponer de los medicamentos en investigación o autorizados, conforme a lodispuesto en el Real Decreto 223/2004, de 6 de febrero.
Permitir en todo momento el acceso a sus instalaciones a los inspectores.
Permitir que la persona cualificada o el director técnico pueda cumplir las fun-ciones establecidas en función de su cargo.
Respetar los principios y directrices de las normas de correcta fabricación demedicamentos previstos en la normativa europea.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
251

2. Para modificar los requisitos establecidos en los apartados 3 y 4 del artículo 3,el titular de una autorización de fabricación deberá solicitarlo a la Agencia Española deMedicamentos y Productos Sanitarios. En el caso de que la modificación se refiera a lostipos y formas farmacéuticas autorizadas o afecte al director técnico, la duración del pro-cedimiento relativo a esta solicitud no será superior a 30 días. En casos excepcionales,este plazo podrá ser prorrogado hasta 90 días.
3. El incumplimiento de los requisitos establecidos será causa de suspensión o re-vocación de la autorización de fabricación de forma parcial o total.
4. Sólo podrán importar medicamentos en investigación los importadores que es-tén en posesión de la autorización de importación prevista en el artículo 3, otorgada porla Agencia Española de Medicamentos y Productos Sanitarios.
ARTÍCULO 5. Adquisición de medicamentos para ensayos clínicos.
1. Medicamentos comercializados en España y en el resto de la Unión Europea:Cuando los medicamentos que se vayan a utilizar en un ensayo clínico estén comerciali-zados en España o en otro Estado miembro de la Unión Europea, el promotor de un en-sayo clínico podrá obtenerlos bien a través de un distribuidor autorizado, tal como unalmacén mayorista, o directamente del titular de la autorización de comercialización, pre-via presentación de la autorización de la Agencia Española de Medicamentos y ProductosSanitarios para ese ensayo clínico.
2. Medicamentos procedentes de un tercer país: Podrá importar medicamentos eninvestigación un importador autorizado por la Agencia Española de Medicamentos y Pro-ductos Sanitarios, previa solicitud de la importación del medicamento en investigación deun ensayo clínico a la Agencia.
Para obtener la autorización de importación de medicamentos procedentes deun tercer país sin un acuerdo de mutuo reconocimiento vigente, es necesario que aporteun certificado del director técnico o persona cualificada en el que se acredite que cadalote importado se ha fabricado en unas condiciones equivalentes a las normas de co-rrecta fabricación de la Unión Europea o, si no se puede emitir ese certificado, un certi-ficado de liberación de estos lotes firmado también por el director técnico, en el que seacredite que se han llevado a cabo, en la Unión Europea, los análisis, pruebas o contro-les necesarios para evaluar su calidad conforme a la información presentada para la au-torización del ensayo.
252

ARTÍCULO 6. Archivo maestro del ensayo y métodos de archivo.
1. La documentación relativa al ensayo clínico constituye el archivo maestro delmismo y constará de los documentos esenciales que permitan la evaluación de la realiza-ción de un ensayo clínico y de la calidad de los datos obtenidos. Estos documentos debe-rán demostrar el cumplimiento por parte del investigador y el promotor de los principiosy directrices de buena práctica clínica y de todos los requisitos aplicables y, en particu-lar, del anexo II sobre normas y protocolos analíticos, farmacotoxicológicos y clínicos re-lativos a la realización de pruebas de medicamentos del Real Decreto 767/1993, de 21de mayo, por el que se regula la evaluación, autorización, registro y condiciones de dis-pensación de especialidades farmacéuticas y otros medicamentos de uso humano fabri-cados industrialmente.
2. El archivo maestro del ensayo proporcionará la base para las auditorias quepueda realizar el auditor independiente del promotor y las inspecciones de las autorida-des competentes.
3. El contenido de los documentos esenciales deberá ajustarse al carácter especí-fico de cada fase del ensayo clínico. Se deberán tener en cuenta las orientaciones suple-mentarias sobre el contenido de dichos documentos publicados por la Comisión Europea.
4. El promotor y el investigador conservarán los documentos esenciales de cadaensayo clínico durante al menos cinco años tras la finalización del ensayo, o durante unperíodo más largo si así lo disponen otros requisitos aplicables, como en el caso de queel estudio se presente como base para el registro de un medicamento en que se deberácumplir el anexo II del Real Decreto 767/1993, de 21 de mayo, o un acuerdo entre el pro-motor, el investigador y el centro.
5. Los documentos esenciales deberán archivarse de forma que se puedan ponerfácilmente a disposición de las autoridades competentes, en caso de que los soliciten.
6. La historia clínica del sujeto del ensayo deberá ser custodiada con arreglo a lodispuesto en la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía delpaciente y de derechos y obligaciones en materia de información y documentación clínica,y conforme al período máximo permitido por el hospital, la institución o la consulta privada.
7. Todos los cambios en la titularidad de los datos y documentos deberán docu-mentarse. El nuevo titular asumirá las responsabilidades de las tareas de archivo y con-servación de los datos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
253

8. El promotor nombrará a la persona de su organización responsable de los archi-vos y el acceso a los mismos deberá limitarse a las personas designadas.
9. Los soportes utilizados para conservar los documentos esenciales deberán ga-rantizar que los documentos permanecen completos y legibles durante el período previstode conservación y que estén a disposición de las autoridades competentes en caso de quelos soliciten. Cualquier modificación de los registros habrá de ser trazable, permitiendo co-nocer el dato inicial y el corregido, así como la fecha y la firma del autor.
ARTÍCULO 7. Titulación, formación y experiencia de los inspectores de buena práctica clínica.
1. Los inspectores deberán tener la debida cualificación y formación universitariaen medicina, farmacia, farmacología, toxicología u otras disciplinas pertinentes.
2. Las administraciones sanitarias garantizarán la formación de los inspectores, ase-gurándose de que:
Reciban la formación adecuada, se evalúen periódicamente sus necesidades de for-mación y se adopten medidas adecuadas para mantener y mejorar su cualificación.
Tengan información referente a:
Los principios y procedimientos referentes al desarrollo de medicamentos y de la in-vestigación clínica.
La legislación europea y nacional aplicable a ensayos clínicos.
Las directrices aplicables a la realización de ensayos clínicos y a la concesión de au-torizaciones de comercialización.
Los procedimientos y sistemas de registro de datos clínicos, así como sobre la or-ganización y regulación del sistema de asistencia sanitaria de su competencia y, cuandoproceda, de terceros países.
3. Las administraciones sanitarias garantizarán que los inspectores tienen experien-cia previa suficiente para llevar a cabo una inspección.
4. Las administraciones sanitarias competentes deberán designar los inspectores.A tal efecto mantendrán un registro actualizado que incluya la titulación, formación y expe-riencia del inspector en temas de inspección de buena práctica clínica.
254

ARTÍCULO 8. Normas comunes para la inspección de buena práctica clínica.
1. Para armonizar la realización de inspecciones por parte de las distintas admi-nistraciones sanitarias competentes, los inspectores deberán seguir para su actuación pro-cedimientos normalizados de trabajo, que se aprobarán en el seno del órgano competentede coordinación técnica de inspecciones de la Agencia Española de Medicamentos y Pro-ductos Sanitarios creado a tal efecto, se actualizarán periódicamente de acuerdo con elavance científico y técnico y se harán públicos.
2. Las inspecciones se realizarán de conformidad con los documentos sobre ins-pecciones elaborados por la Agencia Europea de Medicamentos, para apoyar el reconoci-miento mutuo de las conclusiones de las inspecciones dentro de la Unión Europea. Estosdocumentos serán utilizados a nivel nacional como orientadores de los procedimientosnormalizados de trabajo de inspección, aprobados por el órgano competente de coordina-ción técnica de inspecciones de la Agencia Española de Medicamentos y Productos Sani-tarios creado a tal efecto.
3. Se facilitará a cada inspector un documento con dichos procedimientos norma-lizados de trabajo, así como sus responsabilidades y funciones.
4. Los inspectores de buena práctica clínica deberán firmar un documento deconfidencialidad antes de iniciar las actividades como inspector.
5. La actividad de inspección de buena práctica clínica tendrá carácter confiden-cial. Deberán cumplir los requisitos en cuanto a la protección de los datos personales, es-tablecidos en la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos decarácter personal.
6. Los inspectores contarán con los medios adecuados de identificación facilita-dos por la administración sanitaria correspondiente.
7. El inspector firmará una declaración de intereses, para comunicar cualquier vínculoeconómico o de otro tipo con las partes que serán objeto de inspección. Esta declaración setendrá en cuenta a la hora de designar a los inspectores para una inspección determinada.
8. Sin perjuicio de las incompatibilidades establecidas para los inspectores en elejercicio de sus actividades, asimismo serán incompatibles con cualquier tipo de intere-ses económicos directos derivados de la investigación clínica, así como de la fabricación,elaboración, distribución y comercialización de los medicamentos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
255

9. Se podrán nombrar equipos de inspectores y expertos con cualificaciones y ex-periencia adecuadas para cumplir de forma conjunta la cualificación necesaria para reali-zar la inspección.
ARTÍCULO 9. Inspecciones de buena práctica clínica.
1. Las inspecciones de buena práctica clínica podrán realizarse antes, durante odespués de la realización de los ensayos clínicos y podrán iniciarse como parte de la verifi-cación de las solicitudes de autorización de comercialización, como seguimiento de éstas ocomo consecuencia de una denuncia.
2. Se podrán hacer inspecciones, entre otras, al centro de investigación, al lugar defabricación del medicamento en investigación, a cualquier laboratorio de análisis utilizadoen el ensayo clínico, a las instalaciones del promotor y/o de las organizaciones o empresasde investigación implicadas por contrato en la realización del ensayo y al Comité Ético deInvestigación Clínica.
3. Los objetivos principales de las inspecciones de buena práctica clínica serán:
Verificar que se han protegido los derechos, el bienestar y la seguridad de los suje-tos que participan en los ensayos clínicos.
Garantizar la validez de los datos procedentes de ensayos clínicos que se presentancomo base para la autorización de comercialización de los medicamentos o como segui-miento de ésta.
Garantizar la calidad de los ensayos clínicos que se realicen en centros sanitariospúblicos y privados.
4. Realizada la inspección de buena práctica clínica, se elaborará un informe de ins-pección que deberá enviarse al inspeccionado y poner a disposición del promotor salvaguar-dando los aspectos confidenciales.
5. El informe de la inspección, previa solicitud motivada, se podrá poner a disposi-ción de:
El Comité Ético de Investigación Clínica.
La Agencia Española de Medicamentos y Productos Sanitarios y las autoridades sa-nitarias de las comunidades autónomas.
256

Las autoridades sanitarias de la Unión Europea.
La Agencia Europea de Medicamentos.
6. En caso de que las desviaciones encontradas durante la inspección fueran ca-lificadas de muy graves, de acuerdo con el artículo 101 de la Ley 29/2006, de 26 de ju-lio de garantías y uso racional de los medicamentos y productos sanitarios, se enviará elinforme al Comité Ético de Investigación Clínica, así como a la Agencia Española de Me-dicamentos y Productos Sanitarios o a la comunidad autónoma correspondiente segúnquien haya realizado la inspección.
ARTÍCULO 10. Competencias en materia de inspección de buena práctica clínica.
1. La Agencia Española de Medicamentos y Productos Sanitarios llevará a caboactuaciones de inspección en los siguientes casos:
Para verificar los resultados de los ensayos clínicos presentados en las solicitudesde autorización de medicamentos mediante los procedimientos de reconocimiento mu-tuo, descentralizado y nacional.
Para verificar los resultados de los ensayos clínicos presentados en las solicitudesde autorización de un medicamento mediante el procedimiento centralizado y coordina-das por la Agencia Europea de Medicamentos, dentro del ámbito de aplicación del Regla-mento (CE) nº 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004,por el que se establecen procedimientos comunitarios para la autorización y el control delos medicamentos de uso humano y veterianario y por el que se crea la Agencia Europeade Medicamentos.
Cuando se trate de inspecciones a realizar en el territorio de las comunidades au-tónomas que no hubieran recibido los correspondientes traspasos de la competencia deejecución de la legislación de productos farmacéuticos.
Cuando se trate de ensayos clínicos que carezcan de la debida autorización porparte de la Agencia Española de Medicamentos y Productos Sanitarios.
2. Podrán realizar inspecciones de buena práctica clínica, en todo el territorio es-pañol, los inspectores españoles acreditados, previa conformidad por la autoridad compe-tente de la inspección del centro a inspeccionar. La Agencia Española de Medicamentosy Productos Sanitarios y las comunidades autónomas con competencia en materia de ins-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
257

pección de buena práctica clínica podrán solicitar la colaboración y/o participación deinspectores no adscritos a su ámbito de competencia cuando lo estimen necesario. Si al-guna comunidad autónoma considera necesaria la realización de una inspección de buenapráctica clínica fuera del ámbito territorial de su competencia, lo solicitará a la AgenciaEspañola de Medicamentos y Productos Sanitarios, que dará traslado de la solicitud a lacomunidad autónoma competente de la inspección. Corresponde a la administración sa-nitaria de ésta última, autorizar la inspección y determinar si se lleva a cabo por inspec-tores propios o de la comunidad que la solicitó.
3. Podrán acceder a los centros donde se realizan los ensayos clínicos y tener ac-ceso a los datos para cooperar en inspecciones en España, previa solicitud y de conformi-dad con la Agencia Española de Medicamentos y Productos Sanitarios, los inspectores dela autoridad competente en materia de inspección de los Estados miembros de la Unión Eu-ropea. La Agencia informará de la inspección a la comunidad autónoma donde se encuen-tre el centro a inspeccionar. La inspección se llevará a cabo por el organismo competente.
4. La relación con las autoridades competentes de los Estados miembros de laUnión Europea en materia de inspección de buena práctica clínica, se llevará a cabo a tra-vés de la Agencia Española de Medicamentos y Productos Sanitarios.
5. Las autoridades competentes de terceros países que vayan a realizar inspeccio-nes de buena práctica clínica en España, deberán notificarlo a la Subdirección General deInspección y Control de Medicamentos de la Agencia Española de Medicamentos y Pro-ductos Sanitarios con tiempo suficiente para que pueda acudir a la citada inspección, sifuera pertinente. El promotor será responsable de informar a la autoridad competente deltercer país de esta obligación de notificación.
6. La Agencia Española de Medicamentos y Productos Sanitarios elaborará losprocedimientos pertinentes para las siguientes actividades:
Solicitar inspecciones o ayuda de otros Estados miembros de la Unión Europea ycooperar en las inspecciones realizadas en centros de otro país de la Unión Europea.
Realizar inspecciones en terceros países.
ARTÍCULO 11. Recursos y bases de datos de las inspecciones de buena práctica clínica.
1. Las administraciones sanitarias competentes en materia de inspección debuena práctica clínica proporcionarán recursos personales y materiales suficientes
258

para garantizar la comprobación efectiva del cumplimiento de las normas de buenapráctica clínica.
2. La Agencia Española de Medicamentos y Productos Sanitarios creará una basede datos con las inspecciones nacionales y, en su caso, internacionales, incluyendo la si-tuación de cumplimiento de buena práctica clínica, así como de su seguimiento. Paraello, las administraciones sanitarias competentes en materia de inspección remitirán a laAgencia los siguientes datos de todas las inspecciones de buena práctica clínica que serealicen en su territorio: código EudraCT del ensayo inspeccionado, datos del centro dondese realiza la inspección (nombre y dirección completa), tipo de centro inspeccionado, fe-cha de inspección y situación de cumplimiento de buena práctica clínica.
DISPOSICIÓN ADICIONAL PRIMERA. Normas de buena práctica clínica y directrices dela Unión Europea.
La Agencia Española de Medicamentos y Productos Sanitarios publicará las nor-mas de buena práctica clínica aplicables en los ensayos clínicos realizados en España.
También se tendrán en cuenta las directrices publicadas por la Comisión Europea.
DISPOSICIÓN ADICIONAL SEGUNDA. Exención de la autorización de fabricación paramedicamentos de terapia celular.
En el caso de medicamentos de terapias avanzadas procesados en entidades denaturaleza pública integradas en el Sistema Nacional de Salud, dichas entidades no pre-cisarán autorización de fabricación hasta la publicación de una norma específica.
DISPOSICIÓN FINAL PRIMERA. Título competencial.
Esta Orden se dicta al amparo del artículo 149.1.16 de la Constitución Española,que atribuye al Estado competencia exclusiva en materia de legislación sobre productosfarmacéuticos.
DISPOSICIÓN FINAL SEGUNDA. Incorporación de derecho de la Unión Europea.
Mediante esta Orden se incorpora al derecho español la Directiva 2005/28/CE dela Comisión, de 8 abril de 2005, por la que se establecen los principios y las directricesdetalladas de las buenas prácticas clínicas respecto a los medicamentos en investigaciónde uso humano, así como los requisitos para autorizar la fabricación o importación de di-chos productos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
259

DISPOSICIÓN FINAL TERCERA. Entrada en vigor.
La presente Orden entrará en vigor el día siguiente al de su publicación en el Bo-letín Oficial del Estado.
Madrid, 5 de febrero de 2007.
La Ministra de Sanidad y Consumo,Elena Salgado Méndez.
260

DECLARACION DE HELSINKI DE LA ASOCIACION MÉDICA MUNDIAL
(Tokio 2004)
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
261


DECLARACION DE HELSINKI DE LA ASOCIACION MÉDICA MUNDIAL
Adoptada por la 18ª Asamblea Médica Mundial, Helsinki, Finlandia, junio 1964y enmendada por la
29ª Asamblea Médica Mundial, Tokio, Japón, octubre 1975
35ª Asamblea Médica Mundial, Venecia, Italia, octubre 1983
41ª Asamblea Médica Mundial, Hong Kong, septiembre 1989
48ª Asamblea General Somerset West, Sudáfrica, octubre 1996
52ª Asamblea General, Edimburgo, Escocia, octubre 2000
Nota de Clarificación del Párrafo 29, agregada por la Asamblea General de laAMM, Washington 2002
Nota de Clarificación del Párrafo 30, agregada por la Asamblea General de laAMM, Tokio 2004
59ª Asamblea General, Seúl, Corea, octubre 2008
INTRODUCCION
1. La Asociación Médica Mundial (AMM) ha promulgado la Declaración de Hel-sinki como una propuesta de principios éticos para investigación médica enseres humanos, incluida la investigación del material humano y de informa-ción identificables.
La Declaración debe ser considerada como un todo y un párrafo no debe ser apli-cado sin considerar todos los otros párrafos pertinentes.
2. Aunque la Declaración está destinada principalmente a los médicos, la AMMinsta a otros participantes en la investigación médica en seres humanos aadoptar estos principios.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
263

3. El deber del médico es promover y velar por la salud de los pacientes, inclui-dos los que participan en investigación médica. Los conocimientos y la con-ciencia del médico han de subordinarse al cumplimiento de ese deber.
4. La Declaración de Ginebra de la Asociación Médica Mundial vincula al mé-dico con la fórmula “velar solícitamente y ante todo por la salud de mi pa-ciente”, y el Código Internacional de Etica Médica afirma que: “El médicodebe considerar lo mejor para el paciente cuando preste atención médica”.
5. El progreso de la medicina se basa en la investigación que, en ultimo tér-mino, debe incluir estudios en seres humanos. Las poblaciones que están su-brepresentadas en la investigación médica deben tener un acceso apropiadoa la participación en la investigación.
6. En investigación médica en seres humanos, el bienestar de la persona queparticipa en la investigación debe tener siempre primacía sobre todos los otrosintereses.
7. El propósito principal de la investigación médica en seres humanos es com-prender las causas, evolución y efectos de las enfermedades y mejorar las in-tervenciones preventivas, diagnósticas y terapéuticas (métodos,procedimientos y tratamientos). Incluso, las mejores intervenciones actualesdeben ser evaluadas continuamente a través de la investigación para que seanseguras, eficaces, efectivas, accesibles y de calidad.
8. En la práctica de la medicina y de la investigación médica, la mayoría de lasintervenciones implican algunos riesgos y costos.
9. La investigación médica está sujeta a normas éticas que sirven para promo-ver el respeto a todos los seres humanos y para proteger su salud y sus dere-chos individuales. Algunas poblaciones sometidas a la investigación sonparticularmente vulnerables y necesitan protección especial. Estas incluyena los que no pueden otorgar o rechazar el consentimiento por sí mismos y alos que pueden ser vulnerables a coerción o influencia indebida.
10. Los médicos deben considerar las normas y estándares éticos, legales y jurí-dicos para la investigación en seres humanos en sus propios países, al igualque las normas y estándares internacionales vigentes. No se debe permitirque un requisito ético, legal o jurídico nacional o internacional disminuya o
264

elimine cualquiera medida de protección para las personas que participan enla investigación establecida en esta Declaración.
PRINCIPIOS PARA TODA INVESTIGACION MEDICA
11. En la investigación médica, es deber del médico proteger la vida, la salud, ladignidad, la integridad, el derecho a la autodeterminación, la intimidad y laconfidencialidad de la información personal de las personas que participan eninvestigación.
12. La investigación médica en seres humanos debe conformarse con los princi-pios científicos generalmente aceptados y debe apoyarse en un profundo co-nocimiento de la bibliografía científica, en otras fuentes de informaciónpertinentes, así como en experimentos de laboratorio correctamente realiza-dos y en animales, cuando sea oportuno. Se debe cuidar también del bienes-tar de los animales utilizados en los experimentos.
13. Al realizar una investigación médica, hay que prestar atención adecuada alos factores que puedan dañar el medio ambiente.
14. El proyecto y el método de todo estudio en seres humanos debe describirseclaramente en un protocolo de investigación. Este debe hacer referencia siem-pre a las consideraciones éticas que fueran del caso y debe indicar cómo sehan considerado los principios enunciados en esta Declaración. El protocolodebe incluir información sobre financiamiento, patrocinadores, afiliacionesinstitucionales, otros posibles conflictos de interés e incentivos para las per-sonas del estudio y estipulaciones para tratar o compensar a las personas quehan sufrido daños como consecuencia de su participación en la investigación.El protocolo debe describir los arreglos para el acceso después del ensayo aintervenciones identificadas como beneficiosas en el estudio o el acceso aotra atención o beneficios apropiados.
15. El protocolo de la investigación debe enviarse, para consideración, comenta-rio, consejo y aprobación, a un comité de ética de investigación antes de co-menzar el estudio. Este comité debe ser independiente del investigador, delpatrocinador o de cualquier otro tipo de influencia indebida. El comité debeconsiderar las leyes y reglamentos vigentes en el país donde se realiza la in-vestigación, como también las normas internacionales vigentes, pero no sedebe permitir que éstas disminuyan o eliminen ninguna de las protecciones
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
265

para las personas que participan en la investigación establecidas en esta De-claración. El comité tiene el derecho de controlar los ensayos en curso. El in-vestigador tiene la obligación de proporcionar información del control alcomité, en especial sobre todo incidente adverso grave. No se debe hacer nin-gún cambio en el protocolo sin la consideración y aprobación del comité.
16. La investigación médica en seres humanos debe ser llevada a cabo sólo porpersonas con la formación y calificaciones científicas apropiadas. La investiga-ción en pacientes o voluntarios sanos necesita la supervisión de un médico uotro profesional de la salud competente y calificado apropiadamente. La respon-sabilidad de la protección de las personas que toman parte en la investigacióndebe recaer siempre en un médico u otro profesional de la salud y nunca en losparticipantes en la investigación, aunque hayan otorgado su consentimiento.
17. La investigación médica en una población o comunidad con desventajas ovulnerable sólo se justifica si la investigación responde a las necesidades yprioridades de salud de esta población o comunidad y si existen posibilida-des razonables de que la población o comunidad, sobre la que la investiga-ción se realiza, podrá beneficiarse de sus resultados.
18. Todo proyecto de investigación médica en seres humanos debe ser precedidode una cuidadosa comparación de los riesgos y los costos para las personasy las comunidades que participan en la investigación, en comparación con losbeneficios previsibles para ellos y para otras personas o comunidades afecta-das por la enfermedad que se investiga.
19. Todo ensayo clínico debe ser inscrito en una base de datos disponible al pú-blico antes de aceptar a la primera persona.
20. Los médicos no deben participar en estudios de investigación en seres huma-nos a menos de que estén seguros de que los riesgos inherentes han sido ade-cuadamente evaluados y de que es posible hacerles frente de manerasatisfactoria. Deben suspender inmediatamente el experimento en marcha siobservan que los riesgos que implican son más importantes que los beneficiosesperados o si existen pruebas concluyentes de resultados positivos o bene-ficiosos.
21. La investigación médica en seres humanos sólo debe realizarse cuando la im-portancia de su objetivo es mayor que el riesgo inherente y los costos para lapersona que participa en la investigación.
266

22. La participación de personas competentes en la investigación médica debe servoluntaria. Aunque puede ser apropiado consultar a familiares o líderes de lacomunidad, ninguna persona competente debe ser incluida en un estudio, amenos que ella acepte libremente.
23. Deben tomarse toda clase de precauciones para resguardar la intimidad de lapersona que participa en la investigación y la confidencialidad de su informa-ción personal y para reducir al mínimo las consecuencias de la investigaciónsobre su integridad física, mental y social.
24. En la investigación médica en seres humanos competentes, cada individuopotencial debe recibir información adecuada acerca de los objetivos, métodos,fuentes de financiamiento, posibles conflictos de intereses, afiliaciones ins-titucionales del investigador, beneficios calculados, riesgos previsibles e in-comodidades derivadas del experimento y todo otro aspecto pertinente de lainvestigación. La persona potencial debe ser informada del derecho de parti-cipar o no en la investigación y de retirar su consentimiento en cualquier mo-mento, sin exponerse a represalias. Se debe prestar especial atención a lasnecesidades específicas de información de cada individuo potencial, comotambién a los métodos utilizados para entregar la información. Después deasegurarse de que el individuo ha comprendido la información, el médico uotra persona calificada apropiadamente debe pedir entonces, preferiblementepor escrito, el consentimiento informado y voluntario de la persona. Si el con-sentimiento no se puede otorgar por escrito, el proceso para lograrlo debe serdocumentado y atestiguado formalmente.
25. Para la investigación médica en que se utilice material o datos humanos iden-tificables, el médico debe pedir normalmente el consentimiento para la reco-lección, análisis, almacenamiento y reutilización. Podrá haber situaciones enlas que será imposible o impracticable obtener el consentimiento para dichainvestigación o podría ser una amenaza para su validez. En esta situación, lainvestigación sólo puede ser realizada después de ser considerada y aprobadapor un comité de ética de investigación.
26. Al pedir el consentimiento informado para la participación en la investigación,el médico debe poner especial cuidado cuando el individuo potencial está vin-culado con él por una relación de dependencia o si consiente bajo presión. Enuna situación así, el consentimiento informado debe ser pedido por una personacalificada adecuadamente y que nada tenga que ver con aquella relación.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
267

27. Cuando el individuo potencial sea incapaz, el médico debe pedir el consen-timiento informado del representante legal. Estas personas no deben ser in-cluidas en la investigación que no tenga posibilidades de beneficio paraellas, a menos que ésta tenga como objetivo promover la salud de la pobla-ción representada por el individuo potencial y esta investigación no puederealizarse en personas competentes y la investigación implica sólo un riesgoy costo mínimos.
28. Si un indivividuo potencial que participa en la investigación consideradoincompetente es capaz de dar su asentimiento a participar o no en la in-vestigación, el médico debe pedirlo, además del consentimiento del re-presentante legal. El desacuerdo del individuo potencial debe serrespetado.
29. La investigación en individuos que no son capaces física o mentalmente deotorgar consentimiento, por ejemplo los pacientes inconscientes, se puederealizar sólo si la condición física/mental que impide otorgar el consenti-miento informado es una característica necesaria de la población investi-gada. En estas circunstancias, el médico debe pedir el consentimientoinformado al representante legal. Si dicho representante no está disponi-ble y si no se puede retrasar la investigación, el estudio puede llevarse acabo sin consentimiento informado, siempre que las razones específicaspara incluir a individuos con una enfermedad que no les permite otorgarconsentimiento informado hayan sido estipuladas en el protocolo de la in-vestigación y el estudio haya sido aprobado por un comité de ética de in-vestigación. El consentimiento para mantenerse en la investigación debeobtenerse a la brevedad posible del individuo o de un representante legal.
30. Los autores, directores y editores todos tienen obligaciones éticas con res-pecto a la publicación de los resultados de su investigación. Los autores tie-nen el deber de tener a la disposición del público los resultados de suinvestigación en seres humanos y son responsables de la integridad y exac-titud de sus informes. Deben aceptar las normas éticas de entrega de in-formación. Se deben publicar tanto los resultados negativos e inconclusoscomo los positivos o de lo contrario deben estar a la disposición del pú-blico..En la publicación se debe citar la fuente de financiamiento, afilia-ciones institucionales y conflictos de intereses. Los informes sobreinvestigaciones que no se ciñan a los principios descritos en esta Declara-ción no deben ser aceptados para su publicación.
268

PRINCIPIOS APLICABLES CUANDO LA INVESTIGACION MEDICA SE COMBINA CON LAATENCION MEDICA
31. El médico puede combinar la investigación médica con la atención médica,sólo en la medida en que tal investigación acredite un justificado valor poten-cial preventivo, diagnóstico o terapéutico y si el médico tiene buenas razonespara creer que la participación en el estudio no afectará de manera adversa lasalud de los pacientes que toman parte en la investigación.
32. Los posibles beneficios, riesgos, costos y eficacia de toda intervención nueva de-ben ser evaluados mediante su comparación con la mejor intervención probadaexistente, excepto en las siguientes circunstancias:
• El uso de un placebo, o ningún tratamiento, es aceptable en estudios para losque no hay una intervención probada existente.
• Cuando por razones metodológicas, científicas y apremiantes, el uso de un placeboes necesario para determinar la eficacia y la seguridad de una intervención que noimplique un riesgo, efectos adversos graves o daño irreversible para los pacientesque reciben el placebo o ningún tratamiento. Se debe tener muchísimo cuidadopara evitar abusar de esta opción.
33. Al final de la investigación, todos los pacientes que participan en el estudiotienen derecho a ser informados sobre sus resultados y compartir cualquier be-neficio, por ejemplo, acceso a intervenciones identificadas como beneficiosasen el estudio o a otra atención apropiada o beneficios.
34. El médico debe informar cabalmente al paciente los aspectos de la atenciónque tienen relación con la investigación. La negativa del paciente a participaren una investigación o su decisión de retirarse nunca debe perturbar la relaciónmédico-paciente.
35. Cuando en la atención de un enfermo las intervenciones probadas han resultadoineficaces o no existen, el médico, después de pedir consejo de experto, con elconsentimiento informado del paciente o de un representante legal autorizado,puede permitirse usar intervenciones no comprobadas, si, a su juicio, ello daalguna esperanza de salvar la vida, restituir la salud o aliviar el sufrimiento.Siempre que sea posible, tales intervenciones deben ser investigadas a fin deevaluar su seguridad y eficacia. En todos los casos, esa información nueva debeser registrada y, cuando sea oportuno, puesta a disposición del público.
22.10.2008
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
269


GUÍA DE NORMAS DE BUENA PRÁCTICA CLÍNICA(International Conference on Harmonisation, ICH)
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
271


NORMAS DE LA BUENA PRÁCTICA CLINICA. (CPMP/ICH/135/95)
1. GLOSARIO
A continuación se indicarán los términos por orden alfabético para facilitar sulocalización, indicando entre paréntesis el lugar que ocupa en la versión en inglés parafacilitar la comparación de ambas versiones.
1.1. Acceso Directo (1.21)
Permiso para examinar, analizar, verificar y reproducir cualquier registro e in-forme que sea importante para la evaluación de un ensayo clínico. Cualquiera de laspartes con acceso directo (p. ej. autoridades reguladoras nacionales y extranjeras, mo-nitores y auditores del promotor) deberá tomar todas las precauciones posibles dentrode las restricciones que establece la legislación vigente para mantener la confidencia-lidad de la identidad de los sujetos y de la información propiedad del promotor.
1.2. Acontecimiento Adverso (1.2)
Cualquier experiencia adversa que se presente en un paciente o sujeto de unainvestigación clínica al que se ha administrado un medicamento, y aunque no tenganecesariamente una relación causal con dicho tratamiento. Un acontecimiento ad-verso (AE) puede ser, por tanto, cualquier signo desfavorable y no intencionado (inclu-yendo un hallazgo anormal de laboratorio), síntoma o enfermedad temporalmenteasociada con el uso de un medicamento (en investigación), esté o no relacionado conel medicamento (en investigación).Véase la guía E2A de la ICH para la Gestión de Da-tos de Seguridad Clínica: Definiciones y Normas para Informes Expeditivos.
1.3. Acontecimiento Adverso Grave o Reacción Adversa Grave al Medicamento(1.50)
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
273

Cualquier acontecimiento adverso o reacción adversa que a cualquier dosis2:
Produzca la muerte del paciente,
Amenace la vida del sujeto,
Haga necesaria la hospitalización del sujeto o la prolongación de ésta,
Produzca invalidez o incapacidad permanente o importante, o
Dé lugar a una anomalía o malformación congénita.
Véase la guía E2A de la ICH para la Gestión de Datos de Seguridad Clínica: Defi-niciones y Normas para Informes Expeditivos)
1.4. Aleatorización (1.48)
Procedimiento de asignación al azar de los sujetos del ensayo a los grupos de tra-tamiento o control, reduciéndose así el sesgo.
1.5. “Audit trail” (1.9)
Documentación que permite la reconstrucción del curso de los acontecimientosdel ensayo clínico.
1.6. Auditoria (1.6)
Examen independiente y sistemático de las actividades y los documentos relacio-nados con el ensayo, para determinar si las actividades evaluadas relacionadas con el en-sayo han sido realizadas y si los datos son registrados, analizados y fielmente comunicados,conforme al protocolo, los procedimientos normalizados de trabajo (PNT) del promotor, laBPC y la normativa vigente.
1.7. Autoridades reguladoras (1.49)
Organismos que tienen el poder de legislar. En la guía de la BPC de la ICH, la ex-presión Autoridades reguladoras incluye a las autoridades que revisan los datos clínicospresentados y aquéllas que realizan las inspecciones (véase 1.40). En ocasiones, estos or-ganismos pueden ser denominados también autoridades competentes.
274
2 De acuerdo con el Real decreto 223/2004, Art. 2 letra q) se añade al listado anterior: “A efectos de su notificación,se tratarán también como graves aquellas sospechas que se consideren importantes desde el punto de vista médico,aunque no cumplan los criterios anteriores”.

