
INTRODUCCION orgánicas desde un punto de vista · 2012-06-18 · Este proceso de ciclación es...
12
INTRODUCCION U na gran parte de las publica- ciones científicas en el área de la química se refieren a la sín- tesis de moléculas orgánicas, o al desarrollo de la metodología nece- saria para su síntesis. Los logros alcanzados en esta disciplina son impresionantes y, en principio, las moléculas más complejas son sin- téticamente accesibles. 1 Desde los inicios de la década de los 90 hay dos corrientes de opinión respecto a la síntesis orgánica. Por una parte, hay químicos que opinan que la sín- tesis orgánica ha alcanzado su madurez y que, no cabe esperar demasiadas sorpresas en esta dis- ciplina. 2 Alternativamente, existe la opinión más cercana a la realidad de que, con la tecnología sintética de la que disponemos, solo somos capaces de preparar unos pocos miligramos de una sustancia com- pleja determinada. Normalmente, el éxito solo tiene lugar después de años de duro trabajo por parte de muchas personas. 3 Es cierto que de la lectura de la descripción de la síntesis de moléculas orgánicas se percibe un gran optimismo. Se diría que la habilidad de los autores para plantear rutas sintéticas que con- ducen con éxito a las moléculas objetivo es casi infalible y que, una vez diseñada la estrategia sintética, incluso las moléculas mas comple- jas se preparan sin esfuerzo. Sin embargo, atendiendo a nuestra experiencia cotidiana, nos pregunta- mos hasta que punto esta impresión es reflejo de la realidad. Por razones de diferente índole, no siempre se explica en los artículos de síntesis orgánica porqué la sínte- sis de una molécula se realiza del modo descrito y no de otro. Muchas veces ni siquiera se comenta si la ruta sintética publicada se ha modi- ficado durante el trabajo de labora- torio. Ciertamente, los resultados que se resumen en frases como "descomposición del producto de partida" o "mezcla compleja de reacción" tendrían que aparecer con demasiada frecuencia y de ellos no se extraería ninguna información útil. 4 Sin embargo, en el transcurso de cualquier síntesis hay resultados inesperados o no deseados en los que el químico sintético se basa para culminar con éxito la síntesis total, y por ello muy probablemente, la estrategia final será diferente de la que se propuso inicialmente. En nuestra opinión los resultados inesperados, o no deseados, son los que en definitiva contribuyen al desarrollo de la síntesis orgánica como disciplina. Para la comunidad científica es infinitamente más útil conocer el pensamiento involucrado en la solución de los problemas que aparecen en una síntesis que en el mero relato de una secuencia de reacciones químicas que se desar- rollan según una estrategia magis- tralmente concebida. Por estos motivos, a lo largo de este artículo comentaremos algunas síntesis orgánicas desde un punto de vista diferente al habitual, destacando los problemas que se encontraron y las modificaciones que causaron en el plan sintético original. La ruta defin- itiva de una síntesis es la que con- duce al producto final, pero en ella está contenido el conocimiento adquirido de las aproximaciones que fallaron en la consecución del objetivo. ¡EL PRODUCTO DE PARTIDA SE RECUPERA INALTERADO! Esta es una de las situaciones más frustrantes con las que se enfrenta el químico sintético: la falta de reac- tividad de un intermedio en una sín- tesis. A pesar del enorme arsenal de reactivos de los que disponemos, hay ocasiones en las que una deter- minada transformación no puede llevarse a cabo. En una síntesis, la falta de reactividad en un intermedio avanzado puede suponer simple- mente cambios más o menos sen- cillos (tácticos), o una modificación profunda del esquema sintético (estrategia). El cambio táctico efectuado por Wipf en su síntesis de la (-)-estenina 1.1 es un ejemplo de un cambio inducido por la falta de reactividad de un intermedio clave. 5 En las últi- mas etapas de la síntesis de 1.1, el ciclo de azepina (marcado en rojo) se construiría directamente utilizan- do una ciclación de Mitsunobu en el precursor 1.2. 6 Esta reacción se había producido con buen rendimiento en productos análogos y era de esperar que también ocu- rriese en 1.2. Sin embargo este intermedio sintético no cicló. El problema supuso un cambio en la táctica sintética. En lugar de con- struir el anillo de siete eslabones via una sustitución nucleofila intramole- María C. de la Torre Departamento de Productos Naturales. Instituto de Química Orgánica General. CSIC. Juan de la Cierva 3. 28006-Madrid. Miguel A. Sierra Departamento de Química Orgánica. Facultad de Química. Universidad Complutense. 28040-Madrid. 14 TOPICOS Y REALIDADES EN SINTESIS ORGANICA
Transcript of INTRODUCCION orgánicas desde un punto de vista · 2012-06-18 · Este proceso de ciclación es...

INTRODUCCION
Una gran parte de las publica-ciones científicas en el área dela química se refieren a la sín-
tesis de moléculas orgánicas, o aldesarrollo de la metodología nece-saria para su síntesis. Los logrosalcanzados en esta disciplina sonimpresionantes y, en principio, lasmoléculas más complejas son sin-téticamente accesibles.1 Desde losinicios de la década de los 90 haydos corrientes de opinión respecto ala síntesis orgánica. Por una parte,hay químicos que opinan que la sín-tesis orgánica ha alcanzado sumadurez y que, no cabe esperardemasiadas sorpresas en esta dis-ciplina.2 Alternativamente, existe laopinión más cercana a la realidadde que, con la tecnología sintéticade la que disponemos, solo somoscapaces de preparar unos pocosmiligramos de una sustancia com-pleja determinada. Normalmente, eléxito solo tiene lugar después deaños de duro trabajo por parte demuchas personas.3Es cierto que de la lectura de ladescripción de la síntesis demoléculas orgánicas se percibe ungran optimismo. Se diría que lahabilidad de los autores paraplantear rutas sintéticas que con-ducen con éxito a las moléculasobjetivo es casi infalible y que, unavez diseñada la estrategia sintética,incluso las moléculas mas comple-jas se preparan sin esfuerzo. Sinembargo, atendiendo a nuestraexperiencia cotidiana, nos pregunta-mos hasta que punto esta impresiónes reflejo de la realidad. Por razones de diferente índole, nosiempre se explica en los artículosde síntesis orgánica porqué la sínte-sis de una molécula se realiza delmodo descrito y no de otro. Muchas
veces ni siquiera se comenta si laruta sintética publicada se ha modi-ficado durante el trabajo de labora-torio. Ciertamente, los resultadosque se resumen en frases como"descomposición del producto departida" o "mezcla compleja dereacción" tendrían que aparecer condemasiada frecuencia y de ellos nose extraería ninguna informaciónútil.4 Sin embargo, en el transcursode cualquier síntesis hay resultadosinesperados o no deseados en losque el químico sintético se basapara culminar con éxito la síntesistotal, y por ello muy probablemente,la estrategia final será diferente dela que se propuso inicialmente.En nuestra opinión los resultadosinesperados, o no deseados, sonlos que en definitiva contribuyen aldesarrollo de la síntesis orgánicacomo disciplina. Para la comunidadcientífica es infinitamente más útilconocer el pensamiento involucradoen la solución de los problemas queaparecen en una síntesis que en elmero relato de una secuencia dereacciones químicas que se desar-rollan según una estrategia magis-tralmente concebida. Por estosmotivos, a lo largo de este artículocomentaremos algunas síntesis
orgánicas desde un punto de vistadiferente al habitual, destacando losproblemas que se encontraron y lasmodificaciones que causaron en elplan sintético original. La ruta defin-itiva de una síntesis es la que con-duce al producto final, pero en ellaestá contenido el conocimientoadquirido de las aproximacionesque fallaron en la consecución delobjetivo.
¡EL PRODUCTO DE PARTIDA SERECUPERA INALTERADO!
Esta es una de las situaciones másfrustrantes con las que se enfrentael químico sintético: la falta de reac-tividad de un intermedio en una sín-tesis. A pesar del enorme arsenal dereactivos de los que disponemos,hay ocasiones en las que una deter-minada transformación no puedellevarse a cabo. En una síntesis, lafalta de reactividad en un intermedioavanzado puede suponer simple-mente cambios más o menos sen-cillos (tácticos), o una modificaciónprofunda del esquema sintético(estrategia). El cambio táctico efectuado porWipf en su síntesis de la (-)-estenina1.1 es un ejemplo de un cambioinducido por la falta de reactividadde un intermedio clave.5 En las últi-mas etapas de la síntesis de 1.1, elciclo de azepina (marcado en rojo)se construiría directamente utilizan-do una ciclación de Mitsunobu en elprecursor 1.2.6 Esta reacción sehabía producido con buenrendimiento en productos análogosy era de esperar que también ocu-rriese en 1.2. Sin embargo esteintermedio sintético no cicló. Elproblema supuso un cambio en latáctica sintética. En lugar de con-struir el anillo de siete eslabones viauna sustitución nucleofila intramole-
María C. de laTorre
Departamento deProductos
Naturales. Institutode Química
Orgánica General.CSIC. Juan de la
Cierva 3. 28006-Madrid.
Miguel A. Sierra
Departamento deQuímica Orgánica.
Facultad deQuímica.
UniversidadComplutense.28040-Madrid.