1.8. Bienestar (de los sujetos en un ensayo) (1.62)
Integridad física y mental de los sujetos que participan en un ensayo clínico.
1.9. Buena Práctica Clínica (1.24)
Normas para el diseño, dirección, realización, monitorización, auditoria, regis-tro, análisis e informe del ensayo clínico que garantizan que los datos y los resultadosobtenidos son precisos y creíbles, y que se han protegido los derechos, la integridad yla confidencialidad de los sujetos del ensayo.
1.10. Centro del Ensayo (1.59)
Lugar donde se realizan las actividades relacionadas con el ensayo.
1.11. Certificado de Auditoria (1.7)
Declaración de confirmación, por parte del auditor, de que se ha realizado unaauditoria.
1.12. Ciego/Enmascaramiento (1.10)
Procedimiento por el cual una o más partes del ensayo desconocen la asigna-ción del tratamiento. Habitualmente, simple ciego significa que los sujetos descono-cen el tratamiento asignado y doble ciego hace referencia a que los sujetos, elinvestigador, el monitor y, en algunos casos los que analizan los datos desconocen laasignación del tratamiento
1.13. Código de Identificación del Sujeto (1.58)
Identificador único que asigna el investigador a cada sujeto del ensayo paraproteger su identidad y que se utiliza en vez del nombre del sujeto cuando el inves-tigador comunica de acontecimientos adversos y de otros datos relacionados con elensayo.
1.14. Comité Coordinador (1.18)
Comité que el promotor puede organizar para coordinar la realización de un en-sayo multicéntrico.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
275

1.15. Comité Ético de Investigación Clínica3 (CEIC) (1.27, 1.31)
Organismo independiente constituido por profesionales sanitarios y miembros nosanitarios, encargado de velar por la protección de los derechos, la seguridad y el bienes-tar de los sujetos que participan en un ensayo clínico, y de proporcionar una garantía pú-blica al respecto mediante, entre otras, la evaluación y emisión de un dictamen referenteal protocolo del ensayo, la idoneidad del investigador, la adecuación de las instalaciones,así como los métodos y documentos que serán utilizados en la obtención y la documen-tación del consentimiento informado de los sujetos del ensayo.
La situación legal, composición, funciones, procedimientos y requisitos legalespueden variar en cada país, pero debe permitir que los Comités actúen de acuerdo con lasnormas de BPC descritas en este documento.
1.16. Comité Independiente de Monitorización de Datos (CIMD4) (Consejo de Mo-nitorización de Datos y Seguridad, Comité de Monitorización, Comité de Monitorización deDatos) (1.25)
Comité independiente para la monitorización de los datos que puede ser estable-cido por el promotor para valorar, a intervalos de tiempo determinados, el progreso de unensayo clínico, los datos de seguridad y las variables críticas de eficacia, así como reco-mendar al promotor continuar, modificar o interrumpir el ensayo.
1.17. Comparador (Medicamento) (1.14)
Medicamento comercializado o no (es decir, control activo), o placebo, utilizadoscomo referencia en un ensayo clínico.
1.18. Confidencialidad (1.16)
Conjunto de medidas dirigidas a evitar que individuos no autorizados, tengan ac-ceso a información propiedad del promotor o a la identidad de un sujeto.
1.19. Consentimiento Informado (1.28)
Proceso por el cual un sujeto confirma voluntariamente su decisión de participaren un ensayo determinado después de haber sido informado debidamente de todos los as-
276
3 Denominación que se da en España a los “Independent Ethics Comittee” o a los “Institucional Review Board” (IRB).4 IDMC son las siglas en inglés correspondientes a “Independent Data-Monitoring Committee”

pectos del ensayo que son relevantes para la decisión de participar del sujeto. El consen-timiento informado está documentado por medio del correspondiente documento escritofirmado y fechado.
1.20. Contrato (1.17)
Acuerdo escrito, fechado y firmado entre dos o más partes implicadas que establececualquier disposición sobre la delegación y la distribución de las tareas y obligaciones y, ensu caso, de las cuestiones económicas. El protocolo puede servir como base del contrato.
1.21. Control de Calidad (QC)5 (1.47)
Técnicas y actividades operativas que se emprenden dentro del sistema de Garan-tía de calidad a fin de verificar que se han cumplido los requisitos de calidad en las activi-dades relacionadas con el ensayo.
1.22. Cuaderno de Recogida de Datos (CRD)6, (1.11)
Documento impreso, óptico o electrónico que se ha diseñado para recoger y trans-mitir al promotor toda la información requerida en el protocolo para cada sujeto del ensayo.
1.23. Cumplimiento (en relación a los ensayos) (1.15)
La observancia de todos los requisitos relacionados con el ensayo, de las normasde Buena Práctica Clínica (BPC) y de la normativa vigente.
1.24. Datos fuente (1.51)
Toda la información contenida en los archivos originales y las copias certificadasde los archivos originales referente a hallazgos clínicos, observaciones u otras actividadesdel ensayo clínico que son necesarias para la reconstrucción y la evaluación del ensayo.Los datos fuente están contenidos en los documentos fuente (los archivos originales o lascopias certificadas).
1.25. Dictamen (en relación con el Comité Ético de Investigación Clínica) (1.42)
Juicio o consejo que otorga un Comité Ético de Investigación Clínica (CEIC)
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
277
5 Siglas correspondientes a “Quality Control”6 Case Report Form, CRF en versión inglesa

1.26. Dictamen favorable (en relación con los Comités Éticos de InvestigaciónClínica) (1.5)
El informe favorable emitido por el Comité Ético de Investigación Clínica (CEIC)significa que el ensayo clínico ha sido revisado y puede ser realizado en una instituciónconforme a las normas establecidas por los CEIC, la institución, la Buena Práctica Clínica(BPC) y los requisitos legales pertinentes.
1.27. Documentación (1.22)
Todos los registros, en cualquier formato (incluyendo, entre otros, los registros es-critos, electrónicos, magnéticos y ópticos, los escáneres, radiografías y los electrocardio-gramas), que describan o registren los métodos, la realización y/o los resultados de unensayo, así como los factores que pueden afectarlo y las acciones realizadas.
1.28. Documentos Esenciales (1.23)
Documentos que individual o colectivamente, permiten la evaluación de la reali-zación de un estudio y la calidad de los datos producidos. Véase apartado 8. DocumentosEsenciales para la Realización de un Ensayo Clínico.
1.29. Documentos Fuente (1.52)
Documentos originales, datos y registros (p.ej. historias clínicas, gráficas clínicasy administrativcas, informes de laboratorio, memoranda, diarios de los sujetos o cuestio-narios de evaluación, registros de dispensación de fármacos, datos registrados por instru-mentos informatizados, copias o transcripciones certificadas después de su verificacióncomo copias exactas, microfichas, negativos fotográficos, microfilms o medios magnéticos,radiografías, archivos de los sujetos y registros guardados en la farmacia, en los laborato-rios y en los departamentos medico-técnicos implicados en el ensayo clínico).
1.30. Ensayo Multicéntrico (1.40)
Ensayo clínico realizado en más de un centro de investigación, llevado a cabo pormás de un investigador de acuerdo con un único protocolo.
1.31. Estudio No clínico (1.41)
Estudios biomédicos que no se han realizado en seres humanos.
278

1.32. Estudio o Ensayo Clínico (1.12)
Toda investigación efectuada en seres humanos dirigida a determinar o verificarlos efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o identificarcualquier reacción adversa, y/o estudiar la absorción, distribución, metabolismo y excre-ción de uno o varios medicamentos en investigación con el propósito de determinar suseguridad y/o eficacia. Los términos ensayo clínico y estudio clínico son sinónimos.
1.33. Garantía de Calidad (QA7) (1.46)
Todas aquellas actividades planificadas y sistematizadas que se establecen paraasegurar que el ensayo se realiza y que los datos se generan, documentan (registran) ycomunican conforme a la guía de la la Buena Práctica Clínica (BPC) y la normativa per-tinente.
1.34. Historia Clínica (1.43)
Véase documentos fuente.
1.35. Informe de la Auditoria (1.8)
Evaluación escrita realizada por el auditor del promotor de los resultados de laauditoria.
1.36. Informe de Monitorización (1.39)
Informe escrito dirigido al promotor, realizado por el monitor después de cadavisita a un centro o cualquier otra comunicación relacionada con el ensayo, de acuerdocon los PNT del promotor.
1.37. Informe del Estudio o Ensayo Clínico (1.13)
Descripción escrita del estudio o ensayo de cualquier agente terapéutico, profi-láctico o diagnóstico realizado en seres humanos, donde la descripción estadística y clí-nica, las exposiciones y las evaluaciones están plenamente integradas en un soloinforme. Véase la Guía ICH para la Estructura y Contenido de los Informes de EstudiosClínicos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
279
5 Siglas correspondientes a “Quality Assurance”

1.38. Informe Intermedio del Estudio/Ensayo Clínico (1.32)
Informe referente a los resultados del estudio antes de su finalización y a su eva-luación de acuerdo con el análisis intermedio realizado durante el ensayo.
1.39. Inspección (1.29)
Revisión oficial por una autoridad reguladora competente de los documentos, lasinstalaciones, los archivos y de cualquier otro elemento que considere relacionado con elensayo clínico y que puede encontrarse en el centro de investigación, en las instalacionesdel promotor o en la organización de investigación por contrato (CRO), o en cualquier otroestablecimiento que considere oportuno inspeccionar.
1.40. Institución (sanitaria) (1.30)
Cualquier entidad pública o privada centro sanitario u odontológico, donde se re-alicen ensayos clínicos.
1.41. Investigador (1.34)
Persona8 responsable de la realización del ensayo clínico en un centro de inves-tigación. Si es un equipo el que realiza el ensayo en el centro, el investigador es el res-ponsable del equipo y puede denominarse investigador principal. Véase tambiéninvestigador colaborador.
1.42. Investigador colaborador (1.56)
Cualquier miembro del equipo del ensayo clínico que esté designado y supervisadopor el investigador en un centro de investigación para realizar procedimientos relevantesy/o tomar decisiones importantes relacionados con el ensayo (p.ej. asociados, residentes,becarios de investigación). Véase también Investigador.
1.43. Investigador Coordinador (1.19)
Investigador responsable de la coordinación de los investigadores de los centrosparticipantes en un ensayo multicéntrico.
280
8 De acuerdo con el Real decreto 223/2004, art. 2 letra h) un investigador es un médico o persona que ejerce unaprofesión reconocida para llevar a cabo investigaciones en razón a su formación científica y de su experiencia en laatención sanitaria requerida.

1.44. Investigador/Institución (1.35)
Hace referencia al investigador o a la institución dependiendo de la normativa ala que hace mención.
1.45. Manual del Investigador (1.36)
Recopilación de datos clínicos y no clínicos sobre el medicamento en investiga-ción pertinente para el estudio de dicho medicamento en seres humanos. Véase el punto7. Manual del Investigador.
1.46. Medicamento en Investigación (1.33)
Forma farmacéutica de una sustancia activa o placebo que se investiga o se uti-liza como referencia en un ensayo clínico, incluidos los medicamentos con autorizaciónde comercialización cuando se utilicen o combinen (en la formulación o en el envase) deforma diferente a la autorizada, o cuando se utilicen para tratar una indicación no apro-bada o para obtener más información acerca de un uso autorizado.
1.47. Modificación del protocolo (1.3)
Véase protocolo modificación
1.48. Monitorización (1.38)
Acto de vigilar el desarrollo de un ensayo clínico, y de garantizar que es reali-zado, archivado y publicado de acuerdo con el protocolo, los Procedimientos Normali-zados de Trabajo (PNT), las guías de la Buena Práctica Clínica (BPC) así como a lanormativa vigente.
1.49. Organización de Investigación por Contrato (CRO)9 (1.20)
Persona u organización (comercial, académica u otra)10 contratada por el promo-tor para realizar una o más de las funciones o deberes del promotor en relación con elensayo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
281
9 Siglas correspondientes al término inglés “Contract Research Organization”.10 De acuerdo con el Real decreto 223/2004, art. 2 letra g) una CRO es una persona física o jurídica contratada por
el promotor para realizar una o más de las funciones o deberes del promotor …

1.50. Procedimientos Normalizados de Trabajo (PNT) (1.55)
Instrucciones escritas y detalladas para lograr la uniformidad en la realizaciónde una función específica.
1.51. Promotor (1.53)
Individuo, empresa, institución u organización responsable del inicio, gestióny/o financiación de un ensayo clínico.
1.52. Promotor-Investigador (1.54)
Individuo que inicia y realiza, solo o con otros, un ensayo clínico y bajo cuya di-rección inmediata el medicamento en investigación se administra, dispensa, o utiliza porun sujeto. El término únicamente hace referencia a un individuo (es decir no incluye unacorporación o una agencia). Las obligaciones de un promotor-investigador abarcan tantolas de promotor como las de investigador.
1.53. Protocolo (1.44)
Documento donde se describen los objetivos, el diseño, la metodología, las con-sideraciones estadísticas y la organización de un ensayo. El protocolo habitualmenteproporciona también los antecedentes y la justificación del ensayo, aunque ambos pue-den ser incluidos en otros documentos a los que haga referencia el protocolo. En todala guía de la BPC de la ICH, el término protocolo se refiere tanto al protocolo originalcomo a las sucesivas modificaciones del mismo.
1.54. Protocolo Modificación (1.45)
Descripción escrita de una modificación o una aclaración oficial realizada alprotocolo.
1.55. Reacción Adversa a un medicamento (RAM) (1.1)
Durante la investigación clínica previa a la aprobación de un nuevo medica-mento, o de una nueva indicación, especialmente cuando la dosis terapéutica no estáestablecida, se deberá considerar como reacción adversa cualquier respuesta a un me-dicamento que sea nociva y no intencionada relacionada con el medicamento a cualquierdosis. La expresión “respuesta a un medicamento” significa que existe una posibilidad
282

razonable de que exista una relación causal entre el acontecimiento adverso y el medi-camento, es decir, que no se puede excluir dicha relación.
En relación a los medicamentos comercializados: respuesta a un fármaco nocivay no intencionada y que tenga lugar con la dosis habitualmente usada en el ser humanopara la profilaxis, el diagnostico, o el tratamiento de enfermedades o bien para la11 modi-ficación de funciones fisiológicas Véase la guía E2A de la ICH para la Gestión de Datos deSeguridad Clínica: Definiciones y Normas para Informes Expeditivos.
1.56. Reacción Adversa Inesperada (1.60)
Reacción adversa cuya naturaleza o gravedad no se corresponde con la informacióndisponible sobre el medicamento (por ejemplo, Manual del Investigador para un medica-mento en investigación no autorizado para su comercialización o la ficha técnica del me-dicamento en el caso de medicamento autorizado). Véase la guía E2A de la ICH para laGestión de Datos de Seguridad Clínica: Definiciones y Normas para Informes Expeditivos)
1.57. Representante Legal (1.37)
Individuo, persona jurídica u otra entidad autorizada para consentir, en nombre deun sujeto potencial, su participación en un ensayo clínico.
1.58. Requisitos Legales Pertinentes (1.4)
Cualquier ley o normativa dirigida a la realización de ensayos clínicos con medi-camentos en investigación.
1.59. Sujeto del Ensayo (1.57)
Individuo que participa en un ensayo clínico, recibiendo el medicamento en in-vestigación o actuando como control.
1.60. Sujetos Vulnerables (1.61)
Sujetos cuya disposición para ser voluntarios en un ensayo clínico puede ser in-debidamente influenciada por la expectativa, justificada o no, de los beneficios asociados
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
283
11 Se ha modificado el final de la definición (.. o bien para la restauración, corrección o modificación de funcionesfisiológicas.) de acuerdo con el Real Decreto 711/2002 que incorpora la Directiva 2000/38/CE de la Comisión.

con su participación o de la respuesta como represalia por parte de superiores jerárquicosen el caso de negarse a participar. Ejemplo de ello son los miembros de un grupo con unaestructura jerárquica, tales como los estudiantes de medicina, farmacia, odontología o en-fermería, personal subordinado de un laboratorio u hospital, empleados de una compañíafarmacéutica, miembros de las fuerzas armadas y prisioneros. Otros sujetos vulnerables in-cluyen los pacientes con enfermedades incurables, personas en residencias de ancianos,parados o mendigos, pacientes en situaciones de emergencia, grupos pertenecientes aminorías étnicas, vagabundos, nómadas, refugiados, menores y aquellos que son incapa-ces de dar su consentimiento.
1.61. Testigo Imparcial (1.26)
Persona independiente del ensayo y que no puede ser influenciada indebidamentepor el personal implicado en el mismo, que asiste al proceso del consentimiento informadosi el sujeto o sus representantes legales no pueden leer, y se responsabiliza de leer la hojade información para el sujeto, el documento del consentimiento informado y cualquierotra información escrita dirigida al sujeto.
2. LOS PRINCIPIOS DE LA BPC DE LA ICH
2.1. Los ensayos clínicos deben realizarse de acuerdo con los principios éticos quetienen su origen en la Declaración de Helsinki, y que sean coherentes con la guía de laBPC y la legislación vigente.
2.2. Antes de iniciar un ensayo, deberán considerarse los riesgos e inconvenien-tes previsibles en relación con el beneficio esperado, tanto para el sujeto individual del en-sayo como para la sociedad. Un ensayo deberá iniciarse y continuarse únicamente en elcaso de que los beneficios previstos justifiquen los riesgos.
2.3. Los derechos, la seguridad y el bienestar de los sujetos de un ensayo son lasconsideraciones más importantes y deberán prevalecer sobre los intereses de la ciencia yde la sociedad.
2.4. La información clínica y no clínica disponible sobre un medicamento en in-vestigación deberá ser suficiente para apoyar el ensayo clínico propuesto.
2.5. Los ensayos clínicos deberán estar científicamente justificados y estar des-critos en un protocolo claro y detallado.
284

2.6. El ensayo se deberá realizar de acuerdo con el protocolo que previamente harecibido un dictamen favorable de un CEIC.
2.7. El cuidado médico que reciben los sujetos y las decisiones médicas tomadasen su nombre serán siempre responsabilidad de un médico cualificado o, en su caso, unodontólogo cualificado.
2.8. Cada individuo implicado en la realización de un ensayo deberá estar cuali-ficado, por su titulación, formación y experiencia, para realizar sus tareas y responsabili-dades respectivas.
2.9. Se deberá obtener el consentimiento informado, otorgado de forma libre, decada sujeto antes de su participación en el ensayo clínico.
2.10. Toda la información del ensayo clínico deberá ser registrada, manejada y ar-chivada de forma que permita su comunicación, interpretación y verificación exactas.
2.11. Se deberá proteger la confidencialidad de los registros que pudieran iden-tificar a los sujetos respetando la privacidad y las normas de confidencialidad de acuerdocon los requisitos legislativos pertinentes.
2.12. Los medicamentos en investigación deberán fabricarse, manejarse y alma-cenarse de acuerdo con las Normas de Correcta Fabricación (NCF) pertinentes y se debe-rán utilizar de acuerdo con el protocolo aprobado.
2.13. Se implantarán los sistemas con procedimientos que aseguren la calidad decada aspecto del ensayo.
3. COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA (CEIC)
3.1. Responsabilidades
3.1.1. Un CEIC deberá salvaguardar los derechos, la seguridad y el bienestar detodos los sujetos del ensayo. Deberá prestarse especial atención a los ensayos que pue-dan incluir sujetos vulnerables.
3.1.2. El CEIC deberá estar en posesión de los siguientes documentos:
El protocolo del ensayo y sus modificaciones; el documento del consentimiento in-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
285

formado así como las actualizaciones del mismo propuestas por el investigador, los pro-cedimientos de reclutamiento de sujetos (p.ej. anuncios publicitarios); la hoja de informa-ción para el sujeto; el Manual del Investigador, la información disponible sobre seguridad,la información referente a la remuneración e indemnizaciones previstas para los sujetos,el currículum vitae actualizado del investigador y/u otra documentación que demuestre lacualificación del investigador así como cualquier otro documento que el CEIC pueda ne-cesitar para cumplir con sus responsabilidades.
El CEIC deberá evaluar la documentación que acompaña la solicitud de un ensayoclínico en un tiempo razonable y documentar por escrito sus criterios de evaluación, iden-tificando claramente el ensayo, los documentos revisados así como las fechas de los si-guientes documentos:
• Dictamen favorable,
• Modificaciones requeridas antes del dictamen favorable,
• Dictamen desfavorable y
• Retirada o suspensión de cualquier dictamen favorable previo.
3.1.3. El CEIC deberá considerar la idoneidad del investigador para el ensayo pro-puesto, mediante su currículum vitae actualizado y/o cualquier información pertinenteque el Comité solicite.
3.1.4. El CEIC deberá realizar el seguimiento de cada ensayo en marcha con unaperiodicidad proporcional al riesgo al que se exponen los sujetos del estudio. Este segui-miento será al menos anual.
3.1.5. El CEIC podrá solicitar que se proporcione a los sujetos más informaciónde la señalada en el párrafo 4.8.10 cuando, a su juicio, la información adicional aumentesignificativamente la protección de los derechos, la seguridad y/o el bienestar de los su-jetos.
3.1.6. Cuando se realice un ensayo no terapéutico con el consentimiento del re-presentante legal del sujeto (véase 4.8.12, 4.8.14), el Comité deberá determinar si elprotocolo propuesto y el resto de los documentos contemplan adecuadamente las cuestio-nes éticas y cumplen los requisitos legales para tales ensayos.
3.1.7. En aquellos casos en que el protocolo indique que no es posible obtenerel consentimiento previo del sujeto de ensayo o de su representante legal (véase 4.8.15)
286

el Comité deberá determinar si el protocolo propuesto y/u otros documentos contemplanadecuadamente las consideraciones éticas relevantes y cumplen la normativa vigente paratales ensayos (es decir, en situación de urgencia).
3.1.8. El CEIC deberá revisar tanto la cantidad como la forma de compensacióna los sujetos del ensayo a fin de asegurar que no existe coacción o influencia indebidasobre los mismos. La contraprestación al sujeto se reducirá proporcionalmente a su par-ticipación y no dependerá completamente de que finalice el ensayo.
3.1.9. El Comité deberá asegurar que la información referente a la compen-sación a los sujetos, incluyendo la forma, las cantidades y el calendario de la misma,se refleje en la hoja de información para el sujeto y en alguna información escritaque se le entregue. Se deberá especificar así mismo la forma de prorrateo de la con-traprestación.
3.2. Composición, Funciones y actividades
3.2.1. El CEIC debe estar constituido por un número razonable de miembrosque conjuntamente posean la cualificación y la experiencia necesaria para revisar y eva-luar los aspectos científicos, médicos y éticos del ensayo propuesto. Se recomienda queel CEIC posea12:
Al menos cinco miembros.
Al menos un miembro cuya área principal de interés no sea científica.
Al menos un miembro que sea independiente del centro del ensayo.
Solo aquellos miembros del Comité que sean independientes del investigador ydel promotor del ensayo deberán votar u opinar sobre temas relacionados con el ensayo.
Debe guardarse el listado de los miembros del Comité con sus respectivas cua-lificaciones.
3.2.2. El CEIC deberá realizar sus funciones con arreglo a unos procedimientosnormalizados de trabajo (PNT), mantener registros por escrito de sus actividades y delas actas de sus reuniones así como seguir la guía de la BPC y la normativa vigente.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
287
12 Composición según el artículo 12 del RD 223/04: al menos 9 miembros para asegurar la independencia de lasdecisiones, competencia y experiencia. Compuesto por médicos (al menos un farmacólogo clínico), un farmacéu-tico de hospital, un diplomado universitario en enfermería, al menos un miembro independiente de los centros enque se lleve a cabo el proyecto. Al menos 5 de los miembros deben ser ajenos a las profesiones sanitarias (al me-nos un licenciado en derecho).

3.2.3. El Comité deberá tomar sus decisiones en reuniones convocadas en lasque al menos debe haber quórum tal y como esté estipulado en sus PNT.
3.2.4. Sólo los miembros del Comité que participen en la revisión y discusión delestudio deberán votar, opinar y/o aconsejar.
3.2.5. El investigador puede proporcionar información sobre cualquier aspectodel ensayo pero no debe participar en las deliberaciones ni en la votación/dictamen delComité.
3.2.6. El Comité podrá invitar a expertos, que no sean miembros del mismo, enáreas especiales en las que necesite asesoramiento.
3.3. Procedimientos
El CEIC deberá elaborar, documentar por escrito y seguir para su funcionamientoProcedimientos Normalizados de Trabajo (PNT) que incluyan:
3.3.1. Composición del Comité y los requisitos que deben cumplir sus miembros(nombres y cualificaciones), así como la autoridad competente que les acredita.
3.3.2. Forma de convocar a sus miembros, la periodicidad y la celebración de susreuniones.
3.3.3. Criterios para la evaluación inicial y el sistema de seguimiento de losensayos.
3.3.4. Forma de determinar la frecuencia del seguimiento del ensayo clínico, sifuera necesario.
3.3.5. Casos en que se contemple, conforme a la legislación vigente, la revisiónurgente y el dictamen favorable de cualquier modificación menor en los ensayos iniciadosque tengan un dictamen favorable del CEIC
3.3.6. Especificación de que no deberá ser admitido ningún sujeto en un ensayo,antes de que el CEIC emita un dictamen favorable por escrito del ensayo.
3.3.7. Especificación de que no deberá realizarse ninguna desviación o modifica-ción del protocolo sin un previo dictamen favorable por escrito del CEIC, salvo cuando sea
288

necesario para proteger a los sujetos de cualquier riesgo inmediato o cuando el cambioafecte solo a aspectos logísticos o administrativos del ensayo (p.ej. el cambio de monitor,el número de teléfono) (véase 4.5.2).
3.3.8. Especificación de que el investigador13 deberá informar sin demora alCEIC de:
a) Las desviaciones o modificaciones del protocolo realizadas para proteger a lossujetos de riesgos inmediatos (véase 3.3.7, 4.5.2, 4.5.4)
b) Las modificaciones que supongan un incremento del riesgo para los sujetos y/oafecten de forma significativa la realización del ensayo (véase 4.10.2)
c) Todas las reacciones adversas al medicamento que sean graves e inesperadas.
d) Cualquier información nueva que pueda afectar negativamente la seguridad delos sujetos en la realización del ensayo.
3.3.9. Garantía de que el Comité notificará rápidamente y por escrito al investi-gador14 y a la institución:
a) Los dictámenes relacionados con el ensayo.
b) La justificación de sus dictámenes.
c) Los procedimientos para apelar sus dictámenes.
3.4. Documentos
El Comité deberá archivar todos los documentos relevantes (p.ej. los PNT escri-tos, la lista de miembros, la lista de las ocupaciones y/o afiliaciones de los mismos, losdocumentos presentados, las actas de las reuniones y la correspondencia) durante un pe-riodo de al menos 3 años después de la finalización del ensayo y tenerlos a disposición delas autoridades competentes.
Los investigadores, los promotores o las autoridades competentes pueden soli-citar al CEIC sus procedimientos de trabajo por escrito y la lista de sus miembros.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
289
13 De acuerdo con el Real Decreto 223/2004, es el promotor el responsable de este tipo de notificaciones14 De acuerdo con el Real Decreto 223/2004, el CEIC se lo notifica al promotor, a la AEMPS y si se trata del CEIC
de referencia se lo notificará a los demás CEIC implicados.

4. INVESTIGADOR
4.1. Cualificaciones del Investigador y Acuerdos
4.1.1. El investigador deberá estar cualificado por su titulación, formación y ex-periencia para responsabilizarse de la realización correcta del ensayo clínico y deberácumplir todos los requisitos especificados por la legislación pertinente15. Deberá justificarsu cualificación mediante un currículum vitae actualizado así como la documentación per-tinente que le solicite el promotor, el Comité o las autoridades competentes.
4.1.2. El investigador deberá conocer a fondo las propiedades de los medicamen-tos en investigación, tal como figuren en el protocolo, en el Manual del Investigador ac-tualizado, en la información del medicamento y en otras fuentes de informaciónproporcionadas por el promotor.
4.1.3. El investigador deberá conocer y ajustarse a la guía de BPC y a la norma-tiva vigente.
4.1.4. El investigador y la institución deberán permitir al promotor la monitoriza-ción y la auditoria del ensayo clínico y a las autoridades competentes la realización de lasinspecciones correspondientes.
4.1.5. El investigador deberá archivar el listado de las personas debidamente cua-lificadas en las que haya delegado obligaciones importantes relacionadas con el ensayo.
4.2. Recursos Apropiados
4.2.1. El investigador deberá ser capaz de demostrar (p.ej. basándose en datos re-trospectivos) su capacidad para reclutar el número requerido de sujetos adecuados, den-tro del periodo de reclutamiento establecido.
4.2.2. El investigador deberá disponer de tiempo suficiente para realizar y com-pletar correctamente el ensayo dentro del tiempo convenido.
290
15 De acuerdo con el Real decreto 223/2004, art. 2 letra h) podrá actuar como investigador, un médico o personaque ejerza una profesión reconocida en España para llevar a cabo investigaciones en razón a su formación cien-tífica y de su experiencia en la atención sanitaria requerida. El investigador es responsable de la realización delensayo clínico en un centro. Si es un equipo el que realiza el ensayo en un centro, el investigador es el responsa-ble del equipo y puede denominarse investigador principal.

4.2.3. El investigador deberá disponer del número adecuado de personal cualifi-cado y de las instalaciones necesarias, durante el tiempo previsto del ensayo, para reali-zarlo correctamente y con seguridad.
4.2.4. El investigador deberá garantizar que todas las personas que participan enel ensayo están informadas correctamente del protocolo, los medicamentos en investiga-ción y de sus tareas y obligaciones en relación con el ensayo.
4.3. Asistencia Médica de los Sujetos del Ensayo
4.3.1. Un médico cualificado (o un odontólogo, cuando sea necesario), que seael investigador principal o colaborador del ensayo, será el responsable de todas las deci-siones médicas (u odontológicas) relacionadas con el mismo.
4.3.2. Durante y después de la participación de un sujeto en un ensayo el inves-tigador y la institución deberán asegurarse de que se le proporcione una asistencia mé-dica adecuada ante cualquier acontecimiento adverso, incluyendo la alteraciónclínicamente importante de los valores de laboratorio, que esté relacionado con el ensayo.El investigador o la institución deberán informar al sujeto cuando éste necesite asisten-cia médica para las enfermedades intercurrentes de las que el investigador tenga cono-cimiento.
4.3.3. Se recomienda que el investigador informe al médico de atención prima-ria, de la participación del sujeto en el ensayo siempre que éste tenga médico asignadoy esté de acuerdo con que se le informe.
4.3.4. El sujeto no tiene obligación de justificar su decisión de retirarse prema-turamente del ensayo, pero el investigador deberá hacer un esfuerzo razonable para ave-riguar la razón que le ha llevado a dejar el estudio, siempre y cuando se respeten losderechos del sujeto.
4.4 Comunicación con el CEIC
4.4.1. Antes de iniciar un ensayo, el investigador deberá obtener el dictamen fa-vorable, por escrito y fechado, para el protocolo del ensayo, la hoja de información parael sujeto del consentimiento informado, las actualizaciones de este documento, los pro-cedimientos de reclutamiento (p. ej. anuncios publicitarios) y cualquier otra informaciónescrita que les sea entregada a los sujetos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
291

4.4.2. Como parte de la solicitud escrita al CEIC, el investigador/institución16
deberá proporcionarle una copia actualizada del Manual del Investigador17. Si el Ma-nual del Investigador se modificara durante el ensayo, el investigadordeberá entregaruna copia revisada del Manual del Investigador al Comité.
4.4.3. Durante el ensayo el investigador deberá proporcionar al CEIC todos losdocumentos que hayan sido modificados.
4.5. Cumplimiento del Protocolo
4.5.1. El investigador/institución deberá realizar el ensayo de acuerdo con elprotocolo acordado con el promotor y, autorizado por la AEMPS y que tiene el dicta-men favorable del CEIC. El investigador deberá firmar el protocolo, o un contrato al-ternativo, para confirmar el acuerdo.
4.5.2. El investigador, no deberá realizar ninguna desviación ni modificacióndel protocolo sin el permiso del promotor y la revisión previa y dictamen favorable ala modificación, por escrito, del CEIC, salvo cuando sea necesario reducir un riesgoinminente para los sujetos del ensayo o cuando la modificación implique solamenteaspectos logísticos o administrativos (p.ej. cambio de monitor, o del número deteléfono).
4.5.3. El investigador, o una persona designada por él, deberá documentar yexplicar cualquier desviación del protocolo aprobado.
4.5.4. El investigador puede realizar una modificación o un cambio del proto-colo, sin el dictamen favorable previo del CEIC, a fin de eliminar riesgos inminentesa los sujetos del ensayo. Tan pronto como sea posible, la desviación o la modificaciónrealizada, su justificación y cuando proceda, la modificación del protocolo propuestase deberá presentar:
a) al CEIC para su revisión y dictamen favorable,
b) al promotor para su conformidad y,
c) a las autoridades competentes.
292
16 De acuerdo con el artículo 16 del RD 223/2004, el promotor es quien tiene que solicitar el dictamen del CEICacompañada de la oportuna documentación.
17 De acuerdo con el artículo 25 del RD 223/2004, la solicitud de modificaciones deberá firmarla y fecharla el pro-motor y el investigador.

4.6. Medicamentos en Investigación
4.6.1. La responsabilidad de la contabilidad de los medicamentos en investiga-ción en el centro de investigación reside en el investigador/institución18.
4.6.2. En la medida de lo permitido o requerido, el investigador o la instituciónpodrá asignar alguna o todas sus responsabilidades en relación con la contabilización delos medicamentos en investigación en el centro del ensayo a un farmacéutico u otra per-sona apropiada que esté bajo su supervisión.
4.6.3. El investigador/institución y/o un farmacéutico u otro individuo apropiado,que haya sido designado por el investigador/institución, deberá mantener un registro delenvío del medicamento al centro del ensayo, el inventario, el uso por parte de cada sujetoy la devolución al promotor o eliminación alternativa del preparado no utilizado. Estos re-gistros deberán incluir la fecha, cantidad, número de lote/serie, fecha de caducidad(cuando proceda) y código asignado al medicamento en investigación y a los sujetos delensayo. Los investigadores deberán mantener registros que documenten correctamenteque a los sujetos se les suministraron las dosis especificadas en el protocolo y que los da-tos cuadren con los medicamentos en investigación recibidos del promotor.
4.6.4. Los medicamentos en investigación deberán almacenarse según especifi-que el promotor (véase 5.13.2 y 5.14.3) y conforme a la legislación vigente19.
4.6.5. El investigador deberá garantizar que los medicamentos en investigaciónsólo se utilizarán de acuerdo con el protocolo aprobado.
4.6.6. El investigador, o una persona designada por él, deberá explicar el uso co-rrecto de los medicamentos en investigación a cada sujeto y deberá comprobar, con unaperiodicidad adecuada de acuerdo con cada ensayo, que cada sujeto sigue correctamentelas instrucciones.
4.7. Procedimientos de Aleatorización y Desenmascaramiento
El investigador deberá seguir los procedimientos de aleatorización especificadosen el protocolo del estudio, si procede, y garantizar que únicamente se abrirá el código
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
293
18 La custodia, conservación y dispensación de los medicamentos corresponderá exclusivamente a los Servicios deFarmacia de los Hospitales o de las estructuras de Atención Primaria del Sistema Nacional de Salud para su dis-pensación en dichas instituciones, de acuerdo con el artículo 2.6 de la Ley 29/2006
19 Anexo 13 de las NCF

siguiendo los criterios señalados en el protocolo. Si el estudio es ciego, deberá documen-tar y explicar inmediatamente al promotor cualquier desenmascaramiento de un medica-mento en investigación (por ejemplo, accidentalmente, o por aparición de unacontecimiento adverso grave)
4.8. Consentimiento Informado de los Sujetos del Ensayo
4.8.1. En la obtención y documentación del consentimiento informado, el investi-gador debe cumplir la legislación pertinente, las normas de de BPC y los principios éticosque tienen su origen en la Declaración de Helsinki. Antes del inicio del ensayo, el investi-gador deberá tener el dictamen favorable por escrito del CEIC del documento de consenti-miento informado y de cualquier otra información escrita que se entregue a los sujetos20.
4.8.2. Deberá modificarse el documento del consentimiento así como toda infor-mación escrita que se entregue a los sujetos, cuantas veces se disponga de nueva infor-mación que pueda ser relevante para el consentimiento del sujeto. Cualquier documentode consentimiento informado e información escrita que hayan sido modificadas deberánrecibir el dictamen favorable del CEIC antes de su utilización. Se deberá informar al su-jeto o al representante legal del sujeto de manera oportuna si se dispone de nueva infor-mación que pueda modificar la decisión del sujeto de seguir participando en el ensayo. Lacomunicación de esta información deberá estar documentada.
4.8.3. Ni el investigador ni el personal del ensayo deberán coaccionar o influir in-debidamente al sujeto para que participe o continúe su participación en un ensayo.
4.8.4. La información oral y escrita referente al ensayo, incluyendo la hoja de in-formación al sujeto, no deberá contener ningún tipo de lenguaje que lleve al sujeto o alrepresentante legal del sujeto a renunciar o a parecer que renuncia a cualquier derecholegal, o que libere o parezca liberar al investigador, a la institución, al promotor o a su per-sonal de sus obligaciones en el caso de negligencia.
4.8.5. El investigador o una persona designada por él, deberá informar de formacompleta al sujeto o, si el sujeto es incapaz de proporcionar el consentimiento informado,a su representante legal, de todos los aspectos pertinentes del ensayo, incluyendo la in-formación escrita y el dictamen favorable del CEIC.
294
20 De acuerdo con el art. 17 del RD 223/2004, el CEIC deberá considerar la idoneidad de la información escrita paralos sujetos del estudio y el procedimiento de obtención del consentimiento informado, y la justificación de la in-vestigación en personas incapaces que no pueden dar su consentimiento informado.