14
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:01 PÆgina 14
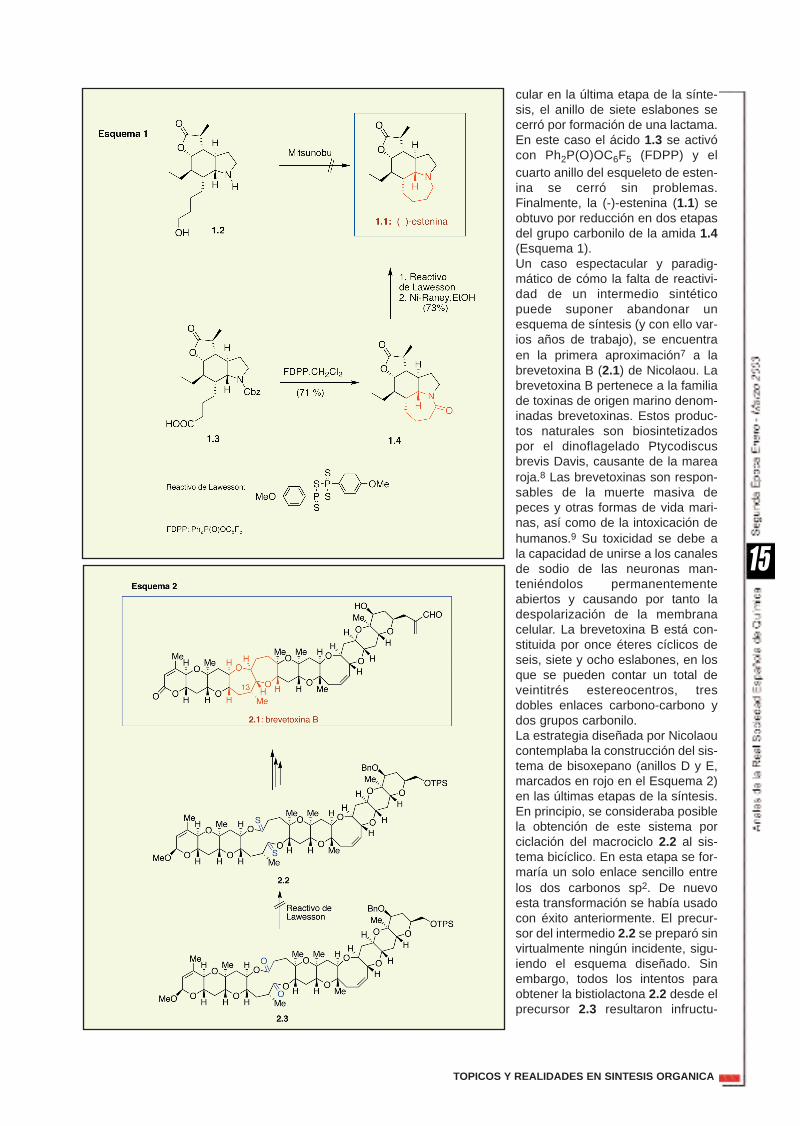
cular en la última etapa de la sínte-sis, el anillo de siete eslabones secerró por formación de una lactama.En este caso el ácido 1.3 se activócon Ph2P(O)OC6F5 (FDPP) y elcuarto anillo del esqueleto de esten-ina se cerró sin problemas.Finalmente, la (-)-estenina (1.1) seobtuvo por reducción en dos etapasdel grupo carbonilo de la amida 1.4(Esquema 1). Un caso espectacular y paradig-mático de cómo la falta de reactivi-dad de un intermedio sintéticopuede suponer abandonar unesquema de síntesis (y con ello var-ios años de trabajo), se encuentraen la primera aproximación7 a labrevetoxina B (2.1) de Nicolaou. Labrevetoxina B pertenece a la familiade toxinas de origen marino denom-inadas brevetoxinas. Estos produc-tos naturales son biosintetizadospor el dinoflagelado Ptycodiscusbrevis Davis, causante de la marearoja.8 Las brevetoxinas son respon-sables de la muerte masiva depeces y otras formas de vida mari-nas, así como de la intoxicación dehumanos.9 Su toxicidad se debe ala capacidad de unirse a los canalesde sodio de las neuronas man-teniéndolos permanentementeabiertos y causando por tanto ladespolarización de la membranacelular. La brevetoxina B está con-stituida por once éteres cíclicos deseis, siete y ocho eslabones, en losque se pueden contar un total deveintitrés estereocentros, tresdobles enlaces carbono-carbono ydos grupos carbonilo.La estrategia diseñada por Nicolaoucontemplaba la construcción del sis-tema de bisoxepano (anillos D y E,marcados en rojo en el Esquema 2)en las últimas etapas de la síntesis.En principio, se consideraba posiblela obtención de este sistema porciclación del macrociclo 2.2 al sis-tema bicíclico. En esta etapa se for-maría un solo enlace sencillo entrelos dos carbonos sp2. De nuevoesta transformación se había usadocon éxito anteriormente. El precur-sor del intermedio 2.2 se preparó sinvirtualmente ningún incidente, sigu-iendo el esquema diseñado. Sinembargo, todos los intentos paraobtener la bistiolactona 2.2 desde elprecursor 2.3 resultaron infructu-
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
15
topicos y realidades.qxd 11/10/2007 14:02 PÆgina 15

osos, hasta el punto de que estaprimera estrategia sintética tuvo queser abandonada (recordemos queesta reacción es muy eficiente enotros sustratos estructuralmentesimilares a 2.3). No obstante, comoconsecuencia de este aparente fra-caso, se desarrolló todo un cuerpode doctrina para la construcción deéteres cíclicos fusionados.10
Como ejemplo del cambio estratégi-co profundo que se tuvo que realizarpor la ausencia de reactividad delcompuesto 2.3, las etapas finales enla síntesis de la brevetoxina B seresumen a continuación. En lugarde construir el sistema de bi-soxepano (anillos D y E) en las últi-mas etapas de la síntesis (ya hemosdiscutido que esto no fue posible),se eligió la construcción del resto dedidehidrooxocano (anillo H, marca-do en azul 2.1) al final de la síntesis,en el intermedio avanzado 3.1. Enesta ruta que tuvo éxito, curiosa-mente, la síntesis del sistema debisoxepano se realizó en las etapastempranas de la síntesis (intermedio3.2) (Esquema 3).
¡PERO SI SÓLO SE DIFERENCIANEN EL GRUPO PROTECTOR!
En la síntesis de cualquier moléculaprobablemente se necesite usargrupos protectores. Este factor daidea del grado de madurez de lasíntesis orgánica. Para realizar unatransformación selectiva en un com-puesto multifuncional, muy fre-cuentemente, por no decir siempre,es necesario bloquear temporal-mente otros centros reactivos. Laelección del grupo protector es unaetapa importante en la secuenciasintética, hasta tal punto que de sucorrecta elección puede dependerel éxito de la síntesis. Aunque sehan desarrollado grupos protectorespara cualquier necesidad imagina-ble,11 los problemas que planteanson mas habituales de lo que seríadeseable. Es frecuente que un grupo protectorno solo evite la reacción en el grupoque está protegiendo, sino que imp-ida la transformación que se pre-tende realizar en su presencia. En lasíntesis de Clardy12 de las octalacti-nas A y B (4.1a-b) se pretendíaobtener el anillo de ocho eslabones
del intermedio 4.3 mediante una oxi-dación de Baeyer-Villiger sobre lacetona 4.2 (Esquema 4). Sin embar-go, en las condiciones ensayadasno fue posible obtener 4.3.Afortunadamente, en este casobastó con eliminar el grupo protec-tor. El tratamiento del alcohol libre4.4 con CF3CO3H si condujo a la
lactona deseada 4.5. Un examendel modelo molecular de la cetonaprotegida 4.2 revela que el grupoTBDPS, no solo impide la reaccióndel grupo hidroxilo, si no queademás bloquea el grupo carbonilo,debido a su elevado volumen estéri-co. Por tanto, ambas caras delgrupo carbonilo están bloqueadas,
16
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:02 PÆgina 16

una por el anillo y la otra por elgrupo TBDPS y por los susti-tuyentes de la cadena lateral .Una situación diferente se da en lasíntesis de la pandamina I (5.1)descrita por Zhu.13 En este caso, elgrupo protector incluso participadirectamente en la reacción quedebería conducir a un precursorinmediato del producto final. Laaproximación de Zhu a la síntesistotal de la pandamina se basa en laformación del enlace éter marcadoen rojo en el intermedio 5.2(Esquema 5). La metodología elegi-da para efectuar esta transforma-ción es una reacción de sustituciónnucleófila aromática (SNAr) sobre elintermedio 5.3. Para efectuar estareacción se desprotegería el grupoalcohol que se encontraba protegi-do como su silil derivado 5.4. Eneste proceso debería ocurrir la for-mación concomitante de un anillode catorce eslabones. Debe obser
varse que el grupo amino del tripep-tido 5.4 esta protegido como N-car-bamato. Aparentemente este tipo deprotección es totalmente compatiblecon las transformaciónes que seestaban buscando. Sin embargo, eltratamiento de 5.4 con TBAF enpresencia de tamiz molecularresultó en la formación de 5.5. Laproximidad entre el grupo hidroxilo yla función N-carbamato en 5.3favorece la formación de la oxazo-lidinona 5.6. La participación deloxígeno de la amida en una SNArpara formar el intermedio 5.7 dedoce eslabones, seguido de tau-tomerización, explica la formacióndel producto de reacción 5.5.Realmente éste no es un problemaque se pueda reconocer en unaprimera aproximación y, una vezmás hay que reconsiderar a poste-riori el grupo protector adecuado enel intermedio clave 5.4. La macroci-clación condujo al producto desea-
do 5.2 en el caso del N-alil derivado5.8. Este proceso de ciclación esnotorio, porque no requiere condi-ciones de alta dilución. Es probableque un proceso de reconocimientointramolecular favorezca cierta pre-organización que facilite la macroci-clación. De hecho, esta mismasituación debe darse en la ciclaciónde las oxazolidinonas que conducena los ciclos de doce eslabones 5.5. Estos pocos ejemplos muestrancomo pueden surgir problemas conlos grupos protectores a pesar de laenorme cantidad de que se dispone,y como claramente un grupo protec-tor nunca es un observador neutro,ya que modifica la distribución elec-trónica del sustrato en mayor omenor medida y por tanto modificasu reactividad.