4.8.6. El lenguaje utilizado en la información oral y escrita sobre el ensayo, inclu-yendo la hoja de información al sujeto, no deberá ser técnico sino práctico y deberá po-der ser entendido por el sujeto o su representante legal y el testigo imparcial, cuandoproceda.
4.8.7. Antes de obtener el consentimiento informado, el investigador o la personadesignada por él, deberá dar al sujeto o a su representante legal oportunidad y tiempo su-ficiente para preguntar acerca de los detalles del ensayo y para decidir si quiere partici-par o no en el ensayo. Deberá responder de forma satisfactoria para el sujeto o surepresentante legal a todas las preguntas sobre el ensayo.
4.8.8. Antes de la participación del sujeto en el ensayo, deberá firmarse y fe-charse el documento del consentimiento informado por el sujeto o su representante legaly por la persona que les informó.
4.8.9. Si el sujeto o su representante legal no saben leer, un testigo imparcial de-berá estar presente durante la información del consentimiento informado. Después de queel documento del consentimiento y cualquier otra información escrita le sea proporcionada,leída y explicada al sujeto o a su representante legal y después de que el sujeto o el re-presentante legal del sujeto otorgue su consentimiento oral a la participación del sujetoen el ensayo y, si es capaz de hacerlo, haya firmado y fechado personalmente el consen-timiento informado, el testigo deberá firmar y fechar también el consentimiento infor-mado. Al firmar el consentimiento el testigo atestigua que la información contenida en elmismo y toda la información escrita fue fielmente explicada y aparentemente entendidapor el sujeto o su representante legal y que el consentimiento fue libremente dado por elsujeto o su representante legal.
4.8.10. Tanto la explicación del consentimiento informado como la hoja de infor-mación escrita del mismo y toda información escrita que se proporcione a los sujetos de-berá incluir la información siguiente:
a) Que el ensayo representa una investigación.
b) El propósito del ensayo.
d) Los tratamientos del ensayo y la probabilidad de asignación aleatoria para cadatratamiento.
d) Los procedimientos a seguir en el ensayo, incluyendo todos los procedimientosinvasivos.
e) Las responsabilidades del sujeto.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
295

f) Aquellos aspectos del ensayo que son experimentales.
g) Los riesgos o inconveniencias razonablemente previsibles para el sujeto y, en sucaso, para el embrión, feto o niño lactante.
h) Los beneficios razonablemente esperados. Se deberá informar claramente al su-jeto en aquellos casos en que no se pretende ningún beneficio clínico específicopara él.
i) Los procedimientos o tratamientos alternativos disponibles para el sujeto y sus po-sibles beneficios y riesgos más importantes.
j) La indemnización y/o tratamiento disponible para el sujeto en caso de cualquierperjuicio relacionado con el ensayo.
k) El prorrateo previsto de pago, si lo hay, al sujeto por su participación en el ensayo.
l) Los gastos previsibles, si los hay, al sujeto por su participación en el ensayo.
m) Que la participación del sujeto en el ensayo es voluntaria y que el sujeto puedenegarse a participar o retirarse del ensayo en cualquier momento, sin ninguna pe-nalización ni pérdida de los beneficios a los que hubieses tenido derecho de otromodo.
n) Que los monitores, auditores, CEIC, y las autoridades competentes tendrán ac-ceso directo a la historia clínica original del sujeto para la verificación de los pro-cedimientos y/o datos del ensayo clínico, sin violar la confidencialidad del sujeto,dentro de lo permitido por la normativa pertinente y que, al firmar el consenti-miento informado, el sujeto o su representante legal está autorizando el accesoa estos datos.
o) Que los registros que identifican al sujeto serán confidenciales y, según lo permi-tido por las leyes y/o regulaciones pertinentes, no estarán a disposición pública.Si se publican los resultados del ensayo, la identidad del sujeto será confiden-cial.
p) Que se informará al sujeto o al representante legal del sujeto en todo momentosi se dispone de nueva información que pueda modificar su decisión de continuaren el ensayo.
q) Las personas de contacto para obtener información adicional del ensayo y de losderechos de los sujetos participantes, y con quien contactar en caso de lesionesrelacionadas con el mismo.
r) Las circunstancias y/o razones previsibles bajo las cuales puede finalizar la par-ticipación del sujeto en el ensayo.
s) La duración esperada de la participación del sujeto en el ensayo.
t) El número aproximado de sujetos implicados en el ensayo.
296

4.8.11. Antes de su participación en el ensayo, el sujeto o el representante legaldel sujeto deberá recibir una copia del consentimiento informado por escrito firmado y fe-chado así como toda la información escrita facilitada a los sujetos. Durante la participa-ción del sujeto en el ensayo deberá recibir una copia de las actualizaciones delconsentimiento firmada y fechada, así como una copia de todas las modificaciones de lainformación escrita, que le haya sido facilitada a los sujetos.
4.8.12. Cuando un ensayo clínico (terapéutico o no terapéutico) incluya sujetosque sólo puedan ser reclutados en el ensayo con el consentimiento de su representantelegal (p.ej. los menores, o los pacientes con una demencia grave), el sujeto deberá ser in-formado del ensayo, teniendo en cuenta su nivel de comprensión y, si es capaz, deberáfirmar y fechar personalmente el consentimiento informado.
4.8.13. Excepto en el caso descrito en el punto 4.8.14, un ensayo no terapéutico(es decir un ensayo en el cual no se espera ningún beneficio clínico directo para el sujeto),deberá ser realizado en los sujetos que den personalmente su consentimiento y que firmeny fechen el consentimiento informado.
4.8.14. Los ensayos sin beneficio terapéutico pueden realizarse en sujetos conel consentimiento de un representante legal siempre que se cumplan las siguientes con-diciones:
a) Que no se puedan obtener los objetivos del ensayo mediante un ensayo con su-jetos que pueden dar su consentimiento informado personalmente.
b) Que el riesgo previsible para el sujeto sea mínimo.
c) Que el impacto negativo sobre el bienestar del sujeto sea bajo y además que seael menor posible.
d) Que el ensayo no esté prohibido por la ley.
e) Que se solicite expresamente el dictamen favorable del CEIC para la inclusiónde tales sujetos y que por tanto el dictamen favorable cubra este aspecto.
Estos ensayos, salvo excepción justificada, deberán ser realizados siempre en pa-cientes que tengan una enfermedad en la que esté indicado el medicamento en investi-gación. Los sujetos en estos ensayos deberán ser monitorizados estrechamente y seránretirados si presentan ansiedad inaceptable.
4.8.15. En situaciones de urgencia, cuando no sea posible obtener el consen-timiento previo del sujeto, se solicitará el consentimiento del representante legal del su-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
297

jeto, si está presente. En los casos en que el consentimiento previo del sujeto no seaposible y el representante legal del sujeto no esté presente, la inclusión del sujeto de-berá cumplir los requisitos descritos en el protocolo o en otra documentación aparte,con el dictamen favorable del CEIC para proteger los derechos, la seguridad y el bien-estar del sujeto y asegurar el cumplimiento de los requisitos legislativos pertinentes21.El sujeto o su representante legal será informado del ensayo en cuanto sea posible y de-berá otorgar su consentimiento para continuar su participación en el mismo si procede.(véase 4.8.10)
4.9. Registros e informes
4.9.1. El investigador deberá garantizar la exactitud, integridad, legibilidad y pun-tualidad en la presentación de los datos al promotor en el plazo de tiempo convenido asícomo de todos los informes que se le requieran.
4.9.2. Los datos incluidos en el CRD, que se deriven de documentos fuente, de-berán ser consistentes con dichos documentos o en caso contrario justificar las discrepan-cias.
4.9.3. Cualquier modificación o corrección de los datos del CRD deberá ser fe-chado, firmado con las iniciales y explicado (si es necesario) y no deberá ocultar los da-tos originales (es decir, se deberá mantener un “audit trail”). Esto es aplicable tanto a loscambios o las correcciones en papel como en soporte electrónico (véase 5.18.4 (n)). Lospromotores facilitarán a los investigadores y a los representantes designados por los inves-tigadores, una guía sobre cómo hacer tales correcciones. Los promotores deberán tenerprocedimientos escritos que garanticen que los cambios o las correcciones realizados enlos CRD por los representantes designados por el promotor están documentados, son ne-cesarios y están confirmados por el investigador. El investigador deberá conservar los re-gistros de los cambios y las correcciones.
4.9.4. El investigador y/o la institución deberán guardar los documentos del en-sayo tal como está especificado en el apartado de Documentos Esenciales para la realiza-ción de un Ensayo Clínico (véase punto 8) de acuerdo con la normativa vigente22. Elinvestigador y/o la institución deberá tomar las medidas necesarias para prevenir la des-trucción accidental o prematura de estos documentos.
298
21 Ver artículo 7 del Real Decreto 223/2004.22 De acuerdo con la Orden Ministerial, durante un periodo de al menos 5 años siempre que el promotor no especi-
fique un periodo superior (ver punto 4.9.5)

4.9.5. Se deberán conservar los documentos esenciales hasta al menos 2 años des-pués de la última aprobación de una solicitud de comercialización en Europa, EEUU o Japóny hasta que no quede ninguna solicitud de comercialización pendiente o en proyecto en di-chas regiones o hasta que hayan pasado al menos 2 años desde la suspensión formal del des-arrollo clínico del medicamento en investigación. Sin embargo, se deberán conservar estosdocumentos durante un periodo de tiempo más prolongado si así lo establecen las normas re-guladoras aplicables o un acuerdo con el promotor. Es responsabilidad del promotor informaral investigador/institución a partir de qué fecha no es necesario conservar estos documentos(véase 5.5.12).
4.9.6. Los aspectos financieros del ensayo deberán estar documentados en unacuerdo entre el promotor, el investigador/ institución.
4.9.7. Ante la solicitud del monitor, auditor, CEIC o autoridad sanitaria, el investiga-dor/institución deberá ser capaz de tener disponibles todos los archivos relacionados con elensayo.
4.10. Informes de seguimiento
4.10.1. El investigador presentará al CEIC un resumen escrito de la situación del en-sayo, anualmente, o con más frecuencia si así se lo requiere.
4.10.2. El investigador facilitará puntualmente informes escritos al promotor, CEIC(véase 3.3.8) y, cuando proceda, a la institución acerca de cualquier cambio que afecte sig-nificativamente la realización del ensayo o incremente el riesgo para los sujetos.
4.11. Informes de seguridad
4.11.1. Se deberá informar inmediatamente al promotor de todos los acontecimien-tos adversos graves con excepción de aquellos que el protocolo u otro documento (p.ej. Manualdel Investigador) indique expresamente que no necesitan una notificación inmediata. La noti-ficación inmediata deberá ir seguida de puntuales informes detallados. El informe inmediatoy de seguimiento deberá identificar a los sujetos mediante un código único asignado al sujetodel ensayo y no por el nombre del sujeto, sus números de identificación personal o su direc-ción. El investigador deberá ajustarse también a la normativa legal referente a notificación delas reacciones adversas graves e inesperadas, a las autoridades pertinentes y al CEIC23.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
299
23 Ver Art. 42 del RD 223/2004

4.11.2. Los acontecimientos adversos o las alteraciones de laboratorio identificadasen el protocolo como críticas para la evaluación de seguridad deberán ser comunicados al pro-motor de acuerdo con los requisitos establecidos y dentro de los periodos de tiempo especifi-cados en el protocolo.
4.11.3. Cuando el investigador notifique un fallecimiento, deberá facilitar al promo-tor y al CEIC toda la información complementaria que le soliciten (p. ej. El informe de la au-topsia y los últimos informes médicos)24.
4.12. Finalización anticipada o Suspensión de un Ensayo
Si el ensayo termina de forma anticipada o se suspende por cualquier razón, el inves-tigador deberá informar puntualmente a los sujetos del ensayo, garantizando el tratamiento yseguimiento apropiado de los mismos. Además:
4.12.1. Si el investigador finaliza o suspende un ensayo sin el acuerdo previo del pro-motor, deberá informar puntualmente a la institución, así como facilitar al promotor y al Co-mité una justificación por escrito de la causa de la finalización o suspensión.
4.12.2. Si el promotor finaliza o suspende un ensayo (véase 5.21), el investigador de-berá informar a la institución donde se realice y el investigador/institución deberán informar rá-pidamente al CEIC presentando una justificación escrita de las razones de la finalización osuspensión25.
4.12.3. Si el Comité finaliza o retira el dictamen favorable de un ensayo (véase 3.1.2y 3.3.9) informará de inmediato a la AEMPS, a la Dirección del centro, al investigador y al pro-motor facilitando una justificación escrita de las razones de la finalización o suspensión.
4.13. Informe Final del Investigador
Al finalizar el ensayo, el investigador, cuando proceda, informará a la institución; el in-vestigador/institución enviará al CEIC un resumen de los resultados del ensayo y enviará a laautoridad reguladora los informes que ésta solicite26.
300
24 Ver Art.42.3 del RD 223/200425 Artículo 27.2 del RD 223/2004: En el caso de finalización anticipada, en el plazo de 15 días el promotor remi-
tirá a la AEMPS y a los CEIC implicados un informe que incluya los datos obtenidos hasta el momento de su con-clusión anticipada, así como los motivos de ésta, y en su caso las medidas adoptadas en relación con los sujetosparticipantes en el ensayo.
26 Ver Art. 27.3.del RD 223/2004: En el plazo de un año desde la finalización del estudio, el promotor remitirá a laAEMPS y a los CEIC implicados, un resumen del informe final sobre los resultados del estudio.

5. PROMOTOR
5.1. Control y Garantía de Calidad
5.1.1. El promotor es el responsable de realizar y mantener un sistema de con-trol y garantía de calidad con procedimientos normalizados de trabajo (PNT) escritosa fin de asegurar que los ensayos sean realizados y los datos sean generados, documen-tados (registrados) y comunicados de acuerdo con el protocolo, la BPC y la normativavigente.
5.1.2. El promotor es el responsable de llegar a un acuerdo entre todas las par-tes implicadas que asegure el acceso directo (véase 1.2.1) a todos los centros del ensayo,a todos los datos/documentos originales y los informes necesarios para garantizar la mo-nitorización y auditoria por parte del promotor y las inspecciones por parte de las autori-dades nacionales y extranjeras.
5.1.3. Se deberá aplicar un control de calidad en cada fase del manejo de datospara asegurar que todos ellos son fiables y que han sido procesados de forma correcta.
5.1.4. Los acuerdos realizados entre el promotor, el investigador/institución o cual-quier otra parte implicada en el ensayo clínico deberán constar por escrito, como parte delprotocolo o en un acuerdo aparte.
5.2. Organización de Investigación por Contrato (CRO)
5.2.1. Un promotor puede transferir alguna o todas las obligaciones y funcionesrelacionadas con el ensayo a una CRO, pero la última responsabilidad sobre la calidad eintegridad de los datos del ensayo siempre recae en el promotor. La CRO deberá imple-mentar la garantía y el control de calidad.
5.2.2. Se deberá especificar por escrito cualquier obligación o función relacio-nada con el ensayo que esté transferida y asumida por una CRO.
5.2.3. Cualquier obligación o función relacionada con el ensayo que no sea trans-ferida y, asumida por una CRO de forma específica, es responsabilidad del promotor.
5.2.4. Todas las referencias que se hacen al promotor en esta guía se deberán apli-car también a la CRO cuando asuma las obligaciones y funciones relacionadas con el en-sayo de un promotor.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
301

5.3. Pericia Médica
El promotor deberá designar el personal médico debidamente cualificado que es-tará fácilmente disponible para aconsejar sobre las cuestiones o problemas médicos rela-cionados con el ensayo. Si es preciso se pueden nombrar, con este propósito, especialistasexternos.
5.4. Diseño del Ensayo
5.4.1. El promotor utilizará personas debidamente cualificadas (p.ej. bioestadís-ticas, farmacólogos clínicos y médicos), cuando proceda, durante todos los estadios delensayo, desde el diseño del protocolo y el cuaderno de recogida de datos así como el plande análisis, hasta el análisis y la preparación de los informes intermedios y finales del en-sayo clínico.
5.4.2. Como guía adicional para la realización del Protocolo del Ensayo Clínico yModificaciones al Protocolo véase el punto 6 (Guía de la ICH sobre la Estructura y Conte-nido de los informes de Estudios Clínicos y otras guías ICH apropiadas para el diseño pro-tocolo y realización del ensayo.
5.5. Dirección del Ensayo, Manejo de Datos y Preparación de Documentos
5.5.1. El promotor deberá utilizar personas debidamente cualificadas para super-visar la realización global del ensayo, manejar y verificar los datos, realizar los análisis es-tadísticos y preparar los informes del ensayo.
5.5.2. El promotor puede establecer un comité independiente de monitorizaciónde datos (IDMC) a fin de evaluar el desarrollo de un ensayo clínico, incluyendo el análisisperiódico de los datos de seguridad y las variables críticas de eficacia con el fin de reco-mendar al promotor la conveniencia de continuar, modificar o detener un ensayo. DichoComité deberá tener procedimientos normalizados de trabajo por escrito y conservar las ac-tas de todas sus reuniones.
5.5.3. Cuando en el ensayo se procesen datos electrónicos del ensayo y/o se uti-licen sistemas informáticos electrónicos remotos, el promotor deberá:
a) Asegurar y documentar que el sistema de procesamiento de datos electrónicosse ajusta a los requisitos establecidos por el promotor en cuanto a la integri-dad, exactitud, fiabilidad y consistencia del proceso (es decir, validación)
302

b) Elaborar y actualizar PNT para la utilización de estos sistemas.
c) Garantizar que el diseño de los sistemas permite la modificación de datos de talmanera que estos cambios queden documentados y que los datos originales nosean eliminados (es decir, mantener un método de seguimiento retrospectivo “au-dit trail”, seguimiento retrospectivo de los datos “data trail”, o seguimiento retros-pectivo de los cambios “edit trail”).
d) Mantener un sistema de seguridad que impida el acceso no autorizado a los da-tos.
e) Elaborar y actualizar una lista de las personas que están autorizadas a hacer cam-bios en los datos ( véase 4.1.5 y 4.9.3)
f) Crear copias de seguridad de los datos.
g) Salvaguardar el enmascaramiento, si lo hay (p.ej mantener el enmascaramientode los datos durante la entrada y procesamiento de los mismos).
5.5.4. Si se modifican los datos durante su procesamiento, siempre deberá ser po-sible comparar los datos originales y las observaciones con los datos procesados.
5.5.5. El promotor deberá utilizar un código de identificación de los sujetos que nosea ambiguo (véase 1.58) que permita la identificación de todos los datos registrados paracada sujeto.
5.5.6. El promotor, u otros propietarios de los datos, conservarán todos los documen-tos esenciales específicos del promotor, que pertenezcan al ensayo (véase 8. DocumentosEsenciales para la Realización de un Ensayo Clínico).
5.5.7. El promotor conservará todos sus documentos esenciales específicos deacuerdo con la normativa pertinente de los países donde el medicamento esté aprobado y/odonde el promotor pretenda solicitar su aprobación.
5.5.8. Si el promotor interrumpe el desarrollo clínico de un medicamento en inves-tigación (es decir, para alguna o todas la indicaciones, vías de administración o forma far-maceútica) el promotor deberá mantener todos los documentos esenciales específicos delpromotor durante por lo menos 2 años después de la interrupción formal o de acuerdo conla normativa vigente de cada país.
5.5.9. Si el promotor interrumpe el desarrollo clínico de un medicamento en inves-tigación, el promotor deberá notificarlo a todos los investigadores e instituciones del ensayo
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
303
27 Ver Art. 27.2 del RD 223/2004 (remitir en el plazo de 15 días informe a AEMPS y CEIC implicados)

así como a las autoridades reguladoras27.
5.5.10. Cualquier transferencia de la propiedad de los datos deberá ser comuni-cada a la autoridad reguladora.
5.5.11. Se deberán conservar los documentos esenciales específicos del promo-tor hasta al menos dos años después de la última aprobación de comercialización en unaregión ICH, y hasta que no haya solicitudes de comercialización pendientes ni en proyectoen una región ICH; o hasta que hayan pasado al menos 2 años desde la interrupción for-mal del desarrollo clínico del medicamento en investigación. Sin embargo, se deberánguardar estos documentos durante un periodo de tiempo mayor si así lo requieren los re-quisitos legales pertinentes o si lo necesita el promotor.
5.5.12. El promotor deberá informar por escrito a los investigadores/institución dela necesidad de guardar los documentos así como la fecha a partir de la cual ya no seanecesario guardar por más tiempo los documentos relacionados con el ensayo.
5.6. Selección del Investigador
5.6.1. El promotor es responsable de la selección de los investigadores y las ins-tituciones. Cada investigador deberá estar cualificado por su formación y experiencia ydeberá tener los recursos adecuados (véase 4.1, 4.2) para realizar correctamente el en-sayo para el que ha sido seleccionado. Si se va a utilizar un comité coordinador o investi-gadores coordinadores en un ensayo multicéntrico, el promotor es responsable de laorganización y selección de cada uno de ellos.
5.6.2. Antes de llegar a un acuerdo con un investigador para realizar un ensayo,el promotor deberá facilitar al investigador/institución el protocolo y el Manual del Inves-tigador actualizado, y darle el tiempo necesario para que revise el protocolo y toda la in-formación facilitada.
5.6.3. El promotor obtendrá un acuerdo con el investigador para que éste :
a) Realice el ensayo de acuerdo con la guía de BPC, los requisitos legales perti-nentes (véase 4.1.3), el protocolo acordado con el promotor y que cuenta conel dictamen favorable del CEIC (véase 4.5.1)28;
304
28 De acuerdo con el Art. 15 del Real Decreto 223/2004 es necesaria también la autorización previa de la AEMPSantes del inicio del estudio.

b) Cumpla con los procedimientos de registro y notificación de datos;
c) Facilite la monitorización, auditoria e inspección del estudio ( véase 4.1.4) y
d) Archive los documentos esenciales del ensayo hasta que el promotor le in-forme de que ya no es necesario archivarlo por más tiempo (véase 4.9.4 y5.5.12).
El promotor y el investigador/institución deberán firmar el protocolo, u otro docu-mento, para confirmar este acuerdo.
5.7. Asignación de las Responsabilidades y Funciones
Previamente al inicio del ensayo, el promotor deberá definir, establecer y asignartodas las obligaciones y funciones relacionadas con el ensayo.
5.8. Indemnización a los Sujetos e Investigadores
5.8.1. Si lo requiere la legislación vigente29, el promotor deberá concertar un se-guro u otra garantía financiera y estos cubrirán las responsabilidades30 del investigador yde la institución, que cubra los daños como consecuencia del ensayo clínico, excepto paraaquellas reclamaciones que surjan por mala práctico o negligencia.
5.8.2. Las pólizas y procedimientos del promotor deberán cubrir los costes de tra-tamiento de los sujetos del ensayo en el caso de producirse daños relacionados con el en-sayo de acuerdo con la legislación vigente31.
5.8.3. Cuando los sujetos del ensayo reciban una indemnización el método y laforma de indemnización deberá cumplir la legislación vigente32.
5.9. Financiación
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
305
29 De acuerdo con el Art. 8.1 del Decreto 223/2004: “Solo podrá realizarse un ensayo clínico con medicamentos eninvestigación si, previamente, se ha concertado un seguro u otra garantía financiera que cubra los daños y perjui-cios que puedan resultar para la persona como consecuencia del ensayo, salvo que el ensayo se refiera únicamentea medicamentos autorizados en España, su utilización en el ensayo se ajuste a las condiciones de uso autoriza-das y el Comité Ético de Investigación Clínica considere que las intervenciones a las que serán sometidos los su-jetos por su participación en el ensayo supongan un riesgo equivalente o inferior al que correspondería a suatención en la práctica clínica habitual”. Nota: El Real Decreto no exceptúa las reclamaciones que surjan por malapráctico o negligencia.
30 De acuerdo con el Art. 8.2 del Real Decreto 223/2004, el seguro también debe cubrir al promotor. 31 Ver Art. 8.3, 4, 5, 6 y 7 del Real Decreto 223/2004.32 Ver Art. 8 del Real Decreto 223/2004.

Los aspectos financieros del ensayo deberán estar documentados en un contratoentre el promotor y el investigador/institución.
5.10. Notificación/Solicitud a las Autoridades Reguladoras
Antes de iniciar el ensayo clínico, el promotor (o el promotor y el investigador silo requiere la legislación) deberá enviar la solicitud de autorización del ensayo clínicoa la autoridad reguladora33 para la revisión, aceptación y aprobación necesaria (deacuerdo con la legislación vigente) para iniciar el estudio. Todas las notificaciones o so-licitudes deberán estar fechadas y contener suficiente información para identificar elprotocolo.
5.11. Confirmación de la evaluación por el CEIC
5.11.1. El promotor deberá obtener del investigador/institución34:
a) El nombre y la dirección del CEIC del investigador/institución.
b) Declaración del CEIC de que está organizado y funciona de acuerdo con la BPCy las normas vigentes.
c) El dictamen favorable del Comité y, si lo requiere el promotor, una copia actua-lizada del protocolo, el consentimiento informado escrito y cualquier otra infor-mación escrita que sea facilitada a los sujetos, los procedimientos dereclutamiento de sujetos y los documentos relacionados con los pagos e in-demnización disponible para los sujetos y todos los documentos que pueda ha-ber solicitado el CEIC.
5.11.2. Si el CEIC condiciona su dictamen favorable a la modificación en cual-quier aspecto del ensayo, tales como modificaciones del protocolo, la hoja del consenti-miento informado escrito y cualquier otra información escrita que sea facilitada a lossujetos y/u otros procedimientos, el promotor deberá obtener del investigador/institución32
una copia de las modificaciones hechas y la fecha en que el Comité emitió el dictamenfavorable.
5.11.3. El promotor deberá obtener del investigador/institución32 la documenta-
306
33 Ver Art. 15 del Real Decreto 223/2004. Para la realización del estudio es necesario el dictamen favorable del CEICy del Director de cada centro donde se va a realizar el estudio así como de la autorización de la AEMPS
34 En España se lo notificará el propio Comité al promotor ya que éste es quien hace la solicitud

ción y las fechas de los dictámenes favorables del Comité relacionados con una nuevaevaluación o aprobación, así como si se produjera la retirada o suspensión del dictamenfavorable.
5.12. Información sobre los Medicamentos en Investigación
5.12.1. Cuando se planifican los ensayos, el promotor garantizará que existen da-tos de seguridad y eficacia, procedentes de estudios no clínicos y/o ensayos clínicos, su-ficientes para avalar su utilización en seres humanos por la vía, a la dosis, para la duracióny en la población que se indica en el estudio.
5.12.2. El promotor deberá actualizar el Manual del Investigador a medida quedisponga de nueva información relevante (véase 7. Manual del Investigador).
5.13. Fabricación, Envasado, Etiquetado y Codificación de los Medicamentos en Investigación
5.13.1. El promotor deberá asegurar que el medicamento en investigación (in-cluyendo el comparador y placebo, si procede) tiene las características apropiadas para lafase de desarrollo del medicamento, es fabricado de acuerdo con las Normas de CorrectaFabricación y está codificado y etiquetado de manera que no se rompe el enmascara-miento, si lo hubiera. Además el etiquetado deberá cumplir la legislación pertinente35.
5.13.2. El promotor deberá determinar, para el medicamento en investigación,la temperatura adecuada de almacenamiento, las condiciones de almacenamiento (p.ej.protección de la luz), el tiempo de almacenamiento, las soluciones necesarias para la re-constitución y procedimiento de la misma y los productos sanitarios necesarios para lainfusión del medicamento si los hubiera. El promotor deberá informar a todas las partesimplicadas (p.ej. monitores, investigadores, farmacéuticos, jefes de almacén) de estasparticularidades.
5.13.3. El medicamento en investigación se empaquetará adecuadamente paraprevenir su contaminación y deterioro durante el transporte y almacenamiento.
5.13.4. En los ensayos ciegos, el sistema de codificación del medicamento en in-vestigación incluirá un sistema de identificación rápida en caso de urgencia médica, pero
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
307
35 Anexo 13 de las NCF, como se recoge en el artículo 33 del Real Decreto 223/2004

que no permita la apertura indetectable del ciego.
5.13.5. Si se hacen cambios relevantes en la formulación farmacéutica del medica-mento en investigación o en el medicamento comparador durante el desarrollo clínico, antesde la utilización de la nueva formulación en ensayos clínicos, se deberá disponer de los resul-tados de cualquier estudio adicional con la nueva formulación del medicamento (por. ej. esta-bilidad, tasa de disolución, biodisponibilidad) que sean necesarios para evaluar si estos cambiospodrían alterar de forma significativa el perfil farmacocinético del medicamento.
5.14 Suministro y Manejo del Medicamento en Investigación
5.14.1. El promotor es responsable de suministrar al investigador/institución elmedicamento en investigación.
5.14.2. El promotor no deberá suministrar a un investigador/institución el medi-camento en investigación hasta que el promotor obtenga toda la documentación necesa-ria (dictamen favorable del CEIC y autorización de la AEMPS).
5.14.3. El promotor deberá asegurar que los procedimientos escritos incluyan lasinstrucciones que el investigador/institución deberá seguir para el manejo y almacenamientodel medicamento en investigación en el ensayo y de la documentación al respecto. Los pro-cedimientos deberán indicar la recepción adecuada y segura, el manejo, el almacenamiento,la dispensación, la recuperación del medicamento no utilizado por los sujetos y la devolu-ción del medicamento en investigación no utilizado al promotor (o procedimientos alterna-tivos si el promotor la autoriza y está de acuerdo con la legislación pertinente).
5.14.4. El promotor deberá:
a) Asegurar la entrega puntual del medicamento en investigación al investigador.
b) Mantener registros de los documentos de envío, recepción, distribución, devo-lución y destrucción del medicamento en investigación (véase punto 8. Docu-mentos Esenciales para la Realización de un Ensayo Clínico)
c) Mantener un sistema documentado de la retirada de medicamentos en inves-tigación (por ej. retirada de medicamentos deficientes, reclamación del medi-camento después de la finalización del ensayo, reclamación del medicamentocaducado).
d) Mantener un sistema documentado para la disponibilidad de medicamentos eninvestigación no utilizados.
308

5.14.5. El promotor deberá
a) Tomar medidas oportunas que aseguren que el medicamento en investigaciónes estable durante el periodo de utilización en el ensayo.
b) Conservar cantidades suficientes del medicamento en investigación que se utilizaen el ensayo para confirmar sus especificaciones, si es necesario, y mantener regis-tros de los análisis y características de las muestras de los lotes. En la medida enque la estabilidad lo permita, las muestras deberán ser conservadas hasta que secompleten los análisis de los datos del ensayo o como marque la normativa vi-gente36, eligiendo de los dos el periodo más largo de los dos.
5.15. Acceso a los documentos
5.15.1. El promotor deberá asegurar que se especifique en el protocolo o cualquierotro acuerdo escrito que el investigador/institución facilitará el acceso a los datos/documentosfuente para la monitorización del ensayo, auditorias, revisiones por el CEIC e inspecciones sa-nitarias.
5.15.2. El promotor deberá verificar que cada sujeto ha dado su consentimiento porescrito para permitir el acceso a los datos de su historia clínica para la monitorización del en-sayo, auditorias, revisiones del CEIC e inspecciones sanitarias.
5.16. Información de Seguridad
5.16.1 El promotor es responsable de la evaluación continua de la seguridad del me-dicamento en investigación.
5.16.2 El promotor deberá notificar rápidamente al investigador/institución implica-dos y a las autoridades sanitarias cualquier información importante que pudiera afectar nega-tivamente la seguridad de los sujetos, la realización del ensayo o modificar el dictamenfavorable del CEIC.
5.17. Notificación de Reacciones Adversas Medicamentosas
5.17.1 El promotor deberá notificar de forma expeditiva a todos los investigadores/ins-tituciones implicados, a los CEIC cuando sea necesario y a las autoridades sanitarias los infor-mes de todas las reacciones adversas al fármaco (RAM) que sean a la vez graves e inesperadas.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
309
36 Anexo 13 de las NCF.