¡ESTAMOS MUY CERCA: SOLOFALTA INSTAL AR EL GRUPOMETILO!
Muchas síntesis están diseñadaspara construir el núcleo hidrocar-bonado de la molécula objetivocompletamente protegido, dejandopara las últimas etapas de la sínte-sis la incorporación de un grupometilo o de una cadena lateral deunos cuantos átomos de carbono.Estas etapas finales, en principiosencillas, pueden representar unabismo insalvable entre el interme-dio en cuestión y el producto final,forzando a un cambio radical deestrategia. La síntesis del (+)-epo-xidictimeno (6.1) de Schreiberpuede ilustrar esta situación.14
El esqueleto tricíclico del (+)-epoxi-dictimeno (6.1) se construye de unaforma magistral mediante unasecuencia de reaccionesNicholas/Pauson-Khand en el inter-medio 6.2 (Esquema 6). Laconstrucción del sistema tetracíclicofusionado 6.3 del producto naturalen una única etapa sintética esespectacular.15,16 Para llegar al pro-ducto final solo queda introducir elgrupo metilo en C(1), invertir la con-figuración de C(12) y reducir la ce-tona de C(14). En primer lugar se abordo la incor-poración del grupo metilo sobreC(1). En los multiples experimentosrealizados, o bien no se conseguíareacción alguna o el grupo metilo se
17
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:03 PÆgina 17

incorporaba sobre C(1) con la con-figuración opuesta a la deseada.Los resultados obtenidos duranteestos experimentaos indicaban queel grupo metilo solo se adicionaríacon la configuración correctadespués de epimerizar el carbonoC(12). Los cálculos de mecánicamolecular indican que los carbonosque forman el puente entre C(1) yC(12) se disponen preferentementeen configuración syn. Por tanto, seabordó entonces el problema decorregir la configuración en C(12)en el intermedio 6.3. Esto se logróen una secuencia de cinco etapassintéticas, después de abrir el anilloC, perdiendo un átomo de carbono.El grupo metilo se introdujo en 6.4como un fragmento de dos átomosde carbono, siendo necesario pre-viamente volver a introducir el car-bono que se había eliminadodurante la corrección de la estereoquímica de C(12). La ciclaciónaniónica de 6.5 condujo a 6.6 quetiene el esqueleto y la estereoquími-ca de todos sus centros quiralesidénticos a los del producto natural6.1. La última etapa sintética para
conseguir el (+)-epoxidictimeno(6.1) fue la eliminación del gruponitrilo en 6.6. Está claro que proble-mas aparentemente tan complejoscomo la construcción del sistematetracíclico del producto natural sepueden resolver de forma tanmagistral como sencilla, con eleva-dos rendimientos. Sin embargo, laincorporación de los átomos de car-bono restantes y de nuevo la co-rrección de la estereoquímica de uncentro estereogénico, que en princi-pio son más sencillos, causanserias complicaciones en el procesosintético.El ejemplo anterior supone la incor-poración de un resto carbonado enun esqueleto base previamente sin-tetizado. En otras ocasiones elgrupo metilo, o en general unacadena lateral, forman parte de losintermedios en los que se efectúanlas etapas clave de la síntesis y elproblema es el contrario. En estecaso las reacciones que transcurrensin dificultad en sustratos desprovis-tos de dichos sustituyentes fallancompletamente en los intermediossintéticos reales.