5.17.2 Tales informes deberán cumplir los requisitos legislativos pertinentes y conla guía E2A de la ICH para la Gestión de Datos de Seguridad Clínica: Definiciones y Normaspara Informes Expeditivos)
5.17.3 El promotor deberá presentar a las autoridades competentes todas las ac-tuaciones de seguridad y los informes periódicos y actualizaciones de seguridad, tal ycomo marquen los requisitos pertinentes37.
5.18. Monitorización
5.18.1 Objetivos
Los objetivos de la monitorización de un ensayo son verificar que:
a) Los derechos y bienestar de los sujetos humanos están protegidos.
b) Los datos obtenidos en el ensayo son exactos, completos y verificables con losdocumentos fuente.
c) La realización del ensayo está de acuerdo con el protocolo y las enmiendasaprobadas, con la BPC y con la normativa vigente.
5.18.2 Selección y cualificación de los monitores
a) Los monitores deberán ser nombrados por el promotor
b) Los monitores deberán estar debidamente formados y tener el conocimientocientífico y/o clínico necesario para monitorizar el ensayo adecuadamente. Lacualificación del monitor estará documentada.
c) Los monitores deberán estar familiarizados con el medicamento en investiga-ción, el protocolo, la hoja de información escrita del consentimiento informadoy cualquier otra información escrita que sea facilitada a los sujetos, los PNT delpromotor, la BPC y la legislación vigente.
5.18.3 Alcance y naturaleza de la monitorización
El promotor se asegurará que la monitorización de los ensayos sea adecuada. Elpromotor deberá determinar el alcance y naturaleza apropiados de la monitorización. Ladeterminación del alcance y la naturaleza de la monitorización deberán basarse en consi-
310
37 Ver Art. 47 del Real Decreto 223/2004

deraciones tales como el objetivo, propósito, diseño, complejidad, enmascaramiento, ta-maño y variables principales del ensayo. En general, es necesaria la monitorización en ellugar del ensayo antes, durante y tras de la finalización del mismo. Sin embargo, en cir-cunstancias excepcionales, el promotor puede determinar que la monitorización centrali-zada junto con otros procedimientos tales como la formación de los investigadores yreuniones con ellos y una guía detallada por escrito, puedan garantizar la realización ade-cuada del ensayo de acuerdo con la BPC. La selección aleatoria puede ser un métodoaceptable para seleccionar los datos que van a ser verificados.
5.18.4. Responsabilidades del monitor
El monitor, de acuerdo con los requisitos del promotor, deberá asegurar que elensayo se realiza y se documenta adecuadamente llevando a cabo las siguientes activida-des, cuando sea importante y necesario para el ensayo y el centro de investigación:
a) Actuar como el principal interlocutor entre el promotor y el investigador.
b) Verificar que el investigador reúne la cualificación y recursos adecuados (véase4.1, 4.2 y 5.6) a lo largo del ensayo. Además, verificar que las instalaciones,incluyendo laboratorios, equipamiento y personal son adecuados para la reali-zación segura y correcta del ensayo a lo largo del ensayo.
c) Verificar para el medicamento en investigación:
i. Que el tiempo y las condiciones de almacenamiento son aceptables y quelos suministros son suficientes durante el ensayo.
ii. Que el medicamento en investigación solamente se suministra a los sujetosseleccionados para recibirlo y a las dosis especificadas en el protocolo.
iii. Que a los sujetos se les proporcionan las instrucciones necesarias para eluso y manejo correcto, almacenamiento y devolución del medicamento eninvestigación.
iv. Que la recepción, utilización y devolución del medicamento en investigaciónen los centros de investigación esté adecuadamente controlado y documen-tado.
v. Que la disponibilidad del medicamento en investigación no utilizado en loscentros del ensayo cumple con los requisitos legales pertinentes y sigue losprocedimientos del promotor.
d) Verificar que el investigador sigue el protocolo aprobado y las modificacionesaprobadas, si las hubiere.
e) Verificar que el consentimiento informado escrito fue obtenido antes de la par-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
311

ticipación de cada sujeto en el ensayo.
f) Garantizar que el investigador recibe el Manual del Investigador actualizado, todoslos documentos y todos los suministros necesarios del ensayo para que lo realice deforma adecuada y de acuerdo con los requisitos legales pertinentes.
g) Garantizar que el investigador y su personal del ensayo están correctamente in-formados acerca del ensayo.
h) Verificar que el investigador y su personal del ensayo realizan las funciones es-pecíficas del ensayo de acuerdo con el protocolo y cualquier otro acuerdo es-crito entre el promotor y el investigador/institución, y que no han delegado estasfunciones a personas no autorizadas.
i) Verificar que el investigador recluta solamente a sujetos que cumplen los crite-rios de selección.
j) Notificar la velocidad de reclutamiento de los sujetos.
k) Verificar que los documentos fuente y los demás registros del ensayo son pre-cisos, completos, actualizados y están correctamente archivados.
l) Verificar que el investigador realiza todos los informes, notificaciones, peticionesy solicitudes, y que estos documentos son exactos, completos, legibles, estándentro del plazo de tiempo estipulado, están fechados e identifican al ensayo.
m) Verificar la exactitud e integridad de los datos incluidos en el CRD, con res-pecto a los documentos fuente y otros registros relacionados con el ensayo. Elmonitor específicamente verificará que:
i. Los datos requeridos en el protocolo están anotados con exactitud en losCRD y concuerdan con los datos fuente.
ii. Todas las dosis y/o cualquier modificación del tratamiento están bien docu-mentadas para cada uno de los sujetos del ensayo.
iii. Los acontecimientos adversos, tratamientos concomitantes y enfermedadesintercurrentes se notifican de acuerdo con el protocolo en los CRD.
iv. Las visitas, pruebas y exploraciones que los sujetos no hayan realizado cons-tan claramente como tales en los CRD.
v. Todas las retiradas y abandonos de los sujetos incluidos en el ensayo cons-tan y están justificadas en los CRD.
n) Informar al investigador de cualquier error, omisión o ilegibilidad en los regis-tros del CRD. El monitor deberá asegurarse de que se han hecho las correccio-nes, adiciones o supresiones apropiadas y que constan la fecha, justificación(en caso necesario) e iniciales del investigador o de un miembro del personaldel ensayo debidamente autorizado para realizar cambios en el CRD en nom-
312

bre del investigador. Esta autorización deberá estar documentada.
o) Determinar si se han comunicado todos los acontecimientos adversos de formaadecuada y dentro de los plazos contemplados en la BPC, el protocolo, el CEIC,el promotor y los requisitos legales pertinentes.
p) Determinar si el investigador mantiene los documentos esenciales (véase epí-grafe 8. Documentos Esenciales para la Realización de un Ensayo Clínico).
q) Comunicar al investigador cualquier desviación del protocolo, PNT, BPC y dela legislación vigente y tomar las medidas necesarias para prevenir que se re-pitan las desviaciones detectadas.
5.18.5. Procedimientos de Monitorización
El monitor deberá seguir los PNT escritos establecidos por el promotor, así comoaquellos procedimientos especificados por el mismo para la monitorización de un ensayoconcreto.
5.18.6.Informe de Monitorización
a) El monitor deberá presentar un informe escrito al promotor después de cada vi-sita al centro de investigación o después de cada comunicación relacionadacon el ensayo.
b) Los informes deberán incluir la fecha, el centro, el nombre del monitor y elnombre del investigador u otra persona con la que se contacte.
c) Los informes deberán incluir un resumen de los aspectos revisados por el mo-nitor y sus comentarios referentes a todos los hallazgos o hechos, desviacioneso deficiencias relevantes así como las conclusiones, y acciones realizadas o allevar a cabo y/o acciones recomendadas con el fin de garantizar el cumpli-miento.
d) La revisión y seguimiento del informe de monitorización con el promotor deberáestar documentada por el representante designado por el promotor.
5.19. Auditoria
Cuando el promotor realiza auditorias como parte de la implementación de la ga-rantía de calidad, deberá considerar:
5.19.1. Objetivo
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
313

El propósito de una auditoria realizada por el promotor, que es independiente yajena de la monitorización rutinaria o de las funciones de control de calidad, deberá eva-luar la realización de un ensayo y el cumplimiento del protocolo, los PNT, la BPC y los re-quisitos legales pertinentes.
5.19.2. Selección y cualificación de los auditores
a) Para realizar auditorias, el promotor deberá nombrar personas que sean inde-pendientes del sistema/ensayos clínicos.
b) El promotor deberá asegurarse de que los auditores estén debidamente cuali-ficados por su formación y experiencia para realizar auditorias de forma ade-cuada. Las cualificación de un auditor deberá estar documentadas.
5.19.3. Procedimiento de las Auditorias
a) El promotor deberá asegurarse de que las auditorias de los sistemas/ensayos clí-nicos se realizan de acuerdo con los procedimientos escritos del promotor so-bre qué y cómo auditar, la frecuencia de las auditorias; así como la forma ycontenido de los informes de las mismas.
b) El plan de auditoria del promotor y los procedimientos para la auditoria de unensayo se establecerán en función de la importancia del ensayo en cuanto a losdatos a presentar a las autoridades reguladoras, el número de sujetos en el en-sayo, el tipo y la complejidad del ensayo, el nivel de riesgos para los sujetos delensayo y cualquier problema que se identifique.
c) Las observaciones y los hallazgos del auditor deberán estar documentadas.
d) Para preservar la independencia y el valor de las auditorias, las autoridades re-guladoras no deberán pedir los informes de auditoria de forma rutinaria. Las au-toridades pueden solicitar el acceso a un informe de auditoria para un casoconcreto cuando exista evidencia de un serio incumplimiento de la BPC o du-rante el curso de una demanda legal.
e) Cuando sea requerido por la ley o normativa pertinente, el promotor deberá pro-porcionar un certificado de auditoria.
5. 20.Incumplimiento
5.20.1. El incumplimiento del protocolo, los PNT, la BPC y/o los requisitos lega-les pertinentes por un investigador/institución o por los miembros del equipo del investi-gador conllevará una intervención rápida por parte del promotor para asegurar el
314

cumplimiento.
5.20.2. Si la monitorización y/o la auditoria identifican un incumplimiento gravey/o persistente por parte de un investigador/institución, el promotor deberá finalizar la par-ticipación de dicho investigador/institución en el ensayo. Cuando finaliza la participaciónde un investigador/institución debido a incumplimiento, el promotor deberá comunicarlorápidamente a las autoridades reguladoras38.
5.21. Finalización Prematura o Suspensión de un Ensayo
Si un ensayo finaliza prematuramente o se suspende, el promotor deberá informarrápidamente al investigador/institución y a las autoridades reguladoras de la finalizacióno suspensión así como de las razones que la motivaron. Además, el promotor o el investi-gador/institución deberán informar rápidamente al CEIC y facilitarle la justificación de lafinalización o suspensión, tal y como especifiquen los requisitos legales pertinentes.
5.22. Informes del Ensayo/ Estudio Clínico:
Tanto si se completa el ensayo como si se finaliza prematuramente, el promotordeberá asegurarse de que los informes del ensayo clínico se preparan y se facilitan a lasagencias reguladoras tal y como especifican los requisitos legales pertinentes. El promo-tor deberá también asegurarse de que los informes de ensayos clínicos en las solicitudesde comercialización cumplen las normas de la Guía ICH para la Estructura y Contenido delos Informes de Estudios Clínicos. (Nota: la Guía ICH sobre la Estructura y Contenido deInformes de Ensayos Clínicos especifica que los informes abreviados del estudio puedenser aceptables en ciertos casos).
5.23. Ensayos Multicéntricos
Para los ensayos multicéntricos, el promotor deberá garantizar que:
5.23.1. Todos los investigadores realizan el ensayo con estricto cumplimiento delprotocolo acordado con el promotor y, cuando proceda, con las autoridades reguladoras,y después de emitido el dictamen favorable del CEIC.
5.23.2. Los CRD se han diseñado para recoger los datos requeridos en todos loscentros en que se realiza el ensayo multicéntrico. Para aquellos investigadores que reco-gen datos adicionales, se deberán facilitar CRD complementarios que están diseñados
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
315
38 En caso de terminación anticipada se deberá informar a la AEMPS y a los CEIC implicados (ver Art. 27.2 del RealDecreto 223/2004).

para recoger dichos datos.
5.23.3. Las responsabilidades de los investigadores coordinadores y de los otrosinvestigadores participantes se documentarán antes del inicio del ensayo.
5.23.4. Todos los investigadores recibirán instrucciones, sobre como seguir el pro-tocolo, y como cumplir con un conjunto uniforme de normas para la evaluación de hallaz-gos clínicos y analíticos, y sobre cómo completar los CRD.
5.23.5. La comunicación entre investigadores sea fluida.
6. PROTOCOLO DEL ENSAYO CLÍNICO Y MODIFICACIONES AL PROTOCOLO
El protocolo del ensayo clínico debe incluir los aspectos que se recogen a conti-nuación. Sin embargo, la información especifica de un centro puede estar en una páginaseparada del protocolo o incluida en un acuerdo aparte y se puede incluir parte de la in-formación listada a continuación en documentos referenciados en el protocolo, tales comoel Manual del Investigador.
6.1. Información General
6.1.1. Título, número de identificación del protocolo y fecha. Cualquier modifica-ción deberá llevar también el número de la modificación y la fecha.
6.1.2. Nombre y dirección del promotor y monitor (si es diferente del promotor).
6.1.3. Nombre y cargo de la persona autorizada por el promotor para firmar elprotocolo y las modificaciones al protocolo.
6.1.4. Nombre, cargo, dirección y números de teléfono de los expertos médicos(u odontólogos cuando proceda) del promotor del ensayo.
6.1.5. Nombre y cargo de todos los investigadores responsables de la realizacióndel ensayo y la dirección y números de teléfono de los centros del ensayo.
6.1.6. Nombre, cargo, dirección y número de teléfono del médico cualificado (uodontólogo cuando proceda) que es responsable de todas las decisiones médicas (u odon-tológicas) en cada centro del ensayo (si es diferente del investigador).
316

6.1.7. Nombre y direcciones de los laboratorios clínicos y los departamentos mé-dicos o técnicos o instituciones implicadas en el ensayo.
6.2. Justificación
6.2.1. Nombre y descripción del medicamento en investigación.
6.2.2. Resumen de los hallazgos de los estudios no clínicos y clínicos que pue-dan ser relevantes para el ensayo actual.
6.2.3. Resumen de los riesgos y beneficios conocidos y potenciales, si los hu-biera, para los seres humanos
6.2.4. Descripción y justificación de la vía de administración, dosis, pauta de do-sificación y periodo de tratamiento.
6.2.5. Declaración de que el ensayo será realizado de acuerdo con el protocolo,la BPC y los requisitos legales pertinentes.
6.2.6. Descripción de la población a estudiar.
6.2.7. Referencias de la literatura y datos que sean relevantes para el ensayo y queproporcionen una justificación del mismo.
6.3. Objetivo y Finalidad del Ensayo
Descripción detallada de los objetivos y propósitos del ensayo.
6.4. Diseño del Ensayo
La integridad científica del ensayo y la credibilidad de los datos obtenidos en elmismo dependen de forma considerable del diseño del ensayo. La descripción del diseñodel ensayo deberá incluir:
6.4.1. Descripción específica de las variables primarias y secundarias, si las hu-biera, que se evaluarán en el ensayo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
317

6.4.2. Una descripción del tipo/diseño del ensayo que se realizará (por ejemplo,doble ciego, controlado con placebo, diseño paralelo) y un diagrama esquemático del di-seño del ensayo, procedimientos y periodos.
6.4.3. Una descripción de las medidas tomadas para minimizar o evitar sesgos,tales como:
a) aleatorización
b) enmascaramiento
6.4.4. Una descripción de los tratamientos del ensayo y de la dosis y pautade tratamiento del medicamento(s) en investigación. Además, deberá incluir una des-cripción de la forma farmacéutica, envasado y etiquetado del medicamento(s) eninvestigación.
6.4.5. La duración esperada de la participación de los sujetos y una descripciónde la secuencia y duración de todos los periodos del ensayo, incluyendo el seguimiento,cuando proceda.
6.4.6. Una descripción de los “criterios de retirada y suspensión” de los sujetos,en partes concretas del estudio o durante todo el estudio.
6.4.7. Los procedimientos para la contabilización del medicamento en investiga-ción, incluyendo el placebo y el comparador, si lo hubiera.
6.4.8. El mantenimiento de los códigos de aleatorización del tratamiento del en-sayo y los procedimientos para la apertura de los mismos.
6.4.9. La identificación de todos los datos que deban ser recogido directamenteen el CRD y que deba ser considerado como dato fuente (es decir, no existe ningún regis-tro escrito o electrónico de los datos).
6.5. Selección y Retirada de Sujetos
6.5.1. Criterios de inclusión de los sujetos
6.5.2. Criterios de exclusión de los sujetos.
6.5.3. Criterios de retirada de los sujetos ( es decir, finalizar el tratamiento del en-
318

sayo) y los procedimientos que especifican:
a) Cuándo y cómo retirar a los sujetos del ensayo o del tratamiento con el medi-camento en investigación.
b) El tipo de datos y el calendario en que se recogerán los datos de los sujetos re-tirados.
c) Si van a ser reemplazados los sujetos y cómo se realizará.
d) El seguimiento de los sujetos retirados del ensayo o del tratamiento con el me-dicamento en investigación.
6.6 Tratamiento de los Sujetos
6.6.1. Los tratamientos que se administrarán, incluyendo el nombre de todos losmedicamentos, la dosis, esquema de dosificación, la vía o modo de administración y losperiodos de tratamiento incluyendo los periodos de seguimiento para los sujetos de cadagrupo o brazo de tratamiento del ensayo.
6.6.2. Los medicamentos o tratamientos permitidos (incluyendo la medicación derescate) y no permitidos antes de y/o durante el ensayo.
6.6.13. Los procedimientos para monitorizar el cumplimiento del sujeto.
6.7 Valoración de la Eficacia
6.7.1. La especificación de los parámetros de eficacia
6.7.2. Los métodos y el calendario para la evaluación, registro y análisis de losparámetros de eficacia.
6.8. Valoración de Seguridad
6.8.1. La especificación de los parámetros de seguridad.
6.8.2. Los métodos y el calendario para la evaluación, registro y análisis de losparámetros de seguridad.
6.8.3. Los procedimientos para obtener los informes de los acontecimientosadversos y enfermedades intercurrentes y para el registro y comunicación de los
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
319

mismos.
6.8.4. El tipo y la duración del seguimiento de los sujetos después de los acon-tecimientos adversos.
6.9. Estadística
6.9.1. Descripción de los métodos estadísticos que se usarán, incluyendo el ca-lendario de todos los análisis intermedios planificados.
6.9.2. El número previsto de sujetos que se incluirán. En los ensayos multicén-tricos, se deberá especificar el número previsto de sujetos que se incluirán en cada cen-tro donde se realizará el ensayo. Justificación del cálculo del tamaño de la muestra,incluyendo la explicación (o cálculo) del poder del ensayo y la argumentación clínica dedicho tamaño.
6.9.3. El nivel de significación que será utilizado.
6.9.4. Los criterios para la finalización del ensayo.
6.9.5. El procedimiento utilizado para la contabilización de los datos perdidos,no usados y erróneos.
6.9.6. Procedimiento de comunicación de todas las desviaciones del plan esta-dístico original (toda desviación del plan estadístico original deberá ser descrito y justifi-cado en el protocolo y/o en el informe final, si fuera necesario).
6.9.7. La selección de los sujetos que se van a incluir en cada análisis (por ejem-plo, todos los sujetos aleatorizados, todos los sujetos tratados, todos los sujetos elegibles,los sujetos evaluables).
6.10. Acceso Directo a los Datos/Documentos Fuente
El promotor garantizará que se especifica en el protocolo u otro documento escritoque el investigador o la institución permitirá el acceso directo a los datos o documentosfuente para la realización de la monitorización, la auditoría, la revisión por el CEIC, asícomo la inspección del ensayo por las autoridades sanitarias.
320

6.11 Control y Garantía de Calidad
6.12 Ética
Descripción de las consideraciones éticas relacionadas con el ensayo
6.13. Manejo de los Datos y Archivo de los Registros
6.14. Financiación y Seguros
La financiación y el seguro, si no se contemplan en un contrato independiente.
6.15. Política de Publicación
La política de publicación, si no se indica en un contrato independiente
6.16. Nota complementaria
(NOTA: Puesto que el protocolo y el informe del estudio o ensayo clínico tienenuna estrecha relación, se puede encontrar información adicional relevante en la Guía ICHpara la Estructura y Contenido de los Informes de Estudios Clínicos)
7. MANUAL DEL INVESTIGADOR (MI)
7.1. Introducción
El manual del investigador (MI) contiene los datos clínicos y no clínicos que sonrelevantes para el estudio de los medicamentos en investigación en el ser humano. Suobjetivo es proporcionar a los investigadores y demás implicados en el ensayo, informa-ción que permita comprender la razón de ser y el motivo de que sea necesario cumplirlos aspectos claves del protocolo, tales como las dosis e intervalo y forma de adminis-tración, y procedimientos para monitorizar la seguridad. El MI también orienta adecua-damente el manejo clínico de los sujetos del estudio durante la realización del ensayoclínico. La información deberá presentarse de forma concisa, sencilla, objetiva, equili-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
321
39 De acuerdo con el documento de aclaraciones sobre la aplicación de la normativa de ensayos clínicos, el manualdel investigador servirá como referencia para la evaluación del carácter esperado o no de las reacciones adversasgraves que pudieran ocurrir durante la realización del ensayo. Por tanto, el MI debe ser el mismo para todos loscentros participantes en el ensayo de todos los países donde éste se realice

brada y no promocional, que permita a los clínicos o posibles investigadores realizar unaevaluación no sesgada de los riesgos y beneficios y de la pertinencia del ensayo clínicopropuesto. Por esta razón, generalmente un médico debe participar en la preparación delMI, aunque sus contenidos deben ser aprobados por los expertos que generaron los da-tos descritos39.
Esta guía define la información mínima que deberá ser incluida en un MI e incluyesugerencias sobre su formato. Se espera que el tipo y extensión de la información dispo-nible varíe con la fase del desarrollo del medicamento en investigación. Si el medica-mento en investigación está comercializado y su farmacología es ampliamente conocidapor los médicos puede no ser necesario un MI extenso. Cuando la legislación lo permita40,la ficha técnica, el prospecto puede ser una alternativa apropiada, siempre que se garan-tice que incluye información actualizada, clara y detallada de todos los aspectos necesa-rios para el estudio. Si un medicamento comercializado está siendo estudiado para unanueva utilización (por ejemplo, una nueva indicación) se deberá preparar un MI especí-fico para esa nueva utilización. El manual del investigador debe revisarse al menos anual-mente y actualizarse cuando sea necesario de acuerdo con los PNT del promotor. Unarevisión más frecuente pude ser necesaria dependiendo del estadio del desarrollo y la re-levancia de la nueva información. Sin embargo, de acuerdo con la BPC, toda nueva infor-mación relevante deberá ser comunicada a los investigadores y a los CEIC implicados y alas autoridades reguladoras antes de incluirse en el MI actualizado.
Generalmente, el promotor es responsable de asegurar que un MI actualizado estédisponible para los investigadores y los investigadores son responsables de proporcionarel MI actualizado a los CEIC responsables. En el caso de un ensayo en el que el promotorsea el investigador, el promotor-investigador deberá determinar si el fabricante disponede un manual. Si el medicamento en investigación es suministrado por el promotor-inves-tigador, entonces éste deberá facilitar la información necesaria al personal del ensayo. Enlos casos en los que la preparación de un MI formal sea poco práctica, como alternativa,el promotor-investigador deberá incluir en el protocolo del ensayo, una sección ampliadade información sobre los antecedentes y que contenga la información mínima actualizadadescrita en esta guía
322
40 En caso de medicamentos autorizados en algún Estado Miembro de la Comunidad, cuando el medicamento seutilice en las condiciones de uso autorizadas el Manual del Investigador podrá reemplazarse por la ficha técnicaautorizada.

7.2. Consideraciones generales
El Manual del Investigador deberá incluir:
7.2.1. Página de Título
Se deberá incluir el nombre del promotor, la identidad de cada medicamentode investigación (por ejemplo el número de investigación, nombre químico o denomi-nación común internacional y nombre comercial cuando sea posible y autorizado porel promotor), y la fecha de edición. También se sugiere que se proporcione un númerode edición y la referencia del número y fecha de la edición a la que reemplaza. Se fa-cilita un ejemplo en el Anexo 1.
7.2.2. Declaración de Confidencialidad
El promotor podrá incluir una declaración en la que se indicará al investiga-dor y a otros destinatarios, que traten el MI como un documento confidencial, exclu-sivamente para la información y uso del equipo investigador y del CEIC.
7.3. Contenido del Manual del Investigador.
El MI deberá incluir las siguientes secciones, cada una con referencias bi-bliográficas cuando sea necesario.
7.3.1. Índice
Se da un ejemplo del Índice en el Apéndice 2
7.3.2. Resumen
Se proporcionará un resumen breve (preferiblemente con una extensión má-xima de dos páginas), destacando la información física, química, farmacéutica, far-macológica, toxicológica, farmacocinética, metabólica y clínica importante ydisponible que sea relevante para la fase del desarrollo clínico del medicamento eninvestigación.
7.3.3. Introducción
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
323

Se proporcionará una breve introducción que contenga el nombre químico (yla denominación común internacional y el nombre comercial cuando estén autoriza-dos) del medicamento en investigación, todas las sustancias activas, la clase farma-cológica del medicamento en investigación y las diferencias con respecto a los de suclase (por ej. ventajas), la justificación para realizar el ensayo clínico propuesto conel medicamento y sus previstas indicaciones profilácticas, terapéuticas y diagnósti-cas. Finalmente, la introducción deberá proporcionar el enfoque general a seguir enla evaluación del medicamento en investigación.
7.3.4. Propiedades Físicas, Químicas y Farmacéuticas y Formulación
Se deberá facilitar una descripción de las sustancias activas del medicamento eninvestigación (incluyendo la fórmula química y/o estructura), y un resumen breve de laspropiedades físicas, químicas y farmacéuticas relevantes.
Para permitir que se tomen medidas de seguridad apropiadas durante el ensayo,se debe facilitar y justificar si es clínicamente relevante, una descripción de la formula-ción que se utilizará, incluyendo los excipientes. Deberá proporcionarse también instruc-ciones sobre el almacenamiento y manejo de las formas farmacéuticas.
Se deberá mencionar cualquier similitud estructural con otros compuestos conocidos.
7.3.5. Estudios No Clínicos
Introducción:
Deberá facilitarse, de forma resumida, los resultados de todos los estudios no clí-nicos relevantes sobre la farmacología, toxicología, farmacocinética y el metabolismo demedicamento en investigación. Este resumen deberá considerar la metodología utilizada,los resultados y una discusión de la relevancia de los hallazgos para la indicación terapéu-tica investigada y los posibles efectos adversos y no intencionados en humanos.
La información facilitada incluirá, lo siguiente, según proceda en caso de ser co-nocido o estar disponible:
• Especies estudiadas
• Número y sexo de los animales en cada grupo
• Unidad de dosis (por ej. miligramo/kilogramo (mg/kg.)
324

• Intervalo de dosis
• Vía de administración
• Intervalo de dosificación
• Información sobre la distribución sistémica
• Duración del seguimiento posterior a la exposición
• Resultados, incluyendo los siguientes aspectos:
– Naturaleza y frecuencia de los efectos farmacológicos o tóxicos
– Gravedad o intensidad de los efectos farmacológicos o tóxicos
– Tiempo transcurrido hasta la aparición de los efectos
– Reversibilidad de los efectos
– Duración de los efectos
– Relación dosis respuesta
Deberán proporcionarse los datos tabulados o listados, siempre que sea posible,para reforzar la claridad de la presentación.
Las siguientes secciones deberán analizar los hallazgos más importantes de los es-tudios, incluyendo la relación dosis respuesta de los efectos observados, su relevancia ycualquier aspecto que debería ser estudiado en humanos. Cuando proceda, se deberáncomparar los hallazgos con las dosis efectivas y no tóxicas en las mismas especies anima-les (es decir, se deberá discutir el índice terapéutico). Deberá valorarse la relevancia deesta información para la dosificación propuesta en humanos. Cuando sea posible, se de-berán comparar los niveles en sangre o tejido más que en base a una relación mg/kg.
a) Farmacología No Clínica
b) Deberá incluirse un resumen de los aspectos farmacológicos del medicamentoen investigación y si es necesario, de los metabolitos más importantes estudia-dos en animales. Dicho resumen deberá especificar los estudios que evalúen laactividad terapéutica potencial (por ej. modelos de eficacia, unión a recepto-res y especificidad) así como aquellos que evalúen la seguridad (por ej. estu-dios especiales para evaluar acciones farmacológicas aparte de los efectosterapéuticos deseados).
c) Farmacocinética y Metabolismo del Medicamento en Animales
d) Deberá incluirse un resumen del metabolismo y eliminación farmacocinética ybiológica del medicamento en investigación en todas las especies estudiadas.La discusión de los hallazgos deberá tratar la absorción y la biodisponibilidadlocal y sistémica del medicamento en investigación y de sus metabolitos, así
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
325

como su relación con los hallazgos farmacológicos y toxicológicos en las espe-cies animales.
e) Toxicología
f) Cuando sea apropiado, deberá incluirse un resumen de los efectos toxicológi-cos hallados en los estudios relevantes realizados en diferentes especies ani-males bajo los siguientes títulos:
– Dosis única
– Dosis repetidas
– Carcinogénesis
– Estudios especiales (por ej. irritabilidad y sensibilización)
– Toxicidad reproductiva
– Genotoxicidad (mutagénesis)
7.3.6. Efectos en Humanos
Introducción:
Deberá facilitarse una discusión rigurosa de los efectos conocidos de los medica-mentos de investigación en humanos incluyendo información sobre farmacocinética, me-tabolismo, farmacodinámica, dosis-respuesta, seguridad, eficacia y otras actividadesfarmacológicas. Deberá proporcionarse, cuando sea posible, un resumen de cada ensayoclínico finalizado. También, se deberá facilitar información en relación a los resultados decualquier uso de los medicamentos de investigación fuera de los ensayos clínicos, talcomo puede ser la experiencia durante la comercialización.
a) Farmacocinética y Metabolismo del Medicamento en Humanos
Deberá presentarse un resumen de información sobre la farmacocinética del me-dicamento en investigación incluyendo lo siguiente, si está disponible:
– Farmacocinética (incluyendo metabolismo, cuando proceda, y absorción, unióna proteínas plasmáticas, distribución y eliminación).
– Biodisponibilidad del medicamento en investigación (absoluta, cuando sea po-sible, y/o relativa) utilizando una forma farmaceutica de referencia.
– Grupos de población (por ej. Por sexo, edad y alteración de la función de unórgano).
– Interacciones (por ej. interacciones de medicamento-medicamento y efecto de
326

la administración conjunta con alimentos).
– Otros datos farmacocinéticos (por ej. resultados de estudios de poblacionesrealizados dentro de ensayos clínicos).
b) Seguridad y Eficacia
Se deberá facilitar un resumen de los datos referentes a la seguridad, farma-codinamia, eficacia así como los estudios dosis respuesta de los medicamentos en in-vestigación (incluyendo metabolitos, si es necesario) obtenidos en ensayos previos enhumanos (voluntarios sanos y/o pacientes). Deberán analizarse las implicaciones queconlleva esta información. En el caso de que se hayan realizado otros ensayos clíni-cos, el uso de resúmenes de la seguridad y eficacia de los distintos estudios por in-dicaciones y subgrupos facilitará la comprensión de los datos. Sería útil realizarresúmenes tabulados de las reacciones adversas aparecidas en todos los ensayos (in-cluyendo los de todas las indicaciones estudiadas). Deberán discutirse las diferenciasimportantes encontradas en los patrones o incidencia de las reacciones adversas enlas diferentes indicaciones o subgrupos.
El MI deberá facilitar una descripción de los posibles riesgos y RAM espera-das en base a la experiencia previa con los medicamentos en investigación y con me-dicamentos relacionados. Asimismo deberán describirse las precauciones o lamonitorización especial a realizar como parte de la investigación de los medicamen-tos.
c) Experiencia durante la comercialización
El MI deberá identificar los países donde el medicamento en investigaciónesté aprobado o comercializado Cualquier información importante que surja del usodel medicamento comercializado debe resumirse (por ej. Formas farmacéuticas, do-sis, vías de administración y RAM). El MI también debe identificar todos los paísesdonde el medicamento en investigación no ha sido autorizado para la comercializa-ción o registro del medicamento o en los que se ha retirado dicha autorización.
7.3.7. Resumen de los Datos y Guía para el Investigador.
Esta sección debe incluir una discusión global de los datos clínicos y no clí-nicos y resumir, siempre que sea posible, la información procedente de varias fuen-tes sobre los diferentes aspectos del medicamento en investigación. De esta manera,el investigador dispondrá de la mejor información de los datos disponibles y de la
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
327

evaluación de las implicaciones que conlleva esta información para futuros ensayosclínicos.
Cuando proceda, se deberán discutir los informes publicados referentes a me-dicamentos relacionados. Estos podrían ayudar al investigador a anticipar reaccionesadversas al medicamento u otros problemas que pudieran surgir en el ensayo clínico.
El objetivo global de esta sección es facilitar al investigador un conocimientoclaro de los posibles riesgos y reacciones adversas, así como de las pruebas especí-ficas, observaciones y precauciones que pueden ser necesarios durante el ensayo clí-nico. Este conocimiento deberá basarse en la información física, química,farmacéutica, farmacológica, toxicológica y clínica disponible referente al medica-mento en investigación. También deberá proporcionarse orientación al investigadorclínico sobre el reconocimiento y tratamiento de posibles sobredosis y reaccionesadversas al fármaco, basándose en la experiencia previa en humanos y en la farma-cología del medicamento en investigación.
7.4. APÉNDICE 1:
TITULO DE LA PAGINA (Ejemplo)
NOMBRE DEL PROMOTOR
Medicamento:
Número de Investigación:
Nombre: Químico, Genérico (si está aprobado)
Nombre Comercial (si está permitido legalmente y lo desea el promotor)
MANUAL DEL INVESTIGADOR
Número de Edición:
328

Fecha de Edición:
Reemplaza al Número de Edición Anterior:
Fecha:7. 5 APÉNDICE 2:
INDICE DEL MANUAL DEL INVESTIGADOR (Ejemplo)
– Declaración de Confidencialidad (opcional)
– Página de firmas (opcional)
1. Índice
2. Resumen
3. Introducción
4. Propiedades Físicas, Químicas y Farmacéuticas y Formulaciones
5. Estudios no clínicos
5.1 Farmacología No Clínica
5.2 Farmacocinética y Metabolismo del Medicamento en animales
5.3 Toxicología
6. Efectos en Humanos
6.1 Farmacocinética y Metabolismo del Medicamento en humanos
6.2 Seguridad y Eficacia
6.3 Experiencia durante la fase de comercialización
7. Resumen de datos y guía para el Investigador
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
329