Esta carencia de reactividad fuerzaen ocasiones a abandonar la rutasintética prevista. La síntesis deDanishefsky de la (±)-mirocina C(7.1) ilustra esta situación.17
El plan sintético original se basabaen el empleo de una cicloadición deDiels-Alder intramolecular en unsustrato como 7.2 (Esquema 7).Esta reacción construiría en las eta-pas iniciales de la síntesis el sis-tema de decalina presente en elproducto final. La hipótesis se basa-ba en el éxito de; reacción análogaefectuada en el sustrato modelo 7.3,que condujo al sistema tricíclico 7.4con elevados rendimientos. El sus-trato análogo 7.5a, con el grupometilo situado correctamente en eldoble enlace, debería producir ladecalina 7.6, con todos los susti-tuyentes sobre el carbono cuater-nario C(4) necesarios para construirla (±)-mirocina C. La reacción deDiels-Alder debería, además, pro-ducir la estereoquímica correcta.Sin embargo, esta cicloadición nose produjo en ninguna de las condi-ciones ensayadas. La sustitucióndel grupo metoxicarbonilo originalpor un grupo ciano menos volumi-noso (sustrato 7.5b) fue igualmenteinfructuosa. Se ensayaron modifica-ciones tácticas del sustrato 7.4,como, por ejemplo, la reducción delgrupo ester a alcohol y la posteriorprotección del hidroxilo. Esta modifi-cación debería minimizar el efectoatractor de electrones en el sistemadiénico, pero tampoco se obtuvoninguna reacción positiva. El con-junto de estos resultados terminaroncon el abandono de esta aproxi-mación dirigida a la construcción delanillo de decalina portando el car-bono cuaternario C(4) de la miroci-na C. El cambio estratégico para prepararla (±)-mirocina C supuso una aprox-imación que empleaba una reacciónde Diels-Alder intermolecular. Eneste caso y en palabras de losautores el componente diénico seha seleccionado más astutamente.La reacción de Diels-Alder del dienocíclico 7.6 y la p-benzoquinona con-dujo al aducto 7.8. Este aducto tienela misma estereoquímica relativadel producto final en los carbonosC(1), C(4) y C(5). El intermedio 7.8se usó como intermedio clave en lasecuencia que condujo, finalmente,
18
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:03 PÆgina 18

a la (±)-mirocina C (7.1).
NOS HEMOS EQUIVOCADO CONLA ESTEREOQUÍMICA.
Actualmente, síntesis asimétrica esprácticamente sinónimo de síntesisorgánica. La necesidad de disponerde compuestos enatiomericamentepuros ha estimulado el desarrollo demetodologías para controlar laestereoquímica de muchos proce-sos básicos. A pesar de los logrosalcanzados en síntesis asimétricatodavía estamos muy lejos dealcanzar un control a voluntad delresultado estereoquímico de unareacción. Por ejemplo, una de las transforma-ciones más frecuentes en síntesisorgánica es la reducción de una ce-tona a un alcohol. El bagaje teórico-experimental sobre este proceso esenorme. Sin embargo, la configu-ración del nuevo centro estere-ogénico formado por reducción deun carbonilo cetónico no siempre esfácil de controlar. Un ejemplo es lasíntesis de Smith de la (±)-breinoli-da (8.1).18 La reducción del interme-dio 8.2 (Esquema 8) con NaBH4
condujo exclusivamente al alcoholendo-8.3 como único producto dereacción. Puesto que el alcohol 8.3tiene configuración errónea se optópor invertir la configuración en dichocentro. Sin embargo, los métodosusuales para invertir la configu-ración de un grupo hidroxilo soloprodujeron trazas del isomerodeseado 8.4. De la misma forma, uncambio drástico en el agente reduc-tor, en este caso el empleo de unsistema metal amoniaco líquido,tampoco produjo resultados acepta-bles. Este fracaso en obtener laestereoquímica deseada forzó a losautores a buscar otra solución alproblema. La topología del sistemabicíclico 8.2, fuerza la adición delhidruro al grupo carbonilo por lacara convexa de la molécula,generando el producto de adicióncon estereoquímica endo-8.3. Lasolución al problema estereoquími-co debe considerar necesariamenteesta característica del sustrato, yaprovecharla para obtener la estere-oquímica deseada. En cualquiercaso, situar el grupo hidroxilo con laestereoquímica deseada no fue en
modo alguno trivial sino que requirióseis etapas adicionales de sínte-sis. El alcohol 8.3 con la estereo-química no deseada se transformóen el dihidrotiofeno 8.5 en cuatroetapas. El grupo hidroxilo con laconfiguración adecuada se introdujo