Nota: Referencias sobre 1. Publicaciones
2. Informes
Se deberán encontrar estas referencias al final de cada capítulo.
Apéndices (si los hay).
8. DOCUMENTOS ESENCIALES PARA LA REALIZACION DE UN ENSAYO CLINICO
8.1. Introducción
Los Documentos Esenciales son aquellos que individualmente y colectivamente per-miten la evaluación del desarrollo de un ensayo y la calidad de los datos producidos. Estos do-cumentos sirven para demostrar el cumplimiento del investigador, promotor y monitor con lasnormas de Buena Práctica Clínica y con todos los requisitos reguladores pertinentes.
Los Documentos Esenciales también sirven para otros propósitos importantes. Lacumplimentación oportuna de los documentos esenciales por parte del investigador / institu-ción y promotor puede ayudar mucho a que el investigador, promotor y monitor conduzcan elensayo con éxito. Estos documentos son también los que auditan habitualmente los audito-res independientes del promotor e inspeccionan las autoridades reguladoras como parte delproceso realizado para confirmar la validez del desarrollo del ensayo y la integridad de los da-tos recogidos.
A continuación aparece una lista mínima de documentos esenciales. Los diferentes do-cumentos han sido agrupados en tres secciones de acuerdo con la fase del ensayo durante lacual serán normalmente generados: 1) antes de la fase clínica del ensayo, 2) durante el des-arrollo clínico del ensayo, y 3) después de completar el ensayo. Se proporciona una descrip-ción del propósito de cada documento y se especifica si debe ser archivado en los archivos delinvestigador / institución, en los del promotor o en ambos. Es aceptable combinar algunos delos documentos dado que los elementos individuales son rápidamente identificables.
Los archivos principales del ensayo deben constituirse al principio del ensayo, tantoen el local del investigador / institución como en la oficina del promotor. La liquidación finaldel ensayo sólo puede hacerse cuando el monitor haya revisado tanto los archivos del investi-gador / institución como los del promotor y haya confirmado que todos los documentos nece-sarios están en los archivos adecuados.
Alguno o todos los documentos indicados en esta guía pueden ser auditados por el au-
330

TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
331
Investigador/Institución Promotor
8.2.1MANUAL DELINVESTIGADOR
Para documentar que se ha facilitado la informacióncientífica relevante y actualizada acerca del producto en investigación.
X X
8.2.4ASPECTOS FINANCIEROS DEL ENSAYO
Documentar el contrato financiero entre elinvestigador / institución yel promotor.
X X
8.2.2
PROTOCOLO FIRMADO YENMIENDAS, SI LAS HUBIERE, Y UN EJEMPLAR DEL CUADERNO DE RECOGIDA DE DATOS(CRD)
Documentar el acuerdoentre promotor e investigador respecto alprotocolo, enmiendas y CRD.
X X
8.2.3 INFORMACION DADA ALSUJETO DEL ENSAYO:• FORMULARIO DE
CONSENTIMIENTOINFORMADO (incluyendo todas lastraducciones posibles)
• CUALQUIER OTRA INFORMACION ESCRITA
• ANUNCIO PARA ELRECLUTAMIENTO DESUJETOS (si es utilizado)
Documentar el consentimiento informadoDocumentar que se dará alos sujetos la informaciónescrita apropiada (contenido y términos)para dar un consentimiento plenamente informado.
Documentar que las medidas de reclutamientoson apropiadas y no coactivas.
X X
X X
X

332
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.2.5DECLARACIÓN DEL SEGURO
Documentar que habráuna indemnización paralos sujetos que sufran da-ños relacionados con elensayo.
X X
8.2.6
CONTRATO FIRMADOENTRE LAS PARTES IM-PLICADAS• Investigador /
institución y promotor• Investigador /
institución y CRO• Promotor y CRO• Investigador /
institución y autoridades
Documentar los acuerdos.
X X
8.2.7
OPINION FAVORABLE /APROBACION DOCUMENTADA Y FECHADA DEL CONSEJO INSTITUCIONAL DE REVISIÓN / COMITEÉTICO INDEPENDIENTE(CIR/CEI) SOBRE LO SIGUIENTE:• Protocolo y cualquier
enmienda• Cuaderno de Recogida
de Datos• Formulario de
Consentimiento Informado
Documentar que el ensayoha sido revisado por elCIR/CEI y que éste hadado la opinión favorable /aprobación.
Identificar el número de laversión y fecha de los do-cumentos.
X X
X X
X X
X X (SR)
X X
X X

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
333
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.2.7
• Cualquier informaciónescrita para los sujetos
• Anuncios para el re-clutamiento de sujetos(si se utilizan)
• Compensación paralos sujetos (si la hay)
• Otros documentosdonde se dé la opiniónfavorable / aprobación
Documentar que el ensayoha sido revisado por elCIR/CEI y que éste hadado la opinión favorable /aprobación.
Identificar el número de laversión y fecha de los do-cumentos.
X X
8.2.8 COMPOSICION DELCIR/CEI
Documentar que elCIR/CEI está constituidode acuerdo con la BPC.
X X (SR)
8.2.9
AUTORIZACIÓN / APROBACION/NOTIFICACION DELPROTOCOLO POR LASAUTORIDADES REGULADORAS
Documentar que se ha obtenido de las autoridades reguladoras laautorización / aprobación /notificación apropiada previamente al inicio delensayo, de acuerdo conlos requisitos reguladorespertinentes.
X (SR) X (SR)
8.2.10
CURRICULUM VITAE Y/OOTROS DOCUMENTOS RELEVANTES QUE EVIDENCIEN LAS CUALIFICACIONES DELOS INVESTIGADORESY SUBINVESTIGADORES
Documentar las cualificaciones y elegibilidad para realizarel ensayo y/o la supervisión médica de lossujetos.
X X
X X
X X
X X
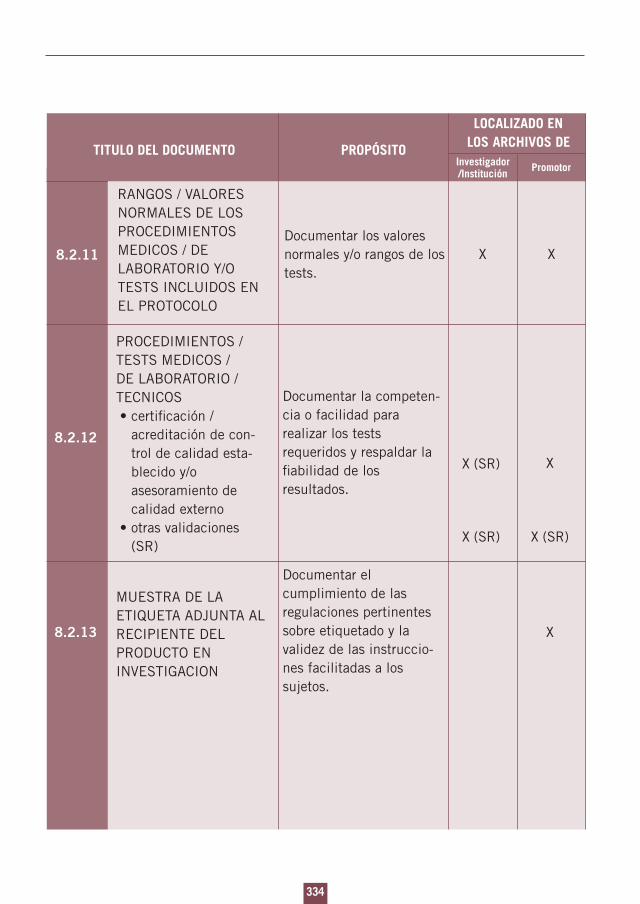
334
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.2.11
RANGOS / VALORESNORMALES DE LOSPROCEDIMIENTOS MEDICOS / DE LABORATORIO Y/OTESTS INCLUIDOS ENEL PROTOCOLO
Documentar los valoresnormales y/o rangos de lostests.
X X
8.2.12
PROCEDIMIENTOS /TESTS MEDICOS / DE LABORATORIO /TECNICOS• certificación /
acreditación de con-trol de calidad esta-blecido y/oasesoramiento de calidad externo
• otras validaciones(SR)
Documentar la competen-cia o facilidad para realizar los tests requeridos y respaldar lafiabilidad de los resultados.
X (SR) X
8.2.13
MUESTRA DE LA ETIQUETA ADJUNTA ALRECIPIENTE DEL PRODUCTO EN INVESTIGACION
Documentar el cumplimiento de las regulaciones pertinentessobre etiquetado y la validez de las instruccio-nes facilitadas a los sujetos.
X
X (SR) X (SR)

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
335
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.2.14
INSTRUCCIONES PARAEL MANEJO DEL PRODUCTO EN INVESTIGACION Y MATERIALES RELACIONADOS CON ELENSAYO (si no están incluidos enel protocolo o en el Manual del Investigador)
Documentar las instrucciones necesariaspara asegurar un adecuado almacenamiento, empaquetado, dispensación y disposiciónde los productos en investigación y de los materiales relacionadoscon el ensayo.
X X
8.2.15
REGISTROS DE ENVIOPARA EL PRODUCTO EN INVESTIGACION Y MATERIALES RELACIONADOS CON EL ENSAYO
Documentar fechas de en-vío, número de lotes y mé-todo de envío de losproductos en investigacióny de los materiales relacionados con el ensayo. Permite controlarel lote del producto, revisar las condiciones deenvío y la responsabilidad.
X X
8.2.16
CERTIFICADO DE ANALISIS DE LOS PRODUCTOS EN INVESTIGACION ENVIADOS.
Documentar la identidad,pureza e intensidad delproducto en investigacióna ser usado en el ensayo.
X
8.2.17
PROCEDIMIENTOS DEDECODIFICACION PARAENSAYOS CIEGOS
Documentar como, en caso de emergencia, la identidad del producto en investigaciónpuede ser revelada sin romperel ciego para los restantes sujetos del tratamiento.
X

336
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.2.18LISTA DE RANDOMIZACION PRINCIPAL
Documentar el método derandomización de la población del ensayo.
X X (TP)
8.2.19INFORME DE MONITORIZACION PRE-ENSAYO
Documentar que el locales adecuado para el ensayo. (puede estarcombinado con el8.2.20).
X
X (TP)
8.2.20INFORME DE MONITORIZACION DELINICIO DEL ENSAYO
Documentar que los procedimientos del ensayofueron revisados con el investigador y el personaldel investigador que participa en el ensayo(puede estar combinadocon el 8.2.19)
X
ditor del promotor e inspeccionados por las autoridades reguladoras y deben estar disponiblespara ello.
8.2. Antes del inicio de la Fase Clínica del Ensayo

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
337
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.3.1ACTUALIZACIONES DELMANUAL DEL INVESTIGADOR
Documentar que el investigador recibe ación relevante que vaapareciendo.
X X
8.3.2
CUALQUIER REVISIONDE:• Protocolo / enmiendas
y CRD• Formulario de
Consentimiento Informado
• Cualquier informaciónescrita para los sujetos
• Anuncios para el reclutamiento de sujetos (si se utilizan)
Documentar las revisionesde estos documentos relacionados con el ensayoque tienen efecto duranteel ensayo.
X X
8.3.3
OPINION FAVORABLE /APROBACION DOCUMENTADA Y FECHADA DEL CONSEJOINSTITUCIONAL DE REVISIÓN / COMITEÉTICO INDEPENDIENTE(CIR/CEI) SOBRE LO SIGUIENTE:• Enmiendas al protocolo• Revisiones de:- formulario de consentimiento informado- cualquier otra información escrita paralos sujetos
Documentar que las enmiendas y/o revisioneshan sido revisadas por elCIR/CEI y que éste hadado su opinión favorable/ aprobación.
Identificar el número de la versión y fecha delos documentos.
X X
X X
X X
X X
X X
X X

338
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.3.3
- anuncios para reclutamiento de sujetos(si se utilizan)• Otros documentos
donde se dé la opiniónfavorable / aprobación.
• Revisión continuadadel ensayo (SR)
Documentar que las enmiendas y/o revisioneshan sido revisadas por elCIR/CEI y que éste hadado su opinión favorable/ aprobación.
Identificar el número de la versión y fecha delos documentos.
X X
8.3.4
AUTORIZACIONES /APROBACIONES / NOTIFICACIONES PORPARTE DE LAS AUTORIDADES REGULADORAS SI SEREQUIEREN PARA:• Enmiendas al
protocolo y otros documentos
Documentar el cumplimiento de los requisitos reguladores pertinentes.
X (SR) X
8.3.5
CURRICULUM VITAE DE NUEVOS INVESTIGADORES / SUBINVESTIGADORES
(Ver 8.2.10). X X
8.3.6
ACTUALIZACIONES DELOS RANGOS / VALORES NORMALESPARA LOS PROCEDIMIENTOS MEDICOS / DE LABORATORIO Y/OTESTS INCLUIDOS ENEL PROTOCOLO
Documentar que los valores y rangos normalesson revisados durante elensayo (ver 8.2.11).
X X
X X
X X

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
339
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.3.7
ACTUALIZACIONES DELOS PROCEDIMIENTOS/ TESTS MEDICOS / DELABORATORIO / TECNICOS• certificación /
acreditación de controlde calidad establecidoy/o asesoramiento decalidad externo
• otras validaciones (SR)
Documentar que los testsson adecuados durantetodo el ensayo (ver8.2.12). X (SR) X
X X
8.3.8
DOCUMENTACION DEENVIO PARA EL PRODUCTO EN INVESTIGACION Y MATERIALES RELACIONADOS CON EL ENSAYO
(Ver 8.2.15). X X
8.3.9
CERTIFICADOS DE ANALISIS PARA LOSNUEVOS LOTES DELPRODUCTO EN INVESTIGACION
(Ver 8.2.16). X
8.3.10INFORMES DE VISITASDE MONITORIZACION
Documentar las visitas realizadas por el monitor alos centros en los que serealiza el ensayo.
X

340
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.3.11
COMUNICACIONES RELEVANTES DIFERENTES A LAS VISITAS A LOS LOCALES• cartas• notas de reuniones• notas de llamadas
telefónicas
Documentar cualquieracuerdo o discusión significativa acerca de laadministración del ensayo,violaciones del protocolo,realización del ensayo, informe de acontecimientos adversos.
X XX X
8.3.12FORMULARIOS DE CONSENTIMIENTO INFORMADO FIRMADOS
Documentar que se ha obtenido el consentimiento deacuerdo con la BPC y elprotocolo y que ha sido fechado previamente a laparticipación de cada sujeto en el ensayo. También para documentarel permiso para el accesodirecto (ver 8.2.3)
X
8.3.13DOCUMENTOS ORIGINALES
Documentar la existenciadel sujeto y la integridadsustancial de los datos recogidos en el ensayo. Incluye documentos originales relacionadoscon el ensayo, el tratamiento médico y la historia del sujeto.
X
X X

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
341
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.3.14
CUADERNOS DE RECOGIDA DE DATOSCOMPLETADOS, FIRMADOS Y FECHADOS
Documentar que el investigador o el miembroautorizado del personaldel investigador confirmalas observaciones registradas.
X (copia) X (original)
8.3.15
DOCUMENTACION DELAS CORRECIONES ENEL CUADERNO DE RECOGIDA DE DATOS
Documentar que se hanregistrado todos los cambios / adiciones o correcciones hechas en elcuaderno de recogida dedatos sobre los datos inicialmente registrados.
X (copia) X (original)
8.3.16
NOTIFICACION POR ELINVESTIGADOR ORIGINANTE AL PROMOTOR DE ACONTECIMIENTOS ADVERSOS GRAVES EINFORMES RELACIONADOS
Notificación por el investigador originante alpromotor de acontecimientos adversosgraves e informes relacionados de acuerdocon 4.11.
X X
8.3.17
NOTIFICACION POR ELPROMOTOR Y/O INVESTIGADOR, A LASAUTORIDADESREGULADORAS YCIR/CEI DE REACCIONES ADVERSAS GRAVES ALFARMACO Y OTRA INFORMACION SOBRESEGURIDAD
Notificación por el promotory/o investigador, cuando seapertinente, a las autoridades reguladoras yCIR/CEI de reacciones adversas al fármaco gravese inesperadas de acuerdocon 5.17 y 4.11.1 y de otra información sobreseguridad de acuerdo con 5.16.2.
X (SR) X

342
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.3.18
NOTIFICACION POR ELPROMOTOR A LOS INVESTIGADORES DE LA INFORMACIONSOBRE SEGURIDAD
Notificación por el promotor a los investigadores de la información sobre seguridad de acuerdo con 5.16.2.
X X
8.3.19INFORMES ANUALES EINTERMEDIOS AL CIR YAUTORIDADES
Informes anuales e intermedios facilitados alCIR/CEI de acuerdo con(SR) 4.10 y a las autoridades de acuerdocon 5.17.3.
X X (SR)
8.3.20SISTEMA DE SELECCION DE SUJETOS
Documentar la identificación de los sujetos que entraron en laselección previa al ensayo.
X X (SR)

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
343
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.4.1
RESPONSABILIDADESSOBRE EL PRODUCTOEN INVESTIGACION ENEL LUGAR DEL ENSAYO
Documentar que el producto en investigaciónha sido utilizado deacuerdo con el protocolo.Documentar la responsabilidad sobre elproducto en investigaciónrecibido en el lugar delensayo, dispensado a lossujetos, devuelto por lossujetos y devuelto al promotor.
X X
8.4.2
DOCUMENTACION DELA DESTRUCCION DELPRODUCTO EN INVES-TIGACION
Documentar la destruccióndel producto en investiga-ción no utilizado por elpromotor o en el lugar delensayo.
X (si es
destruidoen el lugardel ensayo)
X
8.4.3
LISTA COMPLETA DELOS CODIGOS DE INDENTIFICACION DE LOS SUJETOS
Permitir la identificaciónde todos los sujetos incluidos en el ensayo enel caso de que se requieraun seguimiento. La listadebe ser guardada demodo confidencial y durante el tiempo acordado.
X
8.4. Después de la Finalización del Ensayo
8.4.4CERTIFICADO DE AUDITORIA(si está disponible)
Documentar que se hizouna auditoría
X

344
TITULO DEL DOCUMENTO PROPÓSITO
LOCALIZADO ENLOS ARCHIVOS DE
Investigador/Institución Promotor
8.4.5
INFORME DE MONITORIZACIÓN FI-NAL Y DE CIERRE DEL ENSAYO
Documentar que se hancompletado todas las actividades requeridaspara la finalización del ensayo y que las copias delos documentos esencialesestán guardadas en los archivos adecuados.
X
8.4.6
DOCUMENTACION DEASIGNACIÓN DE TRATAMIENTO Y DECODIFICACIÓN
Debe ser devuelta al pro-motor para documentarcualquier decodificaciónque se haya producido.
X
8.4.7
INFORME FINAL DEL INVESTIGADOR ALCIR/CEI CUANDO SEAREQUERIDO Y A LASAUTORIDADES REGULADORASCUANDO SEA PERTINENTE
Documentar la finalizacióndel ensayo
X
8.4.8INFORME DEL ESTUDIOCLÍNICO
Documentar los resultadose interpretaciones del ensayo
X (SR) X
SR - Si es requerido.
TP - Tercera persona, si es pertinente.

8.5. SIGLAS
Sigla Término en castellano Sigla Término en inglés
AA Acontecimiento Adverso AE Adverse Event
AEMPS Agencia Española de Medicamentos
y Productos Sanitarios
BPC Buena Práctica Clínica GCP Good Clinical Practice
CEIC Comité Ético de Investigación Clínica IECIRB Independent Ethics Committee
Institutional Review Board
CI Consentimiento informado
CIMD Comité Independiente de IDMC Independent Data
Monitorización de Datos Monitoring Committee
CRD Cuaderno de Recogida de Datos CRF Case Report Form
CRO Organización de Investigación CRO Contract Research
por contrato Organization
ICH Conferencia Internacional ICH International
de Armonización Conference on
Harmonisation
MI Manual del Investigador IB Investigator’s Brochure
NCF Normas de Correcta Fabricación GMP Good Manufacturing Practice
OMS Organización Mundial de la Salud WHO World Health
Organization
PNT Procedimientos Normalizados SOPs Standard Operating de Trabajo Procedures
QA Garantía de Calidad QA Quality Assurance
QC Control de Calidad QC Quality Control
RAM Reacción Adversa a Medicamentos ADR Adverse Drug Reaction
RD Real Decreto RD Royal Decree
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
345


CONVENIO PARA LA PROTECCIÓN DE LOS DERECHOSHUMANOS Y LA DIGNIDAD DEL SER HUMANO
CON RESPECTO A LAS APLICACIONES DE LA BIOLOGÍAY LA MEDICINA.
CONVENIO SOBRE LOS DERECHOS HUMANOS Y LA BIOMEDICINA.
OVIEDO, 4 DE ABRIL DE 1997
(CONVENIO DE ASTURIAS)
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
347


CONVENIO PARA LA PROTECCIÓN DE LOS DERECHOS HUMANOS Y LA DIGNIDAD DEL SER HUMANO CON RESPECTO A LAS APLICACIONES DE LABIOLOGÍA Y LA MEDICINA
Convenio sobre los Derechos Humanos y la Biomedicina.
Oviedo, 4 de abril de 1997
Preámbulo
Los Estados miembros del Consejo de Europa, los demás Estados y la Comunidad Eu-ropea, signatarios del presente Convenio,
Considerando la Declaración Universal de los Derechos Humanos, proclamada por laAsamblea General de las Naciones Unidas el 10 de diciembre de 1948;
Considerando el Convenio para la Protección de los Derechos Humanos y de las Liber-tades Fundamentales, de 4 de noviembre de 1950;
Considerando la Carta Social Europea de 18 de octubre de 1961;
Considerando el Pacto Internacional de derechos civiles y políticos y el Pacto Interna-cional de derechos económicos, sociales y culturales de 16 de diciembre de 1966;
Considerando el Convenio para la Protección de las Personas con respecto al trata-miento automatizado de datos de carácter personal, de 28 de enero de 1981;
Considerando igualmente la Convención sobre los Derechos del Niño, de 20 de noviem-bre de 1989;
Considerando que la finalidad del Consejo de Europa es la de conseguir una unión másestrecha entre sus miembros y que uno de los medios para lograr dicha finalidad es la salvaguar-dia y el fomento de los derechos humanos y de las libertades fundamentales;
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
349

Conscientes de los rápidos avances de la biología y la medicina,
Convencidos de la necesidad de respetar al ser humano a la vez como persona y comoperteneciente a la especie humana y reconociendo la importancia de garantizar su dignidad;
Conscientes de las acciones que podrían poner en peligro la dignidad humana medianteuna práctica inadecuada de la biología y la medicina;
Afirmando que los progresos en la biología y la medicina deben ser aprovechados en fa-vor de las generaciones presentes y futuras;
Subrayando la necesidad de una cooperación internacional para que toda la Humani-dad pueda beneficiarse de las aportaciones de la biología y la medicina;
Reconociendo la importancia de promover un debate público sobre las cuestiones plan-teadas por la aplicación de la biología y la medicina y sobre las respuestas que deba darse a lasmismas;
Deseosos de recordar a cada miembro del cuerpo social sus derechos y responsabi-lidades;
Tomando en consideración los trabajos de la Asamblea Parlamentaria en este ámbito,comprendida la Recomendación 1160 (1991) sobre la elaboración de un Convenio de Bioética;
Decididos a adoptar las medidas adecuadas, en el ámbito de las aplicaciones de la bio-logía y la medicina, para garantizar la dignidad del ser humano y los derechos y libertades fun-damentales de la persona;
Han convenido en lo siguiente:
CAPÍTULO I. Disposiciones generales.
Artículo 1. Objeto y finalidad.
Las Partes en el presente Convenio protegerán al ser humano en su dignidad y suidentidad y garantizarán a toda persona, sin discriminación alguna, el respeto a su inte-gridad y a sus demás derechos y libertades fundamentales con respecto a las aplicacio-nes de la biología y la medicina.
350

Cada Parte adoptará en su legislación interna las medidas necesarias para daraplicación a lo dispuesto en el presente Convenio.
Artículo 2. Primacía del ser humano.
El interés y el bienestar del ser humano deberán prevalecer sobre el interés exclu-sivo de la sociedad o de la ciencia.
Artículo 3. Acceso equitativo a los beneficios de la sanidad.
Las Partes, teniendo en cuenta las necesidades de la sanidad y los recursos dis-ponibles, adoptarán las medidas adecuadas con el fin de garantizar, dentro de su ámbitojurisdiccional, un acceso equitativo a los beneficios de una sanidad de calidad apropiada.
Artículo 4. Obligaciones profesionales y normas de conducta.
Toda intervención en el ámbito de la sanidad, comprendida la investigación, de-berá efectuarse dentro del respeto a las normas y obligaciones profesionales, así como alas normas de conducta aplicables en cada caso.
CAPÍTULO II. Consentimiento.
Artículo 5. Regla general.
Una intervención en el ámbito de la sanidad sólo podrá efectuarse después deque la persona afectada haya dado su libre e informado consentimiento.
Dicha persona deberá recibir previamente una información adecuada acerca de lafinalidad y la naturaleza de la intervención, así como sobre sus riesgos y consecuencias.
En cualquier momento la persona afectada podrá retirar libremente su consentimiento.
Artículo 6. Protección de las personas que no tengan capacidad para expresar su consen-timiento.
1. A reserva de lo dispuesto en los artículos 17 y 20, sólo podrá efectuarse unaintervención a una persona que no tenga capacidad para expresar su consentimientocuando redunde en su beneficio directo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
351

2. Cuando, según la ley, un menor no tenga capacidad para expresar su consen-timiento para una intervención, ésta sólo podrá efectuarse con autorización de su repre-sentante, de una autoridad o de una persona o institución designada por la ley.
La opinión del menor será tomada en consideración como un factor que será tantomás determinante en función de su edad y su grado de madurez.
3. Cuando, según la ley, una persona mayor de edad no tenga capacidad, a causade una disfunción mental, una enfermedad o un motivo similar, para expresar su consen-timiento para una intervención, ésta no podrá efectuarse sin la autorización de su repre-sentante, una autoridad o una persona o institución designada por la ley.
La persona afectada deberá intervenir, en la medida de lo posible, en el procedi-miento de autorización.
4. El representante, la autoridad, persona o institución indicados en los aparta-dos 2 y 3, recibirán, en iguales condiciones, la información a que se refiere el artículo 5.
5. La autorización indicada en los apartados 2 y 3 podrá ser retirada, en cual-quier momento, en interés de la persona afectada.
Artículo 7. Protección de las personas que sufran trastornos mentales.
La persona que sufra un trastorno mental grave sólo podrá ser sometida, sin suconsentimiento, a una intervención que tenga por objeto tratar dicho trastorno, cuando laausencia de ese tratamiento conlleve el riesgo de ser gravemente perjudicial para su sa-lud y a reserva de las condiciones de protección previstas por la ley, que comprendan losprocedimientos de supervisión y control, así como los de recurso.
Artículo 8. Situaciones de urgencia.
Cuando, debido a una situación de urgencia, no pueda obtenerse el consenti-miento adecuado, podrá procederse inmediatamente a cualquier intervención indispensa-ble desde el punto de vista médico en favor de la salud de la persona afectada.
Artículo 9. Deseos expresados anteriormente.
Serán tomados en consideración los deseos expresados anteriormente con res-pecto a una intervención médica por un paciente que, en el momento de la intervención,no se encuentre en situación de expresar su voluntad.
352

CAPÍTULO III. Vida privada y derecho a la información.
Artículo 10. Vida privada y derecho a la información.
1. Toda persona tendrá derecho a que se respete su vida privada cuando se tratede informaciones relativas a su salud.
2. Toda persona tendrá derecho a conocer toda la información obtenida respectoa su salud. No obstante, deberá respetarse la voluntad de una persona de no ser informada.
3. De modo excepcional, la ley podrá establecer restricciones, en interés del pa-ciente, con respecto al ejercicio de los derechos mencionados en el apartado 2.
CAPÍTULO IV. Genoma humano.
Artículo 11. No discriminación.
Se prohíbe toda forma de discriminación de una persona a causa de su patrimo-nio genético.
Artículo 12. Pruebas genéticas predictivas.
Sólo podrán hacerse pruebas predictivas de enfermedades genéticas o que permi-tan identificar al sujeto como portador de un gen responsable de una enfermedad, o de-tectar una predisposición o una susceptibilidad genética a una enfermedad, con finesmédicos o de investigación médica y con un asesoramiento genético apropiado.
Artículo 13. Intervenciones sobre el genoma humano.
Únicamente podrá efectuarse una intervención que tenga por objeto modificar elgenoma humano por razones preventivas, diagnósticas o terapéuticas y sólo cuando notenga por finalidad la introducción de una modificación en el genoma de la descendencia.
Artículo 14. No selección de sexo.
No se admitirá la utilización de técnicas de asistencia médica a la procreaciónpara elegir el sexo de la persona que va a nacer, salvo en los casos en que sea preciso paraevitar una enfermedad hereditaria grave vinculada al sexo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
353

CAPÍTULO V. Investigación científica.
Artículo 15. Regla general.
La investigación científica en el ámbito de la biología y la medicina se efectuarálibremente, a reserva de lo dispuesto en el presente Convenio y en otras disposiciones ju-rídicas que garanticen la protección del ser humano.
Artículo 16. Protección de las personas que se presten a un experimento.
No podrá hacerse ningún experimento con una persona, a menos que se den lassiguientes condiciones:
1. que no exista un método alternativo al experimento con seres humanos de efi-cacia comparable,
2. que los riesgos en que pueda incurrir la persona no sean desproporcionados conrespecto a los beneficios potenciales del experimento,
3. que el proyecto de experimento haya sido aprobado por la autoridad competentedespués de haber efectuado un estudio independiente acerca de su pertinencia cientí-fica, comprendida una evaluación de la importancia del objetivo del experimento, así comoun estudio multidisciplinar de su aceptabilidad en el plano ético,
4. que la persona que se preste a un experimento esté informada de sus derechosy las garantías que la ley prevé para su protección,
5. que el consentimiento a que se refiere el artículo 5 se haya otorgado expresa yespecíficamente y esté consignado por escrito. Este consentimiento podrá ser librementeretirado en cualquier momento.
Artículo 17. Protección de las personas que no tengan capacidad para expresar su con-sentimiento a un experimento.
1. Sólo podrá hacerse un experimento con una persona que no tenga, conformeal artículo 5, capacidad para expresar su consentimiento acerca del mismo, cuando se denlas siguientes condiciones:
a) que se cumplan las condiciones enunciadas en el artículo 16, párrafos i a iv;
354

b) que los resultados previstos del experimento supongan un beneficio real y di-recto para su salud;
c) que el experimento no pueda efectuarse con una eficacia comparable con su-jetos capaces de prestar su consentimiento al mismo;
d) que se haya dado específicamente y por escrito la autorización prevista en elartículo 6, y
e) que la persona no exprese su rechazo al mismo.
De modo excepcional y en las condiciones de protección previstas por la ley, po-drá autorizarse un experimento cuyos resultados previstos no supongan un beneficio di-recto para la salud de la persona si se cumplen las condiciones enumeradas en los párrafosi, iii, iv y v del apartado 1 anterior, así como las condiciones suplementarias siguientes:
a) el experimento tenga por objeto, mediante una mejora significativa del conoci-miento científico del estado de la persona, de su enfermedad o de su trastorno, contribuira lograr en un determinado plazo resultados que permitan obtener un beneficio para la per-sona afectada o para otras personas de la misma categoría de edad o que padezcan lamisma enfermedad o el mismo trastorno, o que presenten las mismas características,
b) el experimento sólo represente para la persona un riesgo o un inconveniente mínimo.
Artículo 18. Experimentación con embriones in vitro.
1. Cuando la experimentación con embriones in vitro esté admitida por la ley, éstadeberá garantizar una protección adecuada del embrión.
2. Se prohíbe la constitución de embriones humanos con fines de experimentación.
CAPÍTULO VI. Extracción de órganos y de tejidos de donantes vivos para trasplantes.
Artículo 19. Regla general.
1. La extracción de órganos o de tejidos para trasplantes sólo podrá efectuarse deun donante vivo en interés terapéutico del receptor y cuando no se disponga del órgano o
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
355

del tejido apropiados de una persona fallecida ni de un método terapéutico alternativo deeficacia comparable.
2. El consentimiento a que se refiere el artículo 5 deberá ser expresa y específi-camente otorgado, bien por escrito o ante una autoridad.
Artículo 20. Protección de las personas incapacitadas para expresar su consentimiento ala extracción de órganos.
1. No podrá procederse a ninguna extracción de órganos o de tejidos de una per-sona que no tenga capacidad para expresar su consentimiento conforme al artículo 5.
2. De modo excepcional y en las condiciones de protección previstas por la ley, laextracción de tejidos regenerables de una persona que no tenga capacidad para expresarsu consentimiento podrá autorizarse si se cumplen las condiciones siguientes:
a) si no se dispone de un donante compatible capaz de prestar su consentimiento,
b) si el receptor es hermano o hermana del donante,
c) si la donación es para preservar la vida del receptor,
d) si se ha dado específicamente y por escrito la autorización prevista en losapartados 2 y 3 del artículo 6, según la ley y de acuerdo con la autoridad com-petente,
e) si el donante potencial no expresa su rechazo a la misma.
CAPÍTULO VII. Prohibición del lucro y utilización de una parte del cuerpo humano.
Artículo 21. Prohibición del lucro.
El cuerpo humano y sus partes, como tales, no deberán ser objeto de lucro.
Artículo 22. Utilización de una parte extraída del cuerpo humano.
Cuando una parte del cuerpo humano haya sido extraída en el curso de una inter-vención, no podrá conservarse ni utilizarse con una finalidad distinta de aquélla para laque hubiera sido extraída, salvo de conformidad con los procedimientos de información yde consentimiento adecuados.
356

CAPÍTULO VIII. Contravención de lo dispuesto en el Convenio.
Artículo 23. Contravención de los derechos o principios.
Las Partes garantizarán una protección jurisdiccional adecuada con el fin de im-pedir o hacer cesar en breve plazo cualquier contravención ilícita de los derechos y prin-cipios reconocidos en el presente Convenio.
Artículo 24. Reparación de un daño injustificado.
La persona que haya sufrido un daño injustificado como resultado de una intervencióntendrá derecho a una reparación equitativa en las condiciones y modalidades previstas por la ley.
Artículo 25. Sanciones.
Las Partes deberán prever sanciones apropiadas para los casos de incumplimientode lo dispuesto en el presente Convenio.
CAPÍTULO IX. Relación del presente Convenio con otras disposiciones.
Artículo 26. Restricciones al ejercicio de los derechos.
1. El ejercicio de los derechos y las disposiciones de protección contenidos en elpresente Convenio no podrán ser objeto de otras restricciones que las que, previstas porla ley, constituyan medidas necesarias, en una sociedad democrática, para la seguridadpública, la prevención de las infracciones penales, la protección de la salud pública o laprotección de los derechos y libertades de las demás personas.
2. Las restricciones a que se refiere el párrafo precedente no podrán aplicarse alos artículos 11, 13, 14, 16, 17, 19, 20 y 21.
Artículo 27. Protección más amplia.
Ninguna de las disposiciones del presente Convenio deberá interpretarse en elsentido de que limite o atente contra la facultad de cada Parte para conceder una protec-ción más amplia con respecto a las aplicaciones de la biología y la medicina que la pre-vista por el presente Convenio.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
357

CAPÍTULO X. Debate público.
Artículo 28. Debate público.
Las Partes en el presente Convenio se encargarán de que las cuestiones funda-mentales planteadas por los avances de la biología y la medicina sean objeto de un de-bate público apropiado, a la luz, en particular, de las implicaciones médicas, sociales,económicas, éticas y jurídicas pertinentes, y de que sus posibles aplicaciones sean objetode consultas apropiadas.
CAPÍTULO XI. Interpretación y seguimiento del Convenio.
Artículo 29. Interpretación del Convenio.
El Tribunal Europeo de Derechos Humanos podrá emitir dictámenes consultivos,con independencia de todo litigio concreto que se desarrolle ante un órgano jurisdiccional,sobre cuestiones jurídicas relativas a la interpretación del presente Convenio, a solicitud de:
– el Gobierno de una de las Partes, una vez informadas las demás Partes,
– el Comité instituido por el artículo 32, en su composición restringida a los re-presentantes de las Partes en el presente Convenio, mediante decisión adoptada por ma-yoría de dos tercios de los votos emitidos.
Artículo 30. Informes sobre la aplicación del Convenio.
Cualquier Parte, a instancias del Secretario General del Consejo de Europa, pro-porcionará las explicaciones requeridas acerca del modo en que su legislación interna ga-rantiza la aplicación efectiva de todas las disposiciones del presente Convenio.
CAPÍTULO XII. Protocolos.
Artículo 31. Protocolos.
Podrán redactarse protocolos de conformidad con lo dispuesto en el artículo 32, con elfin de desarrollar, en los ámbitos específicos, los principios contenidos en el presente Convenio.
358

Los protocolos quedarán abiertos a la firma de los signatarios del Convenio. Se-rán sometidos a ratificación, aceptación o aprobación. Un signatario no podrá ratificar,aceptar o aprobar los protocolos, sin haber ratificado, aceptado o aprobado el Convenio conanterioridad o simultáneamente.
CAPÍTULO XIII. Enmiendas al Convenio.
Artículo 32. Enmiendas al Convenio.
1. Las tareas encomendadas al «Comité» en el presente artículo y en el artículo29 se llevarán a cabo por el Comité Director para la Bioética (CDBI) o por cualquier otroComité designado a este efecto por el Comité de Ministros.
2. Sin perjuicio de las disposiciones específicas del artículo 29, todo Estado miem-bro del Consejo de Europa, así como toda Parte en el presente Convenio que no sea miem-bro del Consejo de Europa, podrá hacerse representar en el seno del Comité cuando aquéldesempeñe las tareas confiadas por el presente Convenio, y si dispone de voto en el mismo.
3. Todo Estado a que se refiere el artículo 33 o que haya sido invitado a adherirseal Convenio de conformidad con lo dispuesto en el artículo 34, que no sea Parte en el pre-sente Convenio, podrá designar un observador ante el Comité. Si la Comunidad Europeano es Parte, podrá designar un observador ante el Comité.
4. Con el fin de tener en cuenta los avances científicos, el presente Convenio seráobjeto de un estudio en el seno del Comité en un plazo máximo de cinco años a partir desu entrada en vigor, y en lo sucesivo, a intervalos que determinará el Comité.
5. Toda propuesta de enmienda al presente Convenio, así como toda propuesta deProtocolo o de enmienda a un Protocolo, presentada por una Parte, el Comité o el Comitéde Ministros, será comunicada al Secretario General del Consejo de Europa y se transmi-tirá por mediación del mismo a los Estados miembros del Consejo de Europa, a la Comu-nidad Europea, a todo Signatario, a toda Parte, a todo Estado invitado a firmar el presenteConvenio conforme a lo dispuesto en el artículo 33 y a todo Estado invitado a adherirse almismo conforme a lo dispuesto en el artículo 34.
6. El Comité examinará la propuesta no antes de dos meses a partir de que lehaya sido transmitida por el Secretario General, conforme al párrafo 5. El Comité some-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
359

terá a la aprobación del Comité de Ministros el texto adoptado por mayoría de dos terciosde los votos emitidos. Una vez aprobado, este texto será comunicado a las Partes para suratificación, aceptación o aprobación.
7. Toda enmienda entrará en vigor, con respecto a las Partes que la hayan acep-tado, el primer día del mes siguiente a la expiración de un período de un mes a partir dela fecha en que hayan comunicado al Secretario General su aceptación cinco Partes, com-prendidos al menos cuatro Estados miembros del Consejo de Europa.
Para toda Parte que lo acepte posteriormente, la enmienda entrará en vigorel primer día del mes siguiente a la expiración de un período de un mes a partirde la fecha en que la mencionada Parte haya comunicado al Secretario General suaceptación.
CAPÍTULO XIV. Cláusulas finales.
Artículo 33. Firma, ratificación y entrada en vigor.
1. El presente Convenio queda abierto a la firma de los Estados miembros delConsejo de Europa, de los Estados no miembros que hayan participado en su elaboracióny de la Comunidad Europea.
2. El presente Convenio será sometido a ratificación, aceptación o aprobación.Los instrumentos de ratificación, aceptación o aprobación se depositarán en poder del Se-cretario General del Consejo de Europa.
3. El presente Convenio entrará en vigor el primer día del mes siguiente a la ex-piración de un período de tres meses a partir de la fecha en que cinco Estados, que in-cluyan al menos a cuatro Estados miembros del Consejo de Europa, hayan expresado suconsentimiento en quedar vinculados por el Convenio conforme a lo dispuesto en el apar-tado precedente.
4. Para todo Signatario que exprese posteriormente su consentimiento en quedarvinculado por el Convenio, el mismo entrará en vigor el primer día del mes siguiente a laexpiración de un período de tres meses a partir de la fecha del depósito de su instrumentode ratificación, aceptación o aprobación.
360

Artículo 34. Estados no miembros.
1. Una vez entrado en vigor el presente Convenio, el Comité de Ministros del Con-sejo de Europa podrá invitar a adherirse al presente Convenio, previa consulta a las Par-tes, a cualquier Estado no miembro del Consejo de Europa, mediante una decisiónadoptada por la mayoría prevista en el artículo 20, párrafo d, del Estatuto del Consejo deEuropa, y por unanimidad de los votos de los representantes de los Estados Contratantesque tengan derecho a estar representados en el Consejo de Ministros.
2. Para todo Estado adherente, el Convenio entrará en vigor el primer día del messiguiente a la expiración de un período de tres meses a partir de la fecha del depósito delinstrumento de adhesión ante el Secretario General del Consejo de Europa.
Artículo 35. Aplicación territorial.
1. Todo Signatario, en el momento de la firma o en el momento del depósito desu instrumento de ratificación, aceptación o aprobación, podrá designar el territorio o te-rritorios a los que se aplicará el presente Convenio. Cualquier otro Estado podrá formularla misma declaración en el momento de depositar su instrumento de adhesión.
2. Toda Parte, en cualquier momento posterior, podrá extender la aplicación delpresente Convenio, mediante una declaración dirigida al Secretario General del Consejode Europa, a cualquier otro territorio designado en la declaración y del que asuma las re-laciones internacionales o para el que esté habilitado para adoptar decisiones. El Conve-nio entrará en vigor con respecto a este territorio el primer día del mes siguiente a laexpiración de un período de tres meses a partir de la fecha de recepción de la declaraciónpor el Secretario General.
3. Toda declaración hecha en virtud de los dos apartados precedentes podrá serretirada, en lo que se refiere a cualquier territorio designado en dicha declaración, me-diante notificación dirigida al Secretario General. La retirada surtirá efecto el primer díadel mes siguiente a la expiración de un período de tres meses a partir de la fecha de re-cepción de la notificación por el Secretario General.
Artículo 36. Reservas.
1. Cualquier Estado y la Comunidad Europea podrán formular, en el momento dela firma del presente Convenio o del depósito del instrumento de ratificación, una reservacon respecto a una disposición particular del Convenio, en la medida en que una ley vi-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
361

gente en su territorio no sea conforme a dicha disposición. Las reservas de carácter gene-ral no se autorizan según los términos del presente artículo.
2. Toda reserva emitida conforme al presente artículo incluirá un breve informe dela ley pertinente.
3. Toda Parte que extienda la aplicación del presente Convenio a un territorio de-signado en una declaración prevista en aplicación del apartado 2 del artículo 35, podráformular una reserva para el territorio de que se trate, conforme a lo dispuesto en los apar-tados precedentes.
4. Toda Parte que haya formulado la reserva indicada en el presente artículo po-drá retirarla por medio de una declaración dirigida al Secretario General del Consejo deEuropa. La retirada surtirá efecto el primer día del mes siguiente a la expiración de un pe-ríodo de un mes a partir de la fecha de recepción por el Secretario General.
Artículo 37. Denuncia.
1. Toda Parte podrá denunciar el presente Convenio, en cualquier momento, me-diante notificación dirigida al Secretario General del Consejo de Europa.
2. La denuncia surtirá efecto el primer día del mes siguiente a la expiración deun período de tres meses a partir de la fecha de recepción de la notificación por el Secre-tario General.
Artículo 38. Notificaciones.
El Secretario General del Consejo de Europa notificará a los Estados Miembrosdel Consejo, a la Comunidad Europea, a todo Signatario, a toda Parte y a cualquier otroEstado que haya sido invitado a adherirse al presente Convenio:
1. toda firma;
2. el depósito de todo instrumento de ratificación, aceptación, aprobación oadhesión;
3. toda fecha de entrada en vigor del presente Convenio, conforme a sus artícu-los 33 o 34;
4. toda enmienda o Protocolo adoptado conforme al artículo 32, y la fecha en laque dicha enmienda o protocolo entren en vigor;
362

5. toda declaración formulada en virtud de lo dispuesto en el artículo 35;
6. toda reserva y toda retirada de reserva formuladas conforme a lo dispuesto enel artículo 36;
7. cualquier otro acto, notificación o comunicación que tenga relación con el pre-sente Convenio.
En fe de lo cual, los abajo firmantes, debidamente autorizados a estos efectos, hanfirmado el presente Convenio.
Hecho en Oviedo (Asturias), el 4 de abril de 1997, en francés y en inglés, siendoambos textos igualmente auténticos, en un solo ejemplar que será depositado en los Archi-vos del Consejo de Europa. El Secretario General del Consejo de Europa transmitirá copiacertificada conforme del mismo a cada uno de los Estados Miembros del Consejo de Eu-ropa, a la Comunidad Europea, a los Estados no miembros que hayan participado en la ela-boración del presente Convenio y a todo Estado invitado a adherirse al presente Convenio.
(Versión española publicada por el Ministerio de Asuntos Exteriores de España(Depósito Legal M-10.383-1997),
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
363


Normativa de la ComunidadValenciana


LEY 1/2003, DE 28 DE ENERO, DE DERECHOS E INFORMACIÓN AL PACIENTE DE LA
COMUNIDAD VALENCIANA
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
367


LEY 1/2003, DE 28 DE ENERO, DE DERECHOS E INFORMACIÓN ALPACIENTE DE LA COMUNIDAD VALENCIANA
La Constitución española reconoce en su artículo 43 el derecho a la protec-ción de la salud, parcialmente desarrollado por la Ley 14/1986, de 25 de abril, Ge-neral de Sanidad, que se encuentra presidida por el deseo de garantizar unaprestación sanitaria plenamente respetuosa con la dignidad de la persona y con la li-bertad individual.
Hoy, en efecto, se presta una atención cada vez mayor a los derechos de los pa-cientes como fundamento en toda la labor asistencial. Así se ha plasmado, por ejemplo,en el ámbito de la Unión Europea, en la Directiva 95/46, de 24 de octubre, sobre losderechos de los ciudadanos a la intimidad en la información relacionada con su salud,y la Recomendación 5/1997, de 13 de febrero, sobre protección de datos médicos.
A este criterio responde también la presente ley, inspirada en el Convenio Euro-peo sobre Derechos Humanos y Biomedicina, suscrito el 4 de abril de 1997 y que entróen vigor en el reino de España el 1 de enero de 2000, y en cuya elaboración se han se-guido asimismo las recomendaciones del Dictamen de Expertos del Ministerio de Sanidady Consumo.
El objetivo de la ley es proporcionar una clara definición de los derechos y obliga-ciones de los pacientes, potenciando a su vez la participación activa de los profesionalesy de las instituciones sanitarias para lograr una asistencia, promoción, prevención y reha-bilitación cada vez mejores y más humanas, en beneficio de la salud y la calidad de vidade los ciudadanos.
De este modo, el título II recoge una relación de los derechos de los pacientes, al-gunos de ellos constitutivos de derechos fundamentales o contemplados ya en otros tex-tos normativos, pero que parecía conveniente reunir en una enumeración completa comobase de cualquier actuación sanitaria.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
369

El título III articula el derecho a la información tanto para los centros públicos comoprivados, como eje fundamental en la relación médico-paciente. Cabe destacar, como un as-pecto básico de esta ley, la distinción entre el derecho a la información asistencial y el consen-timiento informado. Este último se regula en el título IV, que lo concibe como el derecho delpaciente a obtener una información adecuada a la naturaleza de su enfermedad sobre los efec-tos de la misma y los riesgos y beneficios de los procedimientos diagnósticos y terapéuticos re-comendados, con el fin de poder decidir consciente y libremente acerca de los mismos. En estesentido, y habida cuenta de la complejidad y la trascendencia de las decisiones en este ám-bito, se establece el derecho del paciente a obtener una segunda opinión que le permita con-tar con mayores elementos de juicio para adoptar una decisión adecuada.
En el capítulo II de este mismo título se regula, por primera vez en la Comunidad Va-lenciana, el derecho de los pacientes a emitir voluntades anticipadas, que serán recogidas enel documento conocido vulgarmente como testamento vital, facultando de este modo al pa-ciente a anticipar su voluntad sobre la atención clínica que desea recibir, en el supuesto de quelas circunstancias de su salud no le permita más adelante decidir por sí mismo, y siempre conel máximo respeto a la vida y la dignidad de la persona.
El título V regula la historia clínica, contemplando todos los aspectos relativos a su con-tenido, tratamiento, propiedad y custodia, así como los derechos de acceso a la misma, por losprofesionales e instituciones, los pacientes, o, en los supuestos que proceda, los familiares, alle-gados o representantes legales.
En el título VI se recoge el derecho a la intimidad como derecho de los pacientes a quesea respetada la confidencialidad de los datos referentes a su salud, estableciendo que nadieque no esté autorizado pueda acceder a ellos si no es al amparo de la legislación vigente.
En el título VII se tratan los derechos de participación de los pacientes, reconociendoel decisivo papel que tiene la colaboración de los ciudadanos en la atención sanitaria, comoejercicio de responsabilidad y solidaridad.
Los avances tecnológicos y sociales plantean, con intensidad creciente, nuevos re-tos éticos, que es necesario abordar desde el más profundo respeto a la dignidad de la per-sona y a la autonomía individual. Para proporcionar una respuesta a esta nueva dimensiónde la atención sanitaria, se crean en el título VIII de esta Ley el Consejo Asesor de Bioética,adscrito a la Conselleria de Sanidad, y los comités de bioética asistencial, con el objeto deproteger los derechos de los pacientes y asesorar en la adopción de decisiones complejas. Es-tos foros interdisciplinares permitirán atender aquellas situaciones asistenciales en las quese haga necesaria la emisión de un consejo, formulado desde el prestigio y la autoridad mo-
370

ral, profesional y científica de sus miembros, sin sustituir, en ningún caso, a quien tiene laresponsabilidad de decidir.
Por último, el título IX recoge las obligaciones de los pacientes, reconociendo deeste modo la responsabilidad de una sociedad madura en el cuidado de la salud individualy colectiva.
Esta ley se dicta al amparo del título competencial recogido en el artículo 38.1 del Es-tatuto de Autonomía, que atribuye a la Generalitat Valenciana el desarrollo legislativo de la le-gislación básica del Estado en materia de sanidad interior.
TÍTULO I. Objeto y ámbito de aplicación
Artículo 1. Objeto
La presente ley tiene por objeto reconocer y garantizar los derechos y obligacio-nes que en materia sanitaria tienen los pacientes en el ámbito territorial de la ComunidadValenciana.
Artículo 2. Ámbito de aplicación
La presente ley será de aplicación a todo tipo de actuación sanitaria que se presteen la Comunidad Valenciana, tanto en los centros públicos como privados.
TÍTULO II. Principios generales
Artículo 3. Principios generales
Todo paciente tiene derecho:
1. Al respeto de su dignidad, sin que pueda sufrir discriminación por razones deraza, sexo, económicas, sociales, ideológicas o de edad.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
371

2. A recibir en todo momento un trato humano, amable, comprensivo y res-petuoso.
3. A acceder a todos los servicios asistenciales disponibles en condiciones deigualdad efectiva.
4. A la confidencialidad de los datos sobre su salud, sin que nadie, sin su au-torización, pueda acceder a ellos, salvo en los casos previstos en la legislación vigente.El secreto profesional estará garantizado en todo momento.
5. A obtener las prestaciones sanitarias asistenciales, farmacéuticas y comple-mentarias necesarias para promover, conservar, restablecer su salud y/o paliar el sufri-miento según lo establecido en la normativa vigente.
6. A las prestaciones básicas del sistema nacional de salud. La administraciónsanitaria de la comunidad autónoma puede establecer prestaciones complementariasen las condiciones establecidas por la legislación vigente o normativa que la desarrolley que serán efectivas previa programación expresa y dotación presupuestaria específica.
7. A recibir información sanitaria en la forma más idónea para su comprensióny, especialmente, en la lengua oficial de la comunidad autónoma y asegurarse que aque-lla sea inteligible para los pacientes. No obstante lo dispuesto en este punto, y en la me-dida que la planificación sanitaria lo permita, los centros y servicios sanitariosimplantarán los medios necesarios para atender las necesidades lingüísticas de los usua-rios extranjeros
8. De acuerdo con el espíritu de la Ley 1/1998, de 5 de mayo, establecerá losmecanismos y las alternativas técnicas oportunas para hacer accesible la información alos discapacitados sensoriales.
9. A obtener, dentro de las posibilidades presupuestarias de la Conselleria de Sa-nidad, una habitación individual para garantizar la mejora del servicio y el derecho a laintimidad y confidencialidad de cada usuario.
10. A no ser sometido a procedimientos diagnósticos o terapéuticos de eficaciano comprobada, salvo si, previamente advertido de sus riesgos y ventajas, da su consen-timiento por escrito y siempre de acuerdo con lo legislado para ensayos clínicos. Esteconsentimiento podrá ser revocado en cualquier momento del procedimiento, debiendoquedar constancia en su historia clínica.
372

11. Todos los pacientes de la Comunidad Valenciana tienen derecho a elegir mé-dico/pediatra y centro, tanto en la atención primaria como en la especializada, en los tér-minos y condiciones que se establezcan por la Conselleria de Sanidad.
12. A que se les hagan y faciliten los informes y certificaciones acreditativas de suestado de salud, cuando sean exigidas mediante una disposición legal o reglamentaria.
13. A participar en las actividades sanitarias a través de las instituciones ylos órganos de participación comunitaria y las organizaciones sociales, en los térmi-nos establecidos por la legislación vigente y en todas aquellas disposiciones que ladesarrollen.
14. A disponer de la tarjeta SIP (Sistema de Información Poblacional) y en sucaso la tarjeta solidaria, en las condiciones que se establezcan normativamente, como do-cumento de naturaleza personal e intransferible acreditativa del derecho a la prestaciónsanitaria en el ámbito de la Comunidad Valenciana.
15. A participar a través de las instituciones y órganos de participación comuni-taria y las organizaciones sociales en actividades sanitarias.
16. A que se respete y considere el testamento vital o las voluntades anticipadasde acuerdo con la legislación vigente.
TÍTULO III. Derechos de información
CAPÍTULO I. Información sanitaria y epidemiológica
Artículo 4. Información sanitaria en la Comunidad Valenciana
1. Además de los derechos a la información personalizada, reconocidos en esta ley,todos los ciudadanos en la Comunidad Valenciana así como las asociaciones de enfermos ofamiliares de enfermos con ámbito de actuación tanto Nacional como de la Comunidad Va-lenciana, tendrán derecho a recibir información general referente al sistema de salud de laComunidad Valenciana y la específica sobre los servicios y unidades asistenciales disponibles,así como a su forma de acceso. Para facilitar este derecho todos los centros dispondrán de:
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
373

a) Una guía o carta en la que se especifiquen los derechos y deberes de los pa-cientes, así como las instalaciones, servicios y prestaciones disponibles y las característi-cas asistenciales del centro o servicio.
b) Un servicio específico para la información y atención al paciente que, entre otrasfunciones, orienten al paciente sobre tales servicios y los trámites de acceso a los mismos.
2. La autoridad sanitaria velará por el derecho de los ciudadanos a recibir infor-mación sanitaria clara, veraz, relevante, fiable, actualizada, de calidad y basada en eviden-cia científica, que posibilite el ejercicio autónomo y responsable de la facultad de eleccióny la participación activa del ciudadano en el mantenimiento o recuperación de su salud.
Artículo 5. Derecho a la información epidemiológica
1. Los ciudadanos tienen derecho a recibir información suficiente y adecuada so-bre las situaciones y causas de riesgo que existan para su salud, incluidos los problemasde salud de la comunidad que implican un riesgo para su salud individual.
2. Los ciudadanos tienen también derecho a recibir información epidemiológicasobre los problemas más comunes de salud y sobre aquellos conocimientos que fomen-ten comportamientos y hábitos de vida saludables para el individuo y la comunidad, pre-vención de las enfermedades y la asunción responsable de la propia salud.
3. La Conselleria de Sanidad informará con carácter periódico del análisis epide-miológico de las distintas áreas de salud.
CAPÍTULO II. Información asistencial
Artículo 6. Información asistencial
1. Los pacientes tienen derecho a conocer toda la información obtenida sobre supropia salud en cualquier proceso asistencial; no obstante, deberá respetarse la voluntaddel paciente si no desea ser informado.
2. La información debe formar parte de todas las actuaciones asistenciales. La in-formación será veraz, fácilmente comprensible y adecuada a las necesidades y los reque-rimientos del paciente, con el objeto de ayudarle a tomar decisiones sobre su salud.
374

3. Corresponde al médico responsable del paciente garantizar el derecho de éstea ser informado. Los profesionales asistenciales que le atienden serán también responsa-bles de facilitar la información que se derive específicamente de sus actuaciones.
4. Aquellos pacientes que no deseen ser informados deberán dejar constancia es-crita o indubitada de este deseo, pudiendo designar a un familiar u otra persona a quiense facilite la información. La designación será por escrito o indudable y podrá ser revocadaen cualquier momento.
5. Podrá restringirse el derecho a no ser informado cuando sea estrictamente ne-cesario en beneficio de la salud del paciente o de terceros, o por razones motivadas de in-terés general.
Artículo 7. El titular del derecho a la información asistencial
1. El paciente es el único titular del derecho a la información. La información quese dé a sus familiares o persona que le represente legalmente, será la que él previamentehaya autorizado expresa o tácitamente. En el supuesto del artículo 6.4 se proporcionarátoda la información al familiar o persona que el paciente haya designado.
2. Cuando a criterio del médico, el paciente esté incapacitado, de manera tem-poral o permanente, para comprender la información, se le dará aquella que su gradode comprensión permita, debiendo informarse también a sus familiares, tutores o per-sonas a él allegadas, incluyendo todas aquellas personas vinculadas a las que se re-fiere la Ley 1/2001, de 6 de abril, de la Generalitat Valenciana por la que se regulanlas parejas de hecho.
3. En el caso de menores, se les dará información adaptada a su grado de madu-rez y, en todo caso, a los mayores de doce años. También deberá informarse plenamente alos padres o tutores que podrán estar presentes durante el acto informativo a los menores.
Los menores emancipados y los mayores de dieciséis años son los titulares del de-recho a la información.
4. Constituirá una excepción al derecho a la información sanitaria de los enfermosla existencia acreditada de una necesidad terapéutica. Se entenderá por necesidad tera-péutica la facultad del médico para actuar profesionalmente sin informar antes al pa-ciente, cuando -por razones objetivas- el conocimiento de su propia situación puedaperjudicar su salud de forma grave.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
375

TÍTULO IV. Consentimiento informado, derecho a la libre elección y a la autonomía delpaciente
CAPÍTULO I. Consentimiento informado y libertad de elección
Artículo 8. El consentimiento informado
1. Se entiende por consentimiento informado la conformidad expresa del paciente,manifestada por escrito, previa la obtención de la información adecuada con tiempo sufi-ciente, claramente comprensible para él, ante una intervención quirúrgica, procedimientodiagnóstico o terapéutico invasivo y en general siempre que se lleven a cabo procedimien-tos que conlleven riesgos relevantes para la salud.
2. El consentimiento debe ser específico para cada intervención diagnóstica o te-rapéutica que conlleve riesgo relevante para la salud del paciente y deberá recabarse porel médico responsable de las mismas.
3. En cualquier momento, la persona afectada podrá retirar libremente su consen-timiento.
Artículo 9. Otorgamiento del consentimiento por sustitución
El consentimiento informado se otorgará por sustitución en los siguientes supuestos:
1. Por los familiares o miembro de unión de hecho, y en su defecto por las perso-nas allegadas, cuando el paciente esté circunstancialmente incapacitado para tomarlas.En el caso de los familiares, tendrá preferencia el cónyuge no separado legalmente; en sudefecto, el familiar de grado más próximo y, dentro del mismo grado, el de mayor edad.Si el paciente hubiera designado previamente una persona, a efectos de la emisión en sunombre del consentimiento informado, corresponderá a ella la preferencia.
2. Cuando el paciente sea menor de edad o se trate de un incapacitado legalmente,el derecho corresponde a sus padres o representante legal, el cual deberá acreditar deforma clara e inequívoca, en virtud de la correspondiente sentencia de incapacitación yconstitución de la tutela, que está legalmente habilitado para tomar decisiones que afec-ten a la persona menor o incapacitada por él tutelada. En el caso de menores emancipa-dos, el menor deberá dar personalmente su consentimiento. No obstante, cuando se tratede un menor y, a juicio del médico responsable, éste tenga el suficiente grado de madurez,se le facilitará también a él la información adecuada a su edad, formación y capacidad.
376

En los supuestos legales de interrupción voluntaria del embarazo, de ensayos clí-nicos y de prácticas de reproducción asistida, se actuará según lo establecido con carác-ter general por la legislación civil y, si procede, por la normativa específica que le sea deaplicación.
3. Cuando la decisión del representante legal pueda presumirse contraria a los in-tereses del menor o incapacitado, deberán ponerse los hechos enconocimiento de la au-toridad competente en virtud de lo dispuesto en la legislación civil.
4. En los casos de substitución de la voluntad del afectado, la decisión deberáser lo más objetiva y proporcional posible a favor de la persona enferma y su dignidadpersonal.
5. En el caso de otorgamiento del consentimiento por sustitución, éste podrá serretirado en cualquier momento en interés de la persona afectada.
Artículo 10. Excepciones a la exigencia del consentimiento
Son situaciones de excepción a la exigencia del consentimiento las siguientes:
a) Cuando la no intervención suponga un riesgo para la salud pública, según de-terminen las autoridades sanitarias. En estos supuestos se adoptarán las medidas admi-nistrativas, de conformidad con lo establecido en la Ley Orgánica 3/1986, de 14 de abril,de Medidas Especiales en Materia de Salud Pública.
b) Cuando el paciente no esté capacitado para tomar decisiones y no existan fa-miliares, personas allegadas o representante legal, o estos últimos se negasen injustifica-damente a prestarlo de forma que ocasionen un riesgo grave para la salud del paciente ysiempre que se deje constancia por escrito de estas circunstancias.
c) Ante una situación de urgencia que no permita demoras por existir el riesgo delesiones irreversibles o de fallecimiento y la alteración del juicio del paciente no permitaobtener su consentimiento.
En estos supuestos, se pueden llevar a cabo las intervenciones indispensablesdesde el punto de vista clínico a favor de la salud de la persona afectada.
Tan pronto como se haya superado la situación de urgencia, deberá informarse alpaciente, sin perjuicio de que mientras tanto se informe a sus familiares y allegados.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
377

Artículo 11. La información previa al consentimiento
1. La información deberá ser veraz, comprensible, razonable y suficiente.
2. La información se facilitará con la antelación suficiente para que el pacientepueda reflexionar con calma y decidir libre y responsablemente. Y en todo caso, al menosveinticuatro horas antes del procedimiento correspondiente, siempre que no se trate de ac-tividades urgentes.
En ningún caso se facilitará información al paciente cuando esté adormecido nicon sus facultades mentales alteradas, ni tampoco cuando se encuentre ya dentro del qui-rófano o la sala donde se practicará el acto médico o el diagnóstico.
3. La información deberá incluir:
- Identificación y descripción del procedimiento.
- Objetivo del mismo.
- Beneficios que se esperan alcanzar.
- Alternativas razonables a dicho procedimiento.
- Consecuencias previsibles de su realización.
- Consecuencias previsibles de la no realización.
- Riesgos frecuentes.
- Riesgos poco frecuentes, cuando sean de especial gravedad y estén asociadosal procedimiento por criterios científicos.
- Riesgos y consecuencias en función de la situación clínica personal del pacientey con sus circunstancias personales o profesionales.
Artículo 12. Responsabilidad de la información previa al consentimiento
La obligación de informar incumbe al médico responsable de la atención del pa-ciente, sin perjuicio de aquella que corresponda a los demás profesionales dentro del ám-bito de su intervención.
Artículo 13. Documento formulario
1. El documento de consentimiento informado deberá contener, además de la in-formación a que se refiere el punto 3 del artículo 11, los siguientes datos mínimos:
378

- Identificación del centro.
- Identificación del paciente.
- Identificación de representante legal, familiar o allegado que presta el consen- timiento.
- Identificación del médico que informa.
- Identificación del procedimiento.
- Lugar y fecha.
- Firmas del médico y persona que presta el consentimiento.
- Apartado para la revocación del consentimiento.
2. En el documento de consentimiento informado quedará constancia de que elpaciente o la persona destinataria de la información recibe una copia de dicho documentoy de que ha comprendido adecuadamente la información que se le ha dado. El consenti-miento puede ser revocado en cualquier momento.
Artículo 14. Comisión de Consentimiento Informado
1. A los efectos previstos en esta ley, se constituirá, dependiendo de la Conselle-ria de Sanidad, la Comisión de Consentimiento Informado, a la que corresponderán las si-guientes funciones:
a) Revisión, actualización y publicación periódica de una guía de formularios dereferencia de consentimiento informado.
b) Conocimiento de la implantación de los formularios en las distintas institucio-nes sanitarias.
c) Prestar asesoramiento a los órganos de la Conselleria de Sanidad en las mate-rias relacionadas con sus funciones.
d) Todas aquellas que le sean atribuidas por normas de carácter legal o reglamentario.
2. La composición de la Comisión de Consentimiento Informado será determinadamediante decreto del Gobierno Valenciano.
3. La Comisión de Consentimiento Informado se reunirá, al menos, dos veces alaño y siempre que la convoque su presidente.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
379

4. Corresponde a la propia comisión la elaboración y aprobación de su reglamentode funcionamiento interno, rigiéndose, en todo lo no previsto en él, por lo dispuesto en elcapítulo II del título I de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico delas Administraciones Públicas y del Procedimiento Administrativo Común, en lo que hacereferencia a los órganos colegiados.
Artículo 15. Libertad de elección
Todo paciente tiene derecho, después de una adecuada información, a decidir li-bremente entre las opciones clínicas que le presente el médico responsable de su caso,siendo preciso su consentimiento previo tal y como se regula en la presente ley.
Artículo 16. Derecho a la segunda opinión
Los pacientes en la Comunidad Valenciana tienen derecho, dentro del sistema sa-nitario público, a una segunda opinión, cuando las circunstancias de su enfermedad le exi-jan tomar una decisión difícil. Este derecho a la segunda opinión será ejercido de acuerdoa lo que se establezca normativamente.
CAPÍTULO II. Voluntades anticipadas
Artículo 17. Voluntades anticipadas
1. El documento de voluntades anticipadas es el documento mediante el que unapersona mayor de edad o menor emancipada, con capacidad legal suficiente y libremente,manifiesta las instrucciones que sobre las actuaciones médicas se deben tener en cuentacuando se encuentre en una situación en la que las circunstancias que concurran no lepermitan expresar libremente su voluntad.
En la declaración de voluntades anticipadas, la persona interesada podrá hacerconstar la decisión respecto a la donación de sus órganos con finalidad terapéutica, do-cente o de investigación. En este caso, no se requerirá autorización para la extracción ola utilización de los órganos donados.
En este documento, la persona otorgante podrá también designar a un represen-tante que será el interlocutor válido y necesario con el médico o el equipo sanitario paraque, caso de no poder expresar por si misma su voluntad, la sustituya.
380

2. El documento de voluntades anticipadas deberá ser respetado por los serviciossanitarios y por cuantas personas tengan relación con el autor del mismo. Caso que enel cumplimiento del documento de voluntades anticipadas surgiera la objeción de con-ciencia de algún facultativo, la administración pondrá los recursos suficientes para aten-der la voluntad anticipada de los pacientes en los supuestos recogidos en el actualordenamiento jurídico.
3. Deberá constar, indubitadamente, que este documento ha sido otorgado enlas condiciones expuestas en el apartado anterior. A estos efectos, la declaración devoluntades anticipadas deberá formalizarse mediante alguno de los procedimientossiguientes:
a) Ante notario. En este supuesto no será necesaria la presencia de testigos.
b) Ante tres testigos mayores de edad y con plena capacidad de obrar, de los cua-les dos, como mínimo, no tendrán relación de parentesco hasta el segundo grado ni vin-culación patrimonial con el otorgante.
c) O cualquier otro procedimiento que sea establecido legalmente.
4. Las voluntades anticipadas pueden modificarse, ampliarse o concretarse o de-jarlas sin efecto en cualquier momento, por la sola voluntad de la persona otorgante, de-jando constancia por escrito o indubitadamente.
En estos casos, se considerará la última actuación de la persona otorgante.
5. No podrán tenerse en cuenta voluntades anticipadas que incorporen previsio-nes contrarias al ordenamiento jurídico o a la buena práctica clínica, o que no se corres-pondan exactamente con el supuesto de hecho que el sujeto ha previsto en el momentode emitirlas. En estos casos, quedará constancia razonada de ello en la historia clínica delpaciente.
6. Cuando existan voluntades anticipadas, la persona que las otorga, o cualquierotra, hará llegar el documento al centro sanitario donde esté hospitalizada y/o a cualquierotro lugar donde esté siendo atendida la persona. Este documento será incorporado a lahistoria clínica del paciente.
7. La Conselleria de Sanidad creará un registro centralizado de voluntades anti-cipadas que desarrollará reglamentariamente.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
381

TÍTULO V. Derechos de documentación
CAPÍTULO I. Informe de alta, alta forzosa y otros informes
Artículo 18. Informe de alta
Todo paciente o familiar o persona allegada en los términos establecidos en estaley, al finalizar el proceso asistencial, tiene derecho a recibir un informe de alta con lossiguientes contenidos mínimos: datos del paciente, un resumen de su historial clínico, laactividad asistencial prestada, el diagnóstico y las recomendaciones terapéuticas. Las ca-racterísticas, los requisitos y las condiciones de los informes de alta, se determinarán deforma reglamentaria por la Conselleria de Sanidad.
Artículo 19. Alta forzosa
1. Los pacientes o, en su caso, personas que pueden recibir el informe de alta,estarán obligados a firmar el alta cuando no acepten el tratamiento prescrito; de negarsea ello, la dirección del centro sanitario, a propuesta del médico responsable, podrá orde-nar el alta forzosa.
El hecho de no aceptar el tratamiento prescrito no dará lugar a un alta forzosacuando haya tratamientos alternativos, aunque tengan carácter paliativo y el pacienteacepte recibirlos. Todas estas circunstancias serán y quedarán debidamente docu-mentadas.
2. En el caso de que no se aceptara el alta forzosa, la dirección del centro, unavez comprobado el informe clínico correspondiente, deberá oír al paciente, y si persisteen su negativa lo pondrá en conocimiento del juez para que confirme o revoque ladecisión.
Artículo 20. Información técnica, estadística y administrativa de los profesionales
Los profesionales sanitarios, además de las obligaciones que en materia de infor-mación clínica les corresponde, según se establece en esta ley, tendrán, asimismo, el de-ber de cumplimentar los protocolos, registros, informes estadísticos y demásdocumentación técnica o administrativa, relacionada con los procesos asistenciales en losque intervengan, y que se requiera por las autoridades sanitarias, incluidos los relativos ainvestigaciones médicas e información epidemiológica.
382