en una reacción de hidroboraciónestereoselectiva, aprovechando latendencia del sistema bicíclico areaccionar por la cara convexa. El ejemplo anterior representa unasituación en la que la selectividadintrínseca del sustrato produce el
19
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:03 PÆgina 19

isómero no deseado, pero la confi-guración se puede corregir intro-duciendo etapas adicionales en lasíntesis. Sin embargo, en otras oca-siones no es posible obtener elestereoisómero deseado y la rutasintética tiene que ser abandonada.Esta es una situación mucho másfrecuente de lo que sería deseable.Los dos ejemplos que se discuten acontinuación ilustran este caso.La síntesis del (+)-epoxidictimeno(6.1) descrita por Paquette demues-tra la imposibilidad de corregir laestereoquímica sobre un sistemacíclico, imposibilidad que lleva aabandonar la ruta sintética.19 Laestrategia original empleada porPaquette en la construcción del sis-tema tricíclico del (+)-epoxidicti-meno (6.1) pasaba por el intermedio9.1 (Esquema 9). Desde este inter-medio de síntesis se pretendía lle-var a cabo la epimerización del car-bono C(11) (marcado en rojo), parallegar a la estereoquímica trans, ca-racterística de la fusión de anillosdel hemisferio este del (+)-epoxydic-timeno (6.1). Esta hipótesis se ba-saba en los resultados obtenidosmediante cálculos MM2. Según es-tos cálculos, la enona con fusión deanillos trans 9.2 es 6.7 Kcal.Mol-1más estable que el isómero de par-tida 9.1. Sin embargo, la epimeriza-ción de 9.1 no ocurrió en ningún ca-so. La desprotonación del isómerocis 9.1 producía en todos los casosel enolato extendido 9.3. La reproto-nación de este enolato formaba elintermedio 9.4, en el que no solo nose ha conseguido la epimerizacióndeseada sino que se ha destruido laestereoquímica de uno de los cen-tros quirales clave. La incapacidadpara efectuar la epimerizacióndeseada obligó a cambiar la estrate-gia sintética y el (+)-epoxidictimeno(6.1) se obtuvo finalmente a partirde 9.5.El intento de preparar la goniocina10.1 por Keinan ilustra la situaciónen la que se emplea una reacciónque conduce a la estereoquímicaerrónea per se, obligando a abando-nar incluso la idea de preparar lamolécula objetivo.20 La síntesis delfragmento de goniocina 10.2 (Esquema 10)se intentó medianteuna oxidación en cascada del trienotodo-trans 10.3, inducida por Re(VII). Esta reacción produjo 10.4 co-
20
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:04 PÆgina 20

mo único diastereomero con unbuen rendimiento para un procesoen el que se crean tres anillos ycinco estereocentros. Sin embargo,la estereoquímica de 10.4 no es ladeseada trans-treo-trans-treo-trans-treo de 10.2 necesaria para accedera la goniocina. El producto 10.4tiene una estereoquímica trans-treo-cis-treo-trans-treo que le haceinservible como intermedio en lapreparación de la goniocina (10.1).Como siempre, puede pensarseque el trieno 10.3 no es un buensustrato para efectuar la ciclacióncon la estereoquímica deseada yque, quizás, en un intermedio máselaborado como 10.5 la ciclaciónocurra con la estereoquímica "co-rrecta". De nuevo esta suposiciónfue incorrecta. El sustrato 10.5 cicló con un rendimiento excelente perocon la estereoquímica no deseadaobteniéndose exclusivamente 10.6. En los casos anteriores la selectivi
dad intrínseca de los sustratos dicta el curso estrereoquímico de la reac-ción. ¿Qué ocurre cuando se usaalguno de los métodos de estereo-control por el reactivo que tanexcepcionales resultados han dadoen síntesis durante los últimos trein-ta años? ¿Cabe esperar algún tipode sorpresa en estos casos? Recor-demos que en esta metodología seutilizan reactivos enatiomericamen-te puros para compensar la bajadiastereoselectividad facial del sus-trato. Esto permite controlar la es-tereoquímica de los productos dereacción que es, en general, fácil-mente predecible.21 No obstante, enocasiones, nuestras prediccionesno son las correctas. La síntesis de Evans de la citovarci-na (11.1) sirve como ejemplo paraeste caso.22 La síntesis del frag-mento 11.2 (Esquema 12), consti-tuido por los carbonos C3-C10 de lacitovaricina, se intentó mediante
una condensación aldólica asimétri-ca entre el aldehido 11.3 y el enola-to de boro derivado de 11.4. Segúnel amplio conocimiento de este tipode reacciones, el resultado estereo-químico de la reacción dependeríade la quiralidad del enolato, conindependencia de la quiralidad delaldehido.23