CAPÍTULO II. La historia clínica
Artículo 21. Definición y tratamiento de la historia clínica
1. La historia clínica es el conjunto de documentos en los que está contenida todala información obtenida en todos los procesos asistenciales del paciente. La historia clí-nica tiene como fin principal facilitar la asistencia sanitaria, dejando constancia de todosaquellos datos que permitan el conocimiento veraz y actualizado del estado de salud delpaciente, acumulando toda la información generada en cada episodio asistencial.
2. La historia clínica deberá realizarse bajo criterios de unidad e integración entodos los centros y servicios sanitarios, donde existirá una única historia por paciente, conel fin de facilitar en cualquier momento del proceso asistencial el conocimiento de todoslos datos de un determinado paciente. Estos datos deben estar disponibles para todos losprofesionales que intervengan en el proceso asistencial.
3. El centro debe almacenar las historias clínicas en instalaciones que garanticenla seguridad, confidencialidad, la correcta conservación y la recuperación de la información.
4. Las historias clínicas se pueden elaborar en soporte papel, audiovisual o infor-mático, siempre que esté garantizada la autenticidad del contenido de las mismas y su re-producción futura. En cualquier caso, debe garantizarse que queden debidamenteregistrados todos los cambios e identificados los médicos y demás profesionales asisten-ciales que los han realizado. Se garantizará la confidencialidad de la información conte-nida en ella y se atenderá a lo dispuesto en la normativa vigente sobre tratamientoautomatizado de datos de carácter personal.
5. En la historia clínica deberán ser claramente legibles, evitando, en lo posible,la utilización de símbolos y abreviaturas, y estarán normalizadas en cuanto a su estructuralógica, de conformidad con lo que reglamentariamente se disponga. Cualquier informaciónincorporada deberá ser fechada y firmada de forma que se identifique claramente la per-sona que la realiza.
En las historias clínicas en las que participen más de un médico o un equipo asis-tencial, deberán constar individualizadas las acciones, intervenciones y prescripciones re-alizadas por cada profesional.
6. Los centros sanitarios deberán adoptar todas las medidas técnicas y organiza-tivas necesarias para proteger los datos personales recogidos y evitar su destrucción o su
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
383

pérdida accidental, y también el acceso, alteración, comunicación o cualquier otro trata-miento que no esté autorizado.
7. Los centros sanitarios dispondrán de un modelo normalizado de historia clí-nica, que recoja los contenidos fijados en el artículo siguiente, adaptados al nivel asisten-cial y la clase de prestación que se realice.
8. En caso de traslado urgente y/o obligado del paciente a otro centro asistencial,se remitirá una copia completa de la historia clínica a fin de garantizar a los facultativosdel centro sanitario de destino el pleno conocimiento de la situación clínica actualizadadel paciente.
Artículo 22. Contenido de la historia clínica
1. La historia clínica deberá contener la información suficiente para identificarclaramente al paciente, justificar el diagnóstico y tratamiento y documentar todos los re-sultados con exactitud, para ello la historia clínica tendrá un número de identificación eincluirá los siguientes datos mínimos:
a) Identificación de la institución, del centro, número de tarjeta SIP, si procede,número de historia clínica y nota indicativa de las características de confidencialidad quecontiene.
b) Datos suficientes para la identificación del paciente:
- Nombre y apellidos.
- Fecha y lugar de nacimiento.
- Sexo.
- Domicilio habitual y teléfono.
- Fecha de asistencia y de ingreso si procede.
- Indicación de la procedencia, en caso de derivación desde otro centro asistencial.
- Servicio o unidad en la que se presta la asistencia, si procede.
- Número de habitación y de cama en caso de ingreso.
- Médico responsable del enfermo.
- Todos aquellos documentos básicos que contemple la normativa vigente. Cual-quier otro dato que se establezca normativamente.
384

c) Datos clínico asistenciales:
- Anamnesis y exploración física.
- Descripción de la enfermedad o problema de salud actual y motivos sucesivosde consulta.
- Hoja de interconsulta.
- Procedimientos clínicos diagnósticos y terapéuticos empleados y sus resultados,con los dictámenes correspondientes emitidos en caso de exámenes especiali-zados, y las hojas de interconsulta.
- Hojas de evolución y seguimiento.
- Documento de consentimiento informado, si procede.
- Hoja de voluntades anticipadas, si las hubiere.
- Hoja de autorización.
- Informe quirúrgico y de anestesia, si procede.
- Informe de alta.
- Hoja de problemas.
- Documento firmado de alta voluntaria, si lo hubiere.
- Informe de necropsia, si existe.
- Todos aquellos documentos básicos que contemple la normativa vigente.
d) Datos sociales:
- Informe social, si procede.
2. Para garantizar los usos de la historia clínica, especialmente el asistencial, seconservarán los documentos como mínimo cinco años a partir de la fecha del último epi-sodio asistencial en el que el paciente haya sido atendido o desde su fallecimiento. Aque-llos documentos especialmente relevantes se conservarán indefinidamente o por el tiempoque fije la normativa vigente al respecto. Las historias clínicas que sean prueba en un pro-ceso judicial o procedimiento administrativo se conservarán hasta la finalización del mismo.
Artículo 23. Propiedad y custodia
1. Las historias clínicas son documentos confidenciales propiedad de la adminis-tración sanitaria o entidad titular del centro sanitario cuando el médico trabaje por cuenta
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
385

ajena y bajo la dependencia de una institución sanitaria. En caso contrario, la propiedadcorresponde al médico que realiza la atención sanitaria.
2. La entidad o facultativo propietario es responsable de la custodia de las histo-rias clínicas y habrá de adoptar todas las medidas necesarias para garantizar la confiden-cialidad de los datos y de la información contenida.
3. La gestión de la historia clínica será responsabilidad de la Unidad de Admisióny Documentación Clínica, de manera integrada en un único archivo de historias clínicaspor centro.
4. Toda persona que en el ejercicio de sus funciones o competencias tenga cono-cimiento de los datos e informaciones contenidas en la historia clínica, tiene el deber dereserva y sigilo respecto de los mismos.
5. La obligación de conservar la documentación clínica en condiciones que garan-ticen su correcto mantenimiento, confidencialidad y seguridad, corresponderá al centro enla que se haya generado. En cualquier caso, la conservación de la documentación clínicadeberá garantizar la preservación de la información y no necesariamente del soporte ori-ginal.
6. Todos los profesionales sanitarios deberán cooperar en el mantenimiento deuna documentación clínica ordenada, que refleje, con las secuencias necesarias en eltiempo, la evolución del proceso asistencial del paciente
Artículo 24. Derecho de acceso a la historia clínica de los profesionales sanitarios e ins-tituciones
1. Los profesionales asistenciales del centro implicados en el diagnóstico o el tra-tamiento del enfermo tendrán libre acceso a su historia clínica.
2. Cada centro establecerá los mecanismos que hagan posible el acceso a la his-toria clínica en el momento del proceso asistencial en que sea necesario.
3. Asimismo, se puede acceder a la historia clínica, con finalidades epidemioló-gicas, información estadística sanitaria, actividades relacionadas con el control y evalua-ción de la calidad asistencial, las encuestas oficiales y los programas oficiales deinvestigación o docencia, con sujeción a lo establecido en la Ley orgánica 15/1999, de13 de diciembre, de Protección de Datos de Carácter Personal, y la Ley 14/1986, de 25
386

de abril, General de Sanidad, y las disposiciones concordantes. El acceso a la historia clí-nica con estas finalidades obliga a preservar los datos de identificación personal del pa-ciente, que siempre tendrán que estar separadas de las de carácter clínico asistencial,salvo si el paciente, previamente, ha dado su consentimiento.
4. Podrá accederse a la historia clínica por requerimiento de la autoridad judicial.
5. El personal encargado de tareas administrativas y de gestión de los centros sa-nitarios podrá acceder exclusivamente a los datos de la historia clínica relacionados condichas funciones.
6. La historia clínica estará disponible, con absoluta garantía del derecho a la in-timidad personal y familiar, a efectos de inspección sanitaria, para las actividades de eva-luación, acreditación y comprobación del cumplimiento de los derechos del paciente, yotras debidamente motivadas por la autoridad sanitaria y que tengan por finalidad contri-buir a la mejora de la calidad asistencial. En estos supuestos el acceso a la historia clí-nica estará limitado a la información relacionada con tales fines.
7. Aquel personal que accede en el uso de sus competencias a cualquier clase dedatos de la historia clínica queda sujeto al deber de guardar el secreto de los mismos.
Artículo 25. Derecho del paciente a acceder a su historia clínica
1. El paciente tendrá derecho a acceder a todos los documentos y datos de su his-toria clínica. El derecho de acceso conllevará el de obtener copias de los mencionados do-cumentos. Este acceso nunca será en perjuicio del derecho de terceros a laconfidencialidad de sus datos que figuren en ella.
2. Cuando no sea el paciente quien solicite el acceso a su historia clínica, so-lamente se podrá efectuar si el paciente ha dado expresamente su conformidad porescrito.
3. El derecho de acceso del paciente a la historia clínica puede ejercerse tambiénpor representación legal, siempre que ésta esté debidamente acreditada.
4. En el caso de pacientes fallecidos, sólo se facilitará el acceso a la historia clí-nica a los familiares más allegados o miembro de la unión de hecho, salvo en el supuestode que el fallecido lo hubiese prohibido expresamente, constituyéndose el centro sanita-rio en garante de la información.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
387

5. No se facilitará, en ningún caso, información que afecte a la intimidad del finado,ni los datos que perjudiquen a terceros, tal y como se recoge en el punto 1 de este artículo.
TÍTULO VI. Derecho a la intimidad
Artículo 26. Formulación y alcance del derecho a la intimidad
1. Toda persona tiene derecho a que se respete la confidencialidad de los datosreferentes a su salud. Nadie que no esté autorizado podrá acceder a ellos si no es al am-paro de la legislación vigente.
2. Todos los centros sanitarios adoptarán las medidas oportunas para garantizar losderechos a los que se refiere el punto 1 de este artículo y a tal efecto elaborarán normas in-ternas y procedimientos protocolizados que regulen el acceso a los datos del paciente.
3. Todo paciente tiene derecho a que se preserve la intimidad de su cuerpo conrespecto a otras personas. La prestación de las atenciones necesarias se hará respetandolos rasgos básicos de la intimidad.
TÍTULO VII. Derecho de participación
Artículo 27. Derecho a formular sugerencias, quejas, reclamaciones y agradecimientos
1. Los ciudadanos de la Comunidad Valenciana tienen el derecho a formular su-gerencias, quejas, y reclamaciones cuando consideren que tienen motivo justificado parahacerlo. Estas se deben evaluar y contestar por escrito, en un plazo adecuado, de acuerdocon los términos que se establezcan reglamentariamente.
2. Podrán realizar también manifestaciones de agradecimiento cuando la labordel profesional, el equipo y el centro asistencial que les ha atendido, a su juicio lo me-rece, debiendo llegar esta manifestación de agradecimiento a los profesionales que lahan merecido.
388

Artículo 28. Consejos de salud
A través de las organizaciones sociales, los ciudadanos pueden participar con lasinstituciones sanitarias formando parte de los Consejos de Salud, de acuerdo a lo que lanormativa vigente establece.
Artículo 29. Actividades de voluntariado
Como expresión de la solidaridad, los ciudadanos pueden participar en tareas deapoyo en la atención de los pacientes, dentro del marco legal que establece la Ley del Vo-luntariado de la Comunidad Valenciana.
TÍTULO VIII. Consejo Asesor de Bioética y Comités de Bioética Asistencial
Artículo 30. Consejo Asesor de Bioética y comités de bioética asistencial
1. Con el objeto de dilucidar aspectos de carácter ético relacionados con la prác-tica asistencial, poder establecer criterios generales ante determinados supuestos quepueden aparecer con la incorporación de nuevas modalidades asistenciales y nuevas tec-nologías, fomentar el sentido de la ética en todos los estamentos sanitarios y organizacio-nes sociales o desarrollar cualquier otro tipo de actividad relacionada con la bioética, secrea el Consejo Asesor de Bioética de la Comunidad Valenciana, adscrito a la Conselleriade Sanidad, y los comités de bioética asistencial.
2. Dichos organismos tendrán un carácter consultivo e interdisciplinar. Su com-posición y funciones serán establecidas mediante decreto del Consell de la Generalitat.
TÍTULO IX. Obligaciones de los pacientes
Artículo 31. Obligaciones de los pacientes
Los ciudadanos en el ámbito territorial de la Comunidad Valenciana están sujetosrespecto al sistema de salud al cumplimiento de las obligaciones siguientes:
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
389

1. Hacer buen uso de los recursos, prestaciones asistenciales y derechos, de acuerdocon lo que su salud necesite y en función de las disponibilidades del sistema sanitario.
2. Cumplir las prescripciones de naturaleza sanitaria que con carácter general se es-tablezcan para toda la población con el fin de prevenir riesgos para la salud.
3. Hacer un uso racional y de acuerdo con lo legislado, de las prestaciones farma-céuticas y la incapacidad laboral.
4. Utilizar y cuidar las instalaciones y los servicios sanitarios contribuyendo a suconservación y favoreciendo su habitabilidad y el confort de los demás pacientes.
5. Tratar con consideración y respeto a los profesionales que cuidan de su salud ycumplir las normas de funcionamiento y convivencia establecidas en cada centro sanitario.
6. Facilitar de forma veraz sus datos de identificación y los referentes a su estadofísico y psíquico que sean necesarios para el proceso asistencial o por razones de interésgeneral debidamente justificadas
7. Firmar el documento pertinente o por un medio de prueba alternativo, que encaso de imposibilidad, manifieste claramente su voluntad de negarse a recibir el trata-miento que se le ha prescrito, especialmente cuando se trate de pruebas diagnósticas, me-didas preventivas o tratamientos especialmente relevantes para su salud, teniendo encuenta lo establecido en el artículo 19.1 de esta ley.
8. Aceptar el alta cuando haya finalizado el proceso asistencial.
9. Cumplir las normas y procedimientos de uso y acceso a los derechos que se leotorguen a través de la presente ley.
TÍTULO X. Régimen sancionador
Artículo 32. Régimen sancionador
Sin perjuicio de las exigencias que pudieran derivarse en los ámbitos de la respon-sabilidad civil y penal, o de la responsabilidad profesional o estatutaria, será de aplicación
390

el régimen sancionador previsto en el capítulo VI del título I de la Ley 14/1986, de 25 deabril, General de Sanidad; en el título VII de la Ley 15/1999, de 13 de diciembre, de Pro-tección de Datos de Carácter Personal, y el artículo 5. 2 de la Ley 28/1992, de 24 de no-viembre, de Ordenación Económica y Medidas Presupuestarias Urgentes, referente autilización abusiva de la prescripción de medicamentos.
DISPOSICIONES ADICIONALES
Primera
La Conselleria de Sanidad realizará las inspecciones oportunas a los efectos de ga-rantizar y comprobar que los centros e instituciones sanitarias y el personal a su serviciocumplen las obligaciones establecidas en la presente ley.
Segunda
La Conselleria de Sanidad procederá a agilizar los procedimientos de aprobaciónde los formularios de consentimiento informado para que puedan ser utilizados progresi-vamente en los centros asistenciales públicos y privados.
Tercera
La Conselleria de Sanidad adoptará las medidas adecuadas para la informatiza-ción progresiva de las historias clínicas, garantizando la integridad de la información re-lativa a cada paciente con independencia del soporte en que se encuentre.
DISPOSICIÓN TRANSITORIA
Se dispone de un año, a partir de la entrada en vigor de la presente ley, paraadoptar las medidas técnicas y organizativas necesarias para adaptar el tratamiento delas historias clínicas a lo que esta ley establece, y elaborar los modelos normalizados dehistoria clínica a que se refieren los artículos 21 y 22. Los procesos asistenciales que selleven a cabo transcurrido este plazo deben reflejarse documentalmente de acuerdo conlos modelos normalizados aprobados.
DISPOSICIÓN DEROGATORIA
Quedan derogadas cuantas disposiciones de la Generalitat Valenciana de igual oinferior rango contradigan o se opongan a lo contenido en esta ley.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
391

DISPOSICIONES FINALES
Primera
Se autoriza al Consell de la Generalitat para dictar las normas reglamentarias ne-cesarias para el desarrollo y aplicación de esta ley.
Segunda
La presente ley entrará en vigor el día siguiente al de su publicación en el DiariOficial de la Generalitat Valenciana.
392

Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
393
DECRETO 73/2006, DE 5 DE JUNIO, DEL CONSELL,POR EL QUE SE REGULA LA GESTIÓN DE ENSAYOS
CLÍNICOS Y ESTUDIOS POST-AUTORIZACIÓN OBSERVACIONALES CON MEDICAMENTOS
Y PRODUCTOS SANITARIOS

394

DECRETO 73/2009, DE 5 DE JUNIO, DEL CONSELL, POR EL QUE SE REG-ULA LA GESTIÓN DE ENSAYOS CLÍNICOS Y ESTUDIOS POST-AUTOR-IZACIÓN OBSERVACIONALES CON MEDICAMENTOS Y PRODUCTOSSANITARIOS
Preámbulo
La Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentosy productos sanitarios ha intensificado el impacto de dos ideas-fuerza: la ampliación y re-forzamiento de un sistema de garantías que gire en relación a la autorización del medica-mento y la promoción del uso racional del mismo. El desarrollo tecnológico, la globalización,el acceso a la información así como la pluralidad de agentes que progresivamente inter-vienen en el ámbito de la producción, distribución, dispensación y administración demedicamentos aconsejan ampliar la transparencia y el control de sus resultados en el usode medicamentos y productos sanitarios.
El legislador, tanto europeo, estatal y autonómico, ha diseñado un marco norma-tivo donde a los medicamentos y productos sanitarios se les dota de entidad propia a losefectos de la observación, análisis y control de resultados para la aplicación en la prác-tica clínica asistencial de los conocimientos derivados de la investigación básica enmedicamentos y productos sanitarios. Fruto de ello es la inclusión de los ensayos clínicosdentro del marco competencial y regulatorio de los productos farmacéuticos. La promul-gación de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamen-tos y productos sanitarios vuelve a reafirmar el entorno propio, excluyéndose esta materiadel ámbito de aplicación de la ley 14/2007 de Investigación Biomédica. El marco de ac-tuación y tramitación en ensayos clínicos de medicamentos y productos sanitarios se com-plementa con las directrices estatales en farmacovigilancia y estudios post-autorizaciónobservacionales de interés para la salud pública, ostentando competencias de ejecucióntanto la Agencia Española del Medicamento como el centro coordinador de comités éti-cos de investigación clínica a través de la Dirección General de Farmacia y ProductosSanitarios del Ministerio de Sanidad y Consumo.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
395

La presente norma moderniza la estructura de gestión de los ensayos clínicos y es-tudios postautorización observacionales en medicamentos y productos sanitarios, facili-tando la elección de la Comunitat Valenciana como lugar para el inicio de los estudiosreferenciados, desde el punto de vista de la agilidad en la tramitación y control de la eje-cución de los estudios clínicos en medicamentos y productos sanitarios. La estructura degestión mantiene una separación organizativa de las entidades que coordinan, impulsan,seleccionan o ejecutan las materias terapéuticas objeto de los protocolos de estudio, re-forzando la independencia y transparencia en el control y la toma de decisiones sobre laidoneidad ético-clínica de los referenciados estudios. A los efectos de obtener el mayorgrado de consenso, se ha recabado el apoyo y asesoramiento de las entidades de laGeneralidad Valenciana que impulsan materias de investigación sanitaria, así como de lasfundaciones para la promoción de la investigación, estructuras que impulsan, promueven,favorecen y desarrollan la investigación científico-técnica en los centros sanitarios de laComunitat Valenciana.
La Conselleria de Sanitat viene realizando actividades regulatorias e inspectorasen materia de ensayos clínicos de medicamentos y productos sanitarios desde la promul-gación de la primera ley del medicamento del periodo constitucional.
La Ley 25/1990, de 20 de diciembre, del Medicamento, dedicó su título III a losensayos clínicos con medicamentos, combinando la necesidad de su existencia comomecanismo necesario para los avances científicos y el obligado respeto a los derechos fun-damentales de quienes sean sometidos a los mismos. El Real Decreto 561/1993, de 16 deabril desarrolló la citada ley atribuyendo a las comunidades autónomas la competencia enla especificación de los aspectos económicos de la realización de los ensayos clínicos, enel procedimiento de acreditación de los comités éticos de investigación clínica (en adelanteCEIC) y en las facultades inspectoras en materia de ensayos clínicos respectivamente.
Mediante Orden de 6 de julio de 1994, de la Conselleria de Sanidad y Consumo seregulan estas competencias, unificando los criterios en la acreditación de CEIC y en lainspección sanitaria en materia de ensayos clínicos, estableciendo las medidas que sehabían de adoptar en lo que se refiere a contratos económicos y velando por el cumplimientode las Normas de Buena Práctica Clínica para ensayos clínicos con medicamentos.
En la misma línea, la Ley 29/2006, de 26 de julio, de garantías y uso racional delos medicamentos y productos sanitarios y el Real Decreto 223/2004, de 6 de febrero, porel que se regulan los ensayos clínicos con medicamentos reafirman las competencias delas Comunidades Autónomas en esta materia y la necesidad imperativa de que todos losensayos clínicos tengan un informe previo favorable de un CEIC debidamente acreditado.
396

Aunque los medicamentos han contribuido decisivamente a la mejora de la espe-ranza y al aumento de la calidad de vida, en ocasiones plantean problemas de efectividad yde seguridad, necesitando las Administraciones Públicas adoptar políticas innovadoras queincorporen el concepto de farmacoepidemiología y gestión de los riesgos, así como la garan-tía de seguimiento continuado del balance beneficio/riesgo de los medicamentos autorizados.
La publicación del RD 1344/2007 de 11 de octubre, que regula la farmacovigi-lancia de medicamentos de uso humano persigue agilizar la tramitación de los estudiospost-autorización observacionales de interés para la salud pública con medicamentos yproductos sanitarios. Actualmente se necesita potenciar y facilitar su desarrollo paragenerar un conocimiento que permita evaluar de un modo más efectivo la relación benefi-cio-riesgo de los medicamentos. Estos estudios se llevarán a cabo de acuerdo con lascondiciones que establezcan las Administraciones Sanitarias en el ámbito de su compe-tencia. La Conselleria de Sanidad reguló esta potestad mediante la Orden de 8 de marzode 2004, por la que se crea la Comisión Asesora para la evaluación de los estudios post-autorización observacionales prospectivos.
La publicación en octubre de 2008 de la nueva versión de la declaración deHelsinki de la Asociación Médica Mundial (Seúl 2008) presenta en su articulado algunasnovedades en sus artículos 1, 14, 15 y 25 que afectan tanto a ensayos clínicos y estu-dios observacionales y que se incorporan en la presente regulación.
En el marco expuesto, se hace necesario una ordenación y actualización del marconormativo valenciano, clarificando la participación de la Agencia Valenciana de Salud, através de su Dirección General de Farmacia y Productos Sanitarios, como órgano competentede gestión y control de la información referente a ensayos clínicos autorizados y estudiospost-autorización observacionales de medicamentos y productos sanitarios. La presente dis-posición desarrolla el modelo valenciano mediante el establecimiento del programa de es-tudios clínicos en medicamentos y productos sanitarios de la Comunitat Valenciana (enadelante PECME), modernizando el sistema de información autonómico sobre esta materiay regulando las actividades de formación y coordinación de los CEIC locales. Se constituyeel Comité Autonómico de Estudios Clínicos de medicamentos y productos sanitarios (CAEC),que adicionalmente a las funciones propias de evaluación clínica, se configura como unórgano consultivo encargado de asistir y asesorar en todo aquello relacionado con el desa-rrollo de los procesos de evaluación clínica en medicamentos y productos sanitarios.
En virtud de todo lo anterior, de conformidad con lo dispuesto en el artículo 28de la Ley 5/1983, de 30 de diciembre, del Consell, a propuesta del Conseller de Sanidad,previa audiencia de los sectores implicados, conforme con el dictamen del Consejo Ju-
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
397

rídico Consultivo y tras la deliberación del Consell de la Generalidad en la reunión de 5de junio de 2009
DISPONGO
Artículo 1. Ámbito y objeto de aplicación
La presente norma tiene por objeto establecer la estructura organizativa para larealización de ensayos clínicos y estudios post-autorización de tipo observacional conmedicamentos y productos sanitarios de uso humano en la Comunitat Valenciana deacuerdo con la normativa estatal en la materia.
Artículo 2. Definiciones
1. Ensayo clínico: Estudio efectuado en seres humanos para determinar o confir-mar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o de de-tectar las reacciones adversas, y/o de estudiar la absorción, distribución, metabolismo yexcreción de uno o varios medicamentos en investigación con el fin de determinar su se-guridad y/o su eficacia.
2. Ensayo clínico multicéntrico: ensayo clínico realizado de acuerdo con un pro-tocolo único pero en más de un centro y, por tanto, realizado por más de un investigador.
3. Estudio observacional: estudio en el que los medicamentos se prescriben de lamanera habitual, de acuerdo con las condiciones normales de la práctica clínica (aque-llas establecidas en la autorización de comercialización). La asignación de un paciente auna estrategia terapéutica concreta no estará decidida de antemano por un protocolo deensayo, sino que estará determinada por la práctica habitual de la medicina, y la decisiónde prescribir un medicamento determinado estará claramente disociada de la decisión deincluir al paciente en el estudio. No se aplicará a los pacientes ninguna intervención, yasea diagnóstica o de seguimiento, que no sea la habitual de la práctica clínica, y se uti-lizarán métodos epidemiológicos para el análisis de los datos recogidos
3a. Estudio observacional prospectivo: aquel estudio en el que los pacientes son se-leccionados por su exposición a un determinado medicamento y son después seguidos du-rante un tiempo suficiente, siendo el período de estudio posterior al inicio de la investigación.
3b. Estudio observacional retrospectivo: aquel estudio en el que los pacientes sonseleccionados por su exposición a un determinado medicamento y son después seguidos
398

durante un tiempo suficiente, siendo todo el período de estudio anterior al inicio de la in-vestigación.
3c. Estudio observacional transversal: aquel estudio en el que se registran obser-vaciones sobre numerosos factores en el mismo momento y luego se comparan entre ellas.
4. Estudio post-autorización: Cualquier estudio clínico o epidemiológico realizadodurante la comercialización de un medicamento según las condiciones autorizadas en suficha técnica, o bien en condiciones normales de uso, en el que el medicamento o losmedicamentos de interés son el factor de exposición fundamental investigado. Este estu-dio podrá adoptar la forma de un ensayo clínico o un estudio observacional. Los estudiospost-autorización deberán tener como finalidad el complementar la información obtenidadurante el desarrollo clínico de los medicamentos, previo a su autorización, y no podráncondicionar directa o indirectamente la prescripción o dispensación. No se planificarán,realizarán o financiarán estudios post-autorización con la finalidad de promover la pres-cripción o dispensación de los medicamentos
4a. Estudio post-autorización observacional: Estudio epidemiológico que cumplelas condiciones de ser post-autorización y observacional.
4b. Estudio post-autorización observacional de seguimiento: Todo aquel estudiopost-autorización de tipo observacional en el que los pacientes son seleccionados por suexposición a un determinado medicamento y son después seguidos durante un período detiempo suficiente, en relación con el acontecimiento de interés. Se consideran prospec-tivos cuando el período de estudio es posterior al inicio de la investigación y retrospectivoscuando el período de estudio es todo él anterior al inicio de la investigación. Cuando serealice un estudio post-autorización observacional de seguimiento prospectivo, el/los pro-motor/es e investigador/es deberán expresar específicamente en el protocolo los proce-dimientos que se emplearán para garantizar que la realización del estudio no modificarálos hábitos de prescripción del médico o de dispensación del farmacéutico (en caso demedicamentos que no requieran prescripción).
5. Reacción adversa: Cualquier respuesta a un medicamento que sea nociva y nointencionada, y que tenga lugar a dosis que se apliquen normalmente en el ser humanopara la profilaxis, el diagnóstico o el tratamiento de enfermedades, o para la restauración,corrección o modificación de funciones fisiológicas. Este término incluye también todaslas consecuencias clínicas perjudiciales derivadas de la dependencia, abuso y uso inco-rrecto de medicamentos, incluyendo las causadas por el uso fuera de las condicionesautorizadas y las causadas por errores de medicación.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
399

5a. Reacción adversa grave: Cualquier reacción adversa que ocasione la muerte,pueda poner en peligro la vida, exija la hospitalización del paciente o la prolongación dela hospitalización ya existente, ocasione una discapacidad o invalidez significativa o per-sistente o constituya una anomalía congénita o defecto de nacimiento. A efectos de su no-tificación, se tratarán también como graves aquellas sospechas de reacción adversa quese consideren importantes desde el punto de vista médico, aunque no cumplan los crite-rios anteriores, como las que ponen en riesgo al paciente o requieren una intervenciónpara prevenir alguno de los desenlaces anteriores. Así mismo, a efectos de su notificación,se tratarán como graves todas las sospechas de transmisión de un agente infeccioso através de un medicamento.
5b. Reacción adversa inesperada: Cualquier reacción adversa cuya naturaleza,gravedad o consecuencias no sean coherentes con la información descrita en la ficha técnica.
6. Medicamento en investigación: forma farmacéutica de un principio activo oplacebo, que se investiga o utiliza como referencia en un ensayo clínico, incluidos los pro-ductos con autorización cuando se utilizan o combinen (en la formulación o en el envase)de forma diferente a la autorizada, o cuando se utilicen para tratar una indicación noautorizada., o para obtener más información sobre un uso autorizado.
Artículo 3. Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios en laComunitat Valenciana (PECME)
1. El Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios(PECME), dependiente de la Agencia Valenciana de Salud, gestiona las funciones citadasen esta disposición propias de ensayos clínicos y estudios post-autorización observa-cionales, así como las establecidas en esta materia en el RD 223/2004, de 6 de febrero,por el que se regulan los ensayos clínicos con medicamentos, el RD 1344/2007, de 11de octubre, por el que se regula la farmacovigilancia de medicamentos de uso humano yel RD 1143/2007, de 31 de agosto, por el que se modifican los Reales Decretos634/1993, de 3 de mayo, sobre productos sanitarios implantables activos; 414/1996, de1 de marzo, por el que se regula los productos sanitarios; y 1662/2000, de 29 de sep-tiembre, sobre productos sanitarios para diagnóstico «in vitro».
2. El Programa tendrá los objetivos siguientes:
a) Promover la correcta realización de ensayos clínicos y estudios post-autorizaciónobservacionales de medicamentos y productos sanitarios tanto en la aplicación de la nor-mativa vigente como desde el punto de vista científico, así como el adecuado conocimiento
400

de los aspectos éticos y metodológicos por parte de todos los agentes implicados en larealización y evaluación de dichos ensayos y estudios.
b) Garantizar que los miembros de los CEIC acreditados en la Comunitat Valen-ciana están capacitados para evaluar los protocolos de los ensayos clínicos y estudios post-autorización observacionales en sus aspectos metodológico, ético y legal.
c) Establecer y organizar sistemas de información adecuados y fluidos entre laAdministración Sanitaria autonómica, la Agencia Española del Medicamento, el CentroCoordinador de CEIC, los CEIC y los promotores e investigadores de los ensayos clínicosy estudios post-autorización observacionales.
3.- El Programa se articula con la intervención de las siguientes entidades:
El Consejo de Dirección y la Comisión Delegada del PECME.
El Coordinador del PECME.
El Comité Autonómico de Estudios Clínicos de Medicamentos y Productos Sani-tarios de la Comunitat Valenciana (CAEC).
El Comité Autonómico de Estudios Post-autorización Observacionales de Medica-mentos y Productos sanitarios de la Comunitat Valenciana (CAEPO)
Los CEIC locales y corporativos acreditados en el ámbito de la Comunitat Valen-ciana (CEIC).
Artículo 4. Consejo de Dirección y la Comisión Delegada del Programa de Estudios Clíni-cos de Medicamentos y Productos Sanitarios en la Comunitat Valenciana
1. El Consejo de Dirección del PECME tutela las actividades y acciones para el de-sarrollo de los objetivos del Programa en la Comunitat Valenciana. El Consejo conocerá yaprobará el plan anual y la memoria de actividades del PECME.
2. El Consejo de Dirección del PECME estará constituido por los miembrossiguientes:
Presidente: El Director Gerente de la Agencia Valenciana de Salud.
VicePresidente: El Director General de Farmacia y Productos Sanitarios.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
401

Secretario: El Coordinador del PECME.
El Director General de Ordenación, Inspección e Investigación Sanitaria
El Director General de Asistencia Sanitaria
El Director General de Calidad y Atención al Paciente.
El Jefe de Área de Farmacia y Productos Sanitarios.
El Jefe de Área de Inspección, Ordenación e Investigación Sanitaria.
El Jefe de la Oficina de Investigación Sanitaria
Los presidentes de cada uno de los CEIC acreditados en la Comunitat Valenciana.
El Consejo de Dirección se reunirá con carácter al menos semestral, o cuando seprecise con carácter de urgencia.
3. La Comisión Delegada del PECME mantendrá el nivel de decisiones operativasentre periodos de reuniones del Consejo de Dirección de PECME, realizando jornadas detrabajo preceptivamente con carácter bimestral. Estará presidida por el Jefe de Área deFarmacia y Productos Sanitarios, actuando como secretario el Coordinador del PECME. LaComisión delegada estará integrada por aquellos miembros del Consejo designados por elPresidente del Consejo de Dirección del PECME.
Artículo 5. Coordinador del Programa de Estudios Clínicos de Medicamentos y ProductosSanitarios en la Comunitat Valenciana
1. El Coordinador del PECME será nombrado por el Director-Gerente de la Agen-cia Valenciana de Salud a Propuesta del Director General de Farmacia y Productos Sani-tarios de la Agencia Valenciana de Salud.
2. Serán funciones del Coordinador del PECME:
Recibir, informar y tramitar en la Dirección General de Farmacia y Productos Sa-nitarios de la Agencia Valenciana de Salud las solicitudes de acreditación/reacreditaciónde los CEIC, el ámbito geográfico de actuación y comunicar el resultado de dicha acred-itación al centro solicitante. Comunicar a la Agencia Española del Medicamento y Pro-ductos Sanitarios las acreditaciones y reacreditaciones de los CEIC.
402

Comunicar a los centros la necesidad de reacreditación de cada CEIC en los pla-zos pertinentes o cuando así se disponga, recibir las memorias para reacreditación de loscomités, tramitar la solicitud de la reacreditación y comunicar el resultado de la misma.
Archivar copia de los informes de aprobación o desestimación de los CEIC acercade los protocolos recibidos. También se recogerán y archivarán las modificaciones rele-vantes de los protocolos aprobados y los certificados emitidos de adecuación deontoló-gica de protocolos de investigación en humanos.
Regular los trámites para el traslado de un ensayo clínico de un centro de la Co-munitat Valenciana a otro y recibir y archivar copia de los documentos tramitados, una vezautorizado por la Agencia Española del Medicamento.
Recibir y evaluar las comunicaciones de reacciones adversas graves o inesperadasque acontezcan durante el desarrollo del ensayo clínico, conocidas por el CEIC, y comu-nicarlas al resto de centros implicados en el mismo ensayo o en otros con el mismo pro-ducto de estudio.
Recibir, informar y tramitar a la Dirección General de Farmacia y Productos Sa-nitarios de la Agencia Valenciana de Salud la propuesta de los CEIC de suspensión caute-lar de un ensayo clínico que se esté llevando a cabo, o realizar propuestas de suspensión.
Proponer la realización de inspecciones en materia de ensayos clínicos, colaboraren la elaboración y comunicación de los protocolos de dichas inspecciones, recibir yarchivar una copia de los resultados de las mismas e informar de ellas a las autoridadesde la Conselleria de Sanidad.
Solicitar, recoger y archivar la notificación por el promotor de la finalización delensayo clínico, o de la justificación de su suspensión.
Realizar revisiones bibliográficas de las publicaciones de ensayos clínicos realiza-dos en centros sanitarios de la Comunitat, comprobando que han sido llevados a cabo enlos términos que establece la normativa vigente. Asimismo, aconsejar la publicación o no-tificación de resultados tanto positivos como negativos de los ensayos clínicos realizados.
Ser vehículo de coordinación con la Agencia Española del Medicamento y otras co-munidades autónomas en materia de desarrollo y realización de Ensayos Clínicos. En estesentido, recibirá la comunicación sobre autorización, denegación, suspensión o modifi-cación de ensayos clínicos de la Agencia Española de Medicamentos.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
403

Recepción de los CEIC acreditados en la Comunitat Valenciana de todos los estu-dios por ellos autorizados. Esta función será sustituida si la información residente en laAgencia del medicamento se encuentra disponible con medios electrónicos.
Gestión de una base de datos y un archivo documental donde se recoja toda la in-formación relativa a su actividad.
Elaboración de una Memoria anual de sus actividades.
3.- La Agencia Valenciana de Salud, a través de su Dirección General de Farma-cia y Productos Sanitarios, impulsará los medios telemáticos y electrónicos necesariospara agilizar todos los trámites descritos en el apartado anterior.
Artículo 6. Procedimiento de acreditación de los CEIC
1. El responsable o responsables de los centros, servicios y establecimientos sa-nitarios de la Comunitat Valenciana autorizados por la Conselleria de Sanidad, podrán so-licitar la acreditación de los CEIC de dichos centros ante la Dirección General de Farmaciay Productos Sanitarios de la Agencia Valenciana de Salud.
2. La acreditación del CEIC tendrá una validez de 3 años. Según los términos in-dicados en la legislación vigente los miembros del CEIC serán provistos de la correspon-diente acreditación de pertenencia al CEIC expedida por la Dirección General de Farmaciay Productos Sanitarios.
3. La renovación de la acreditación de los CEIC deberá realizarse cada tresaños. La Dirección General de Farmacia y Productos Sanitarios de la Agencia Valencianade Salud podrá revocar la acreditación concedida a cualquier CEIC que no cumpla losrequisitos mínimos exigidos en la presente disposición y en el resto de la normativaaplicable.
Artículo 7. Requisitos de acreditación de los CEIC
1. El ámbito geográfico e institucional de actuación de cada CEIC será determi-nado por la Dirección General de Farmacia y Productos Sanitarios de la Agencia Valencianade Salud, y se establecerá en función del número de CEIC acreditados, abarcando, nece-sariamente, el departamento de salud correspondiente y, en su caso, otros departamentosde salud, oído el centro o centros interesados, y teniendo en cuenta la distribución geográ-fica de éstos.
404

2. Sus miembros serán nombrados por el Director General de Farmacia y Produc-tos Sanitarios de la Agencia Valenciana de Salud a propuesta de la Dirección de la insti-tución de que se trate, tras informe del la coordinación de PECME, de acuerdo con loscriterios establecidos en la presente disposición en su artículo 5, 2 a). La renovación dela composición de los CEIC se realizará por el mismo procedimiento, tras consulta con elpropio CEIC.
3. Los requisitos mínimos para la acreditación de un CEIC serán los siguientes:
Estar formado como mínimo por once miembros, y en todo caso formar parte de él:
- El director del centro.
- Médicos:
- Un farmacólogo clínico
- Dos médicos especialistas con labor asistencial activa en un centro hospitalario
- Un médico con labor asistencial activa en un Centro de Atención Primaria
- Un pediatra
- Farmacéuticos:
- Un farmacéutico de Hospital
- Un farmacéutico de Atención Primaria
- Un diplomado en Enfermería.
- Dos miembros ajenos a las profesiones sanitarias, debiendo ser uno de ellos li-cenciado en Derecho. Uno de estos miembros será una persona independiente de la orga-nización asistencial, entendiendo por independiente la desvinculación laboral en relacióna los centros en los que se lleven a cabo los proyectos de investigación que requieran laevaluación ética por parte del Comité, a ser posible propuesto por el Consejo de Salud.
b) Tener garantía explícita, por parte del titular del centro, de que el CEIC cuentacon los medios necesarios para poder realizar su cometido. Los medios básicos a disponerson:
- Instalaciones específicas que permitan la realización de su trabajo, en condi-ciones que garanticen la confidencialidad. Deberán disponer de un espacio apropiado parala secretaría del comité, para la realización de las reuniones y para el manejo y archivo dedocumentos confidenciales.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
405

- Equipamiento informático con capacidad suficiente para manejar toda la infor-mación generada por el comité y disponibilidad de un sistema rápido de transmisión deinformación.
- Personal administrativo y técnico que permita al comité poder ejercer de ma-nera apropiada sus funciones.
c) Disponer de los correspondientes procedimientos normalizados de trabajo.
4. La renovación de la acreditación de los CEIC deberá cumplir los requisitos mí-nimos siguientes:
a) Los de su previa acreditación.
b) Realizar con carácter mensual al menos 11 reuniones anuales, con evaluaciónde ensayos clínicos y seguimiento de los aprobados.
c) Superar favorablemente las inspecciones de la Conselleria de Sanidad.
Artículo 8. Funcionamiento de los CEIC
1.- Además de las funciones señaladas en el RD 223/2004, los CEIC en la Comu-nitat Valenciana llevarán a cabo las funciones siguientes:
Evaluar la información escrita sobre las características del estudio que se dará alos posibles sujetos de la investigación, así como la forma en que dicha información seaproporcionada y el tipo de consentimiento que fuera a obtenerse.
Conocer y evaluar el alcance de las compensaciones ofrecidas a los investigadoresy a los sujetos de la investigación, así como la forma en que dicha información sea pro-porcionada.
Recabar el asesoramiento de personas expertas no pertenecientes al Comité en lossupuestos a que se refiere el artículo 14.4 del Real Decreto 223/2004, que respetarán elprincipio de confidencialidad.
Realizar un informe acerca de las modificaciones de los protocolos de los ensayos,velando por la corrección de los protocolos propuestos desde el punto de vistametodológico, ético y legal
406

Informar al coordinador del PECME de todos aquellos protocolos de investigaciónque aprueben y que se eleven al órgano competente para su autorización, y también delos que finalmente se desestimen.
Remitir al coordinador del PECME una copia de los certificados que se emitansobre la adecuación deontológica de los protocolos que impliquen la investigación en hu-manos.
Comunicar al coordinador del PECME las reacciones adversas graves o inesperadasque acontezcan durante el desarrollo del ensayo, mediante copia de la notificación del in-vestigador o promotor.
Proponer a la Conselleria de Sanidad, a través del coordinador del PECME, la sus-pensión cautelar de un ensayo clínico en los supuestos previstos en la legislación vigente.
El CEIC hará explícitos los criterios de idoneidad de los investigadores y del Cen-tro para ser objetivos en la evaluación.
2. El CEIC desarrollará sus funciones con independencia y según sus procedimien-tos normalizados de trabajo, de conformidad con lo previsto en el artículo 6 de la Ley29/2006 de garantías y uso racional de medicamentos y productos sanitarios y en elartículo 14.2 del Real Decreto 223/2004 de ensayos clínicos con medicamentos de usohumano. Los miembros del Comité deberán respetar el principio de confidencialidad, enlo que respecta a la documentación recibida para la evaluación del protocolo y la identi-dad de los pacientes.
2. La pertenencia a un CEIC será incompatible con cualquier clase de interesesderivados de la fabricación y venta de medicamentos y productos sanitarios. Losmiembros harán constar ante la Dirección General de Farmacia y ProductosSanitarios de la Agencia Valenciana de Salud antes de su toma de posesión lano concurrencia de dicha circunstancia. El investigador principal o los colab-oradores de un estudio, no podrán participar ni en la evaluación ni en el dicta-men de su propio protocolo, aun cuando sean miembros del CEIC. Esta normaes extensiva a los promotores y monitores de los ensayos clínicos cuyo proyectosea sometido al estudio del comité. Ni el CEIC ni ninguno de sus miembros po-drán percibir directa o indirectamente remuneración alguna del promotor delensayo o estudio observacional.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
407

Artículo 9. Organización de los CEIC
1. Los CEIC estarán compuestos de un presidente, un vicepresidente, un secre-tario y los vocales. Corresponde al Presidente del CEIC:
a) Presidir las reuniones del Comité.
b) Elaborar, junto con el Secretario, las actas y el orden del día de las reunionescorrespondientes.
c) Elaborar, junto con el Secretario, la memoria anual del Comité.
d) Velar por la consecución de los objetivos asignados al Comité.
e) Realizar cuantas funciones sean inherentes a su condición de Presidente.
2. El Vicepresidente del CEIC asistirá en sus funciones al Presidente del Comitéy le sustituirá en caso de vacante, ausencia o enfermedad.
3. Son funciones del Secretario del CEIC:
a) Efectuar la convocatoria de las reuniones por orden del Presidente y las cita-ciones correspondientes.
b) Redactar las actas de las reuniones.
c) Expedir certificaciones de los dictámenes y acuerdos adoptados.
d) Redactar y firmar junto con el Presidente la memoria anual del Comité, asícomo las funciones generales de Secretario contempladas en la Ley 30/1992,de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas ydel Procedimiento Administrativo Común.
e) Realizar cuantas otras funciones sean inherentes a su condición de Secretario.
4. Son funciones de los vocales del CEIC:
a) Asistir a las reuniones a las que hayan sido convocados.
b) Evaluar la documentación que reciban correspondiente a la posterior valoraciónpor el Comité.
c) Realizar aquellas tareas que les sean asignadas por el Presidente.
5. Para la válida constitución del órgano, a efectos de celebración de sesiones, de-liberaciones o toma de acuerdos, se requerirá la presencia del Presidente, o en su caso del
408

Vicepresidente, y del Secretario, o, en su caso, de quien le sustituya, y de la mitad más unode sus miembros. En el caso de no concurrir suficiente número de miembros para consid-erarse válidamente constituida la sesión, transcurrida media hora, o el tiempo establecidoen la convocatoria, se tendrá por realizada una segunda convocatoria, en cuyo caso paraque se constituya válidamente bastará con la concurrencia del Presidente, o en su caso elVicepresidente, del Secretario o persona que le sustituya, y, al menos, tres miembros más.
6. En lo no previsto expresamente en este artículo se aplicará lo dispuesto en elCapítulo II del Título II de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico delas Administraciones Públicas y del Procedimiento Administrativo Común.
Artículo 10. Funciones de la dirección del centro o establecimiento sanitario con CEICacreditado en la Comunitat Valenciana.
1.- La dirección del centro llevará a cabo las funciones siguientes:
a) Solicitar la acreditación del CEIC.
b) Firmar el documento correspondiente de conformidad con la realización delensayo clínico en el centro y con el contrato firmado con el promotor en el quese especifican todos los aspectos económicos de dicho ensayo clínico.
c) Garantizar los medios necesarios para la correcta realización de las funcionesdel CEIC, descritos en el artículo 7, apartado 3.b) de esta disposición.
d) Coordinar las actuaciones del CEIC con las distintas Comisiones existentes enel centro.
e) Solicitar, en los casos que así se determine, la autorización del centro para larealización de ensayos clínicos sin finalidad terapéutica.
Artículo 11. El Comité Ético Autonómico de Estudios Clínicos de Medicamentos y Produc-tos Sanitarios de la Comunitat Valenciana (CAEC) y los CEIC de ámbito corporativo.
1. El Comité Ético Autonómico de Estudios Clínicos de Medicamentos y Produc-tos Sanitarios (CAEC) es un órgano colegiado de carácter técnico sobre ensayos clínicos yestudios post-autorización observacionales, con ámbito de actuación en la Comunitat Va-lenciana.
2. El CAEC de la Comunitat Valenciana tendrá las siguientes atribuciones y activi-dades:
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
409

a) Evaluación de las solicitudes de ensayos clínicos con medicamentos/productossanitarios o estudios post-autorización observacionales que de forma ordinariao extraordinaria requieran su consideración, remitidos por los directores de loscentros sanitarios de la Agencia Valenciana de Salud o por los CEIC acredita-dos en la Comunitat Valenciana o por fundaciones dependientes de los centrossanitarios de la Agencia Valenciana de Salud.
b) Establecer los requisitos mínimos curriculares de los miembros de un CEIC, elcontenido de la memoria de acreditación y el sistema de evaluación de las ac-tuaciones de los CEIC acreditados en la Comunitat Valenciana.
c) Realizar la evaluación de la actividad de los CEIC locales acreditados, garanti-zando la unidad de criterio y dictamen y prestando el apoyo y asesoramientotécnico que requieran en el ejercicio de sus funciones.
d) Velar por la correcta aplicación de los principios éticos, metodológicos y legalesde los CEIC acreditados en la Comunitat Valenciana, realizando propuestaspara la normalización de los procedimientos de trabajo y criterios de evalu-ación.
e) Proponer medidas para agilizar los trámites burocráticos y reducir los tiemposde evaluación y respuesta en los ensayos clínicos y proyectos de investigación.
f) Promover reuniones periódicas para abordar temas de interés común que apoyeny aumenten la calidad de los ensayos clínicos y de los proyectos de investi-gación.
g) Promover las relaciones entre las instituciones y personas relacionadas con losensayos clínicos y proyectos de investigación.
3. El CAEC estará constituido por: presidente, vicepresidente, secretario y vocales.Podrán asistir a las reuniones del Comité expertos que asesoran cuando sean requeridospara ello. El CAEC adoptará los procedimientos de trabajo que a tal efecto se establezcan yactuará de acuerdo con lo que se determine en sus normas internas de funcionamiento. ElComité se reunirá con carácter mensual al menos once veces al año. De cada reunión se lev-antará acta en al que se consignarán los acuerdo adoptados y los miembros asistentes.
4. La designación de los miembros será realizada por el Director General de Far-macia y Productos Sanitarios de la Agencia Valenciana de Salud, a propuesta del Coordi-nador del PECME que actúa como secretario del CAEC. El Director General de Farmaciay Productos Sanitarios de la Agencia Valenciana de Salud nombrará entre ellos al Presi-dente y Vicepresidente. El CAEC estará formado por un mínimo de trece miembros, en-tre los que figurarán:
410

a) Médicos, entre los cuales habrá al menos un farmacólogo clínico, dos médicosespecialistas con labor asistencial en un centro hospitalario, un médico con la-bor asistencial en un centro de Atención Primaria, un pediatra y un experto enepidemiología clínica.
b) Farmacéuticos, entre los cuales al menos habrá un farmacéutico de hospital,un farmacéutico de atención primaria y un farmacéutico del Centro Valencianode Información de Medicamentos.
c) Un diplomado en enfermería.
d) Al menos dos miembros ajenos a la profesión sanitaria, debiendo ser uno deellos Licenciado en Derecho.
e) Una persona independiente de la organización asistencial.
f) Un representante del Centro de Farmacovigilancia de la Comunitat Valenciana.
g) El coordinador del PECME.
5. A los efectos de agilizar la evaluación de los ensayos clínicos y estudios post-autorización observacionales en determinados ámbitos asistenciales, se conforman losCEIC corporativos de Atención Primaria de la Comunitat Valenciana y de Salud Pública(Centro Superior de Investigaciones en Salud Pública), de ámbito multicéntrico, facili-tando el fomento de la investigación en sus respectivos ámbitos sanitarios.
6. El CEIC corporativo de Salud Pública del Centro Superior de Investigación enSalud Pública se regirá, en cuanto a su régimen de creación, composición y fun-cionamiento por lo dispuesto en los artículos 6, 7, 8, 9 y 10 de la presente norma.
7. El CEIC corporativo de Atención Primaria de la Comunitat Valenciana Públicase regirá, en cuanto a su régimen de creación, composición y funcionamiento por lo dis-puesto en los artículos 6, 7, 8, 9 y 10 de la presente norma. La designación de los miem-bros será realizada por el Director General de Farmacia y Productos Sanitarios de laAgencia Valenciana de Salud, a propuesta del Coordinador del PECME. El Director Gen-eral de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud nombrará en-tre los miembros al Presidente y Vicepresidente.
Artículo 12. El Comité Autonómico de Estudios Post-autorización Observacionales deMedicamentos y Productos sanitarios de la Comunitat Valenciana (CAEPO)
1. El Comité Autonómico de Estudios Post-autorización Observacionales deMedicamentos y Productos sanitarios de la Comunitat Valenciana (CAEPO) es un órgano
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
411

consultivo para la autorización de la realización de dichos estudios en su ámbito de com-petencias.
2. El CAEPO estará formada por:
• Un presidente que será el jefe del Área de Farmacia y Productos Sanitarios
• Un vicepresidente que será el jefe del Servicio de Provisión y Asistencia Farma-céutica
• Los jefes de Servicio de prestación ortoprotésica y ordenación y control demedicamentos y productos sanitarios.
• Un profesional del Centro Autonómico Valenciano de Información de Medica-mentos que actuará como secretario
• Dos profesionales, preferentemente médicos, que pertenezcan o hayanpertenecido a los CEIC de atención primaria acreditados por la Conselleria deSanidad
• Dos profesionales, preferentemente médicos, que pertenezcan o hayanpertenecido a los comités éticos de investigación clínica de hospital acredita-dos por la Conselleria de Sanidad
• Un farmacéutico de atención primaria
• Un farmacéutico de atención especializada
• Un especialista en oncología.
• Un especialista de medicina interna
• Dos médicos de atención primaria
• Un farmacólogo clínico
• Un experto en epidemiología y estadística
3. Son funciones del CAEPO:
• Evaluar la pertinencia de los estudios post-autorización presentados en la Di-rección General de Farmacia y Productos Sanitarios.
• Establecer los criterios de valoración por los que se regirá CAEPO para la eval-uación de la pertinencia de los estudios post-autorización.
• Emitir informe motivado al director general de Farmacia y Productos Sanitariossobre la pertinencia de los estudios post-autorización.
412

4. Los miembros del CAEPO serán nombrados por el Director General de Farma-cia y Productos Sanitarios. El nombramiento tendrá una duración inicial de dos años, pu-diendo renovarse sucesivamente y por una sola vez, por otro período adicional de 2 años.El comité establecerá sus normas de funcionamiento. Todo lo que no esté dispuesto enesta norma de creación se ajustará a las disposiciones que regulan los órganos colegiados.
Artículo 13. Aspectos legales y económicos de los ensayos y estudios post-autorización ob-servacionales con medicamentos y productos sanitarios
1.- Todos los estudios en humanos con medicamentos y productos sanitarios, in-cluyendo los supuestos donde sólo se recoge información de la historia clínica, deben serpresentado en forma de protocolo, y evaluado por un CEIC acreditado por la Conselleriade Sanitat. Se incluyen tanto los ensayos clínicos como los estudios observacionales tantoprospectivos como retrospectivos. El protocolo debe incluir información sobre finan-ciamiento, patrocinadores, afiliaciones institucionales, otros posibles conflictos de interése incentivos para las personas del estudio.
2. De conformidad con lo previsto en la Ley 41/2002, de 14 de noviembre, reg-uladora de la autonomía de los pacientes, en los ensayos clínicos y estudios post-autor-ización de tipo observacional prospectivos es imprescindible que el sujeto otorgue libre yvoluntariamente su consentimiento informado, antes de ser incluido en el estudio, debi-endo ser informado sobre la naturaleza, importancia, implicaciones y riesgo del ensayoclínico. En el caso de menores de edad o personas incapaces, este consentimiento lo debeotorgar su representante legal.
3.- El consentimiento debe obtenerse por escrito. Si el sujeto del ensayo o estu-dio no está en condiciones de escribir, podrá dar, en casos excepcionales, su consen-timiento verbal en presencia de, al menos, un testigo mayor de edad y con capacidad deobrar. El sujeto participante en un ensayo clínico o estudio o su representante podrá re-vocar, en todo momento, su consentimiento sin expresión de causa.
4.-En los estudios post-autorización de tipo observacional retrospectivos y trans-versales que requieran entrevistar al sujeto o en aquellos en los que, utilizando otrasfuentes de información, no sea posible adoptar un procedimiento de disociación seguro,se solicitará el consentimiento informado de los sujetos. Podrá haber situaciones en lasque será imposible o impracticable obtener el consentimiento para dicha investigación opodría ser una amenaza para su validez .En esta situación, la investigación sólo puede serrealizada después de ser considerada y aprobada por un CEIC.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
413

5. Todos los aspectos contractuales y económicos relacionados con el ensayoclínico y estudios post-autorización observacionales quedarán reflejados en un contratoentre el promotor y quien con personalidad jurídica pueda comprometer al centro dondese vaya a realizar los estudios con el fin de conseguir la mayor transparencia económicade los pactos existentes. Para ello, la Conselleria de Sanidad especificará los requisitos co-munes, condiciones de financiación y contratación de investigadores, así como los sis-temas adecuados que se han de utilizar para la percepción de la remuneración acordadaen el contrato por parte del centro u hospital y por el investigador principal u otros colab-oradores en los ensayos y estudios con medicamentos y productos sanitarios.
Artículo 14. Sistema de información autonómico en materia de ensayos clínicos y estu-dios post-autorización observacionales de medicamentos y productos sanitarios
1. Para facilitar la comunicación con los CEIC y disponer de un repositorio de in-formación sobre ensayos clínicos y estudios post-autorización observacionales con medica-mentos y productos sanitarios, la Dirección General de Farmacia y Productos Sanitarios dela Agencia Valenciana de Salud dispondrá de un portal de trabajo exclusivo de PECME,garantizando la confidencialidad en todo momento.
2. La Agencia Valenciana de Salud facilitará a PECME un sistema de gestión op-erativo y bases de datos para realizar la gestión de la información y trámites administra-tivos de ensayos clínicos y estudios post-autorización observacionales en medicamentos yproductos sanitarios en la Comunitat Valenciana. El sistema facilitará la integración o lacomunicación con los sistemas de información de la Agencia Española del Medicamentoy Productos Sanitarios, en especial SIC-CEIC.
3. Las historias clínicas de los pacientes dispondrán de un sistema permanente,ágil y rápido para identificar que un paciente participa o ha participado en un ensayoclínico o estudio post-autorización observacional. En la historia clínica del paciente searchivará una copia del consentimiento informado. Por parte de la Agencia Valenciana deSalud se potenciará la identificación y el consentimiento informado electrónico por partedel paciente en el sistema de información sanitario de la Agencia Valenciana de Salud. Encualquier caso, y en ausencia de firma electrónica del paciente, en aquellos centros san-itarios que dispongan de historia clínica electrónica, deberá constar su firma de consen-timiento en formato de papel
4. Para facilitar la comunicación y el manejo de datos en los proyectos de estu-dios de medicamentos y productos sanitarios, la documentación deberá ser presentada porel promotor en formato digital.
414

Artículo 15. Formación y normalización en la toma de decisiones de los miembros de losCEIC
1. PECME impulsará las actividades periódicas de formación y jornadas de nor-malización de criterios, entre los profesionales que integran los CEIC acreditados en la Co-munitat Valenciana, para minimizar las variaciones en la toma de decisiones intentandofomentar, en el ámbito de la Comunitat Valenciana, unos criterios de evaluación homogé-neos y adecuados.
2. Para agilizar el proceso de evaluación, PECME promoverá jornadas de coordi-nación con todos los agentes implicados, evaluadores, promotores e investigadores con lacolaboración del Centro Coordinador de Comités Éticos de Investigación Clínica de la Di-rección General de Farmacia y Productos Sanitarios del Ministerio de Sanidad y el Comitéde Coordinación de Estudios Post-autorización de la Agencia Española del Medicamento.
Artículo 16. Inspección de PECME por la Conselleria de Sanidad
1. Se entiende por inspección la revisión oficial por una autoridad competente delos documentos, las instalaciones, los archivos, los sistemas de garantía de la calidad ycualesquiera otros elementos que la autoridad competente considere relacionado con elensayo clínico/proyecto de investigación y que puedan encontrarse en el lugar del ensayo,en las instalaciones del promotor y/o de la organización de investigación por contrato, oen cualquier otro establecimiento que la autoridad competente considere oportuno in-speccionar. Tras la inspección se elaborará un informe que estará a disposición de los in-speccionados.
2. En un proceso de inspección de un ensayo clínico/proyecto de estudio, se au-ditará al menos:
a) Hoja de información al paciente.
b) Aprobación del estudio y/o modificaciones del protocolo por parte del CEIC yAutoridades sanitarias.
c) Grado de adhesión al protocolo: cumplimiento de los criterios de selección,asignación al tratamiento, administración y dosificación de fármacos, preser-vación del ciego y pruebas complementarias.
d) Control de la medicación suministrada: almacenamiento, dispensación,recogida y recuento medicación sobrante.
e) Verificación de los datos de las fuentes originales.
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
415

f) Validación de las variables eficacia y seguridad
g) Registro de las notificaciones de efectos adversos.
3. La Inspección de Servicios Sanitarios de la Conselleria de Sanidad llevará acabo las actuaciones necesarias para verificar el cumplimiento de lo dispuesto en la nor-mativa vigente en la materia, poniendo especial empeño en verificar la observancia de lasNormas de Buena Práctica Clínica en los ensayos clínicos. Los datos relativos a las inspec-ciones serán remitidos a PECME y a la Agencia Española del Medicamento para su intro-ducción en la base de datos europea de ensayos clínicos EUDRACT de acuerdo con lasdirectrices de la Comisión Europea.
Artículo 17. Infracciones
El incumplimiento de lo dispuesto en la presente disposición será sancionado deconformidad con el régimen previsto en el Capítulo II del Título VIII de la Ley 29/2006,de 26 de julio, de garantías y uso racional del medicamentos y productos sanitarios.
Disposición adicional primera. Indemnizaciones y gratificaciones por servicios extraordi-narios
Los profesionales que formen parte de los Comités referenciados en esta disposi-ción participarán en ellos mientras que PECME se encuentre en desarrollo. Su actuaciónserá exclusivamente de colaboración asesora, sin que origine creación de puesto de tra-bajo, si bien la Conselleria de Sanidad abonará, en su caso, las indemnizaciones o gastosque su asistencia a la Comisión pueda ocasionar, de acuerdo con lo establecido en el De-creto 24/1997, de 11 de febrero, del Gobierno Valenciano, sobre indemnizaciones porrazón del servicio y gratificaciones por servicios extraordinarios
Disposición transitoria primera. Plazo de adaptación de los CEIC
Los CEIC acreditados en la Comunitat Valenciana deberá acomodar sus actua-ciones a la presente disposición en el plazo de 90 días.
Disposición transitoria segunda. Comités de Ética de la Investigación
En tanto se desarrolle la normativa regulada por la Ley 14/2007 de InvestigaciónBiomédica, los CEIC de las fundaciones gestoras y promotoras de investigación biomédicaque habitualmente no evalúen protocolos de ensayos clínicos de Medicamentos y Produc-
416

tos Sanitarios, sino estudios comprendidos dentro del campo de aplicación de la citadaley, podrán cambiar su denominación a Comité de Ética de la Investigación, sin cambiarsu composición ni su ámbito geográfico de influencia. Esta acreditación tendrá validezhasta que se publiquen nuevas normas en este sentido por las autoridades competentes.
Disposición Derogatoria Única
Quedan derogadas la Orden de 8 de marzo de 2004 de la Conselleria de Sanidadpor la que se crea la Comisión Asesora para la evaluación de los estudios post-autorizaciónobservacionales prospectivos, la Orden de 6 de julio de 1994, de la Conselleria de Sanidady Consumo por la que se regulan los ensayos clínicos en la Comunitat Valenciana. Quedanderogadas cuantas disposiciones de igual o inferior rango se opongan a lo dispuesto en esteDecreto
Disposición final
Se faculta al Conseller de Sanidad para dictar las disposiciones precisas para eldesarrollo y ejecución del presente decreto.
El presente decreto entrará en vigor al día siguiente de su publicación en el Di-ario Oficial de la Comunitat Valenciana
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
417

418

Bibliografía


1. Código de Nüremberg del Tribunal Internacional de Nüremberg de1946
2. Declaración Universal de Derechos Humanos de la Asamblea General de las NacionesUnidas, del 10 de diciembre de 1948
3. Declaración de Helsinki de la Asociación Médica Mundial. Versiones:
• Helsinki, Finlandia. 1964
• Tokio, Japón, octubre 1975.
• Venecia, Italia, octubre 1983
• Hong Kong, septiembre 1989
• Somerset West, Sudáfrica, octubre 1996
• Edimburgo, Escocia, octubre 2000
• Nota de Clarificación del Párrafo 29, Washington 2002
• Nota de Clarificación del Párrafo 30, Tokio 2004
• Seúl, Corea, octubre 2008
4. Directiva 65/65/CEE del Consejo Europeo, de 26 de enero de 1965 relativa a la apro-ximación de las disposiciones legales, reglamentarias y administrativas sobre medi-camentos
5. Convención Internacional de las Naciones Unidas sobre la Eliminación de todas lasFormas de Discriminación Racial del 21 de diciembre de 1965
6. Directiva 75/318/CEE del Consejo Europeo, de 20 de mayo de 1975 relativa a laaproximación de las legislaciones de los Estados miembros sobre normas y protoco-los analíticos, toxicofarmacológicos y clínicos en materia de pruebas de medicamen-tos
7. Pacto Internacional de Derechos Civiles y Políticos adoptados el 16 de diciembre de1966. Pautas éticas internacionales para investigación biomédica que involucra se-res humanos del Consejo de Organizaciones Internacionales de Ciencias Médicas,aprobadas en 1982 y enmendadas en 1993 y 2002(CIOMS)
8. Principios de ética médica de la Asamblea General de Naciones Unidas de 1982
9. Convención de las Naciones Unidas sobre los Derechos del Niño del 20 de noviem-bre de 1989
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
421

10. Ley 25/1990, de 20 de diciembre, del medicamento
11. Código de Reglamentos Federales de los EE.UU., Título 45, sección 46 (45cfr46), re-visión de junio de 1991
12. Directiva de la Comisión 91/507/CEE de 19 de julio de 1991 por la que se modificael Anexo de la Directiva 75/318/CEE del Consejo relativa a la aproximación de las le-gislaciones de los Estados miembros sobre normas y protocolos analíticos, toxicofar-macológicos y clínicos en materia de pruebas de medicamentos
13. Real Decreto 561/1993 de 16 de abril, por el que se establecen los requisitos parala realización de ensayos clínicos con medicamentos.
14. Normas uniformes de las Naciones Unidas sobre la igualdad de oportunidades para laspersonas con discapacidad aprobadas por la Asamblea General de las Naciones Uni-das en 1993
15. Declaración Universal sobre el Genoma Humano y los Derechos Humanos aprobada porla Conferencia General de la UNESCO el 11 de noviembre de 1997
16. Declaración de la UNESCO sobre las Responsabilidades de las Generaciones Actua-les para con las Generaciones Futuras del 12 de noviembre de 1997,
17. Convención sobre los derechos humanos y la biomedicina del Consejo de Europa,Oviedo, 4 de abril de 1997
18. Guía de Normas de Buena Práctica Clínica de la de la Conferencia Internacional deArmonización (ICH), de 1997. (CPMP/ICH/135/95/Step5).
19. Principios Éticos en la Investigación: Declaración sobre la ciencia y el uso del sabercientífico. (Declaración de Budapest) de la Conferencia mundial sobre la ciencia.UNESCO - ICSU. 1 de Julio de 1999
20. Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter per-sonal
21. Declaración bioética de Gijón, de 24 de junio de 2000 del Comité Científico de la So-ciedad internacional de Bioética (SIBI).
22. Guías operacionales para Comités de Ética que evalúan investigación biomédica, dela Organización Mundial de la Salud (OMS), del año 2000.
23. Directiva 2001/20/CE del Parlamento Europeo y del Consejo, de 4 de abril de 2001relativa a la aproximación de las disposiciones legales, reglamentarias y administra-tivas de los Estados miembros sobre la aplicación de buenas prácticas clínicas en larealización de ensayos clínicos de medicamentos de uso humano
24. Declaración Universal de la UNESCO sobre la Diversidad Cultural del 2 de noviembrede 2001
422

25. Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente yde derechos y obligaciones en materia de información y documentación clínica.
26. Ley 1/2003, de 28 de enero, de derechos e información al paciente de la comunidadvalenciana
27. Declaración Internacional sobre los Datos Genéticos Humanos aprobada por la Con-ferencia General de la UNESCO el 16 de octubre de 2003
28. Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicoscon medicamentos
29. Directiva 2005/28/CE de la Comisión de 8 de abril de 2005 por la que se establecenlos principios y las directrices detalladas de las buenas prácticas clínicas respecto alos medicamentos en investigación de uso humano, así como los requisitos para au-torizar la fabricación o importación de dichos productos
30. Declaración universal sobre bioética y derechos humanos. Conferencia General de laUNESCO. 33ª sesión de 19 de octubre de 2005
31. Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y pro-ductos sanitarios
32. Código Internacional de Ética Médica de la Asamblea general de la Asociación MédicaMundial. Pilanesberg. Sudáfrica. Octubre 2006
33. Orden SCO/256/2007, de 5 de febrero, por la que se establecen los principios y lasdirectrices detalladas de buena práctica clínica y los requisitos para autorizar la fa-bricación o importación de medicamentos en investigación de uso humano
34. Decreto 73/2009, de 5 de junio, del Consell, por el que se regula la gestión de ensa-yos clínicos y estudios post-autorización observacionales con medicamentos y pro-ductos sanitarios
Guía para el Investigador de Ensayos Clínicos en la Comunidad Valenciana
423



