Sorprendentemente, el aldol anti-11.5 se obtuvo como único diaste-reoisómero (el aldol esperado era eladucto sin-11.2). Este resultado es-tereoquímico no tenía precedentescon el enolato quiral utilizado. Apa-rentemente, la configuración delcarbono C(9) está definida por elenolato, y la selectividad inherentedel aldehido quiral determina la for-mación del estereocentro C(8),según predice el modelo de Felkin-Anh.24 Todos los intentos para inver-tir la selectividad diastereofacial delaldehido resultaron infructuosos.Para situar la estereoquímica co-rrecta en C(8) fue necesario dar unrodeo de tres etapas que condujo alintermedio sintético 11.6, a partir delcual se sintetizó la citovaricina(11.1).Los ejemplos que hemos discutidoen esta sección ilustran las dificul-tades que todavía existen en el con-trol de la estereoquímica, incluso enprocesos aparentemente sencillos yen reacciones bien establecidas.Existe un cuerpo doctrinal bienestablecido sobre los factores queregulan la estereoquímica de losprocesos más relevantes. La efi-ciencia del control estereoquímico,que se obtiene con muchos reac-tivos catalíticos y estequiométricos,es impresionante. Sin embargo aúnqueda mucho por descubrir sobre lageneralidad y el control estereo-químico de estas transformaciones.Entre otros, problemas como elestereocontrol del acoplamiento dedos fragmentos quirales, o cómoinvertir la selectividad facial intrínse-ca de un sustrato cíclico (una meto-dología que podríamos denominarestereocontrol cíclico) están aunpor resolver.25
ESTE SUSTITUYENTE ESTÁ MÁSCERCA DE LO QUE PARECE.Desde el principio de cualquier sín-tesis total, se elige la estrategia másadecuada. Las transformaciones
21
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:04 PÆgina 21

clave se estudian cuidadosamente,muy a menudo en compuestosmodelo. Sin embargo, algunasveces una transformación amplia-mente ensayada falla en un inter-medio real de la síntesis, debido a lapresencia de un substituyenteremoto que no estaba presente enel compuesto modelo, y que en prin-cipio no debería influir en el resulta-do sintético. La síntesis del (±)-lubiminol (Esquema 12) deCrimmins puede ilustrar estepunto.26
La estrategia original consideraba lasíntesis del sistema de ciclopentanoespiránico utilizando un reorde-namiento radicálico en el intermedioavanzado 12.2 (Esquema 12). Estatransformación se había ensayadocon éxito en el sustrato modelo 12.3idéntico a 12.2, excepto por laausencia del grupo metilo en el car-bono C(4). La calefacción de 12.3en presencia de TBTH/AIBN induceuna expansión de anillo via el reor-denamiento Dowd-Bekwith27 y ge-nera el espirano 12.4. El mismotratamiento en el intermedio clave12.2 condujo a una mezcla de com-puestos en los que 12.5 era el pro-ducto mayoritario. En ningún casose detectó la formación del produc-to deseado, análogo a 12.4. Estosresultados indican que el radicalintermedio derivado de 12.5 noexperimenta el reordenamiento deDowd-Bekwith. La única diferenciaestructural entre 12.3 y 12.2 es lapresencia del grupo metilo en el car-bono C(4). La estrategia sintéticaoriginal tuvo que ser modificada y elgrupo metilo en C(4), se introdujo enuna etapa posterior de la síntesis,una vez se hubo construido el sis-tema espiránico vía el reorde-namiento de Dowd-Bekwith. Esinnecesario comentar que estamodificación alargó considerable-mente la síntesis.La síntesis del (±)-lubiminol 12.1 esun ejemplo de cómo un substi-tuyente remoto inhibe una determi-nada transformación (en este casoprobablemente por las restriccionesestéricas que impone en el estadode transición de dicha transforma-ción). Otra situación diferente surgecuando el sustituyente remoto par-ticipa directamente en la etapa clavede la síntesis modificando el resulta-do de la reacción. Esto sucede en la
22
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:05 PÆgina 22

síntesis de Grieco de la (-)-chaparri-nona (13.1).28 En este caso lapreparación del triol 13.2 (Esquema13), intermedio clave en la síntesisde 13.1, se abordó por aperturaácida del epóxido 13.3. Esta trans-formación es, en principio, directa yno debería presentar complica-ciones. Como dato adicional, lareacción se había usado con éxitoen la síntesis de klaineanona13.4,29 un compuesto con estruc-tura similar a 13.1. La reacción de13.3 en medio ácido condujo, sinembargo a la deoxichaparrinona13.5. En este caso, el fracaso de latransformación de 13.3 en 13.2 esfácilmente explicable a posteriori, yaque el grupo hidroximetilo protegidocomo silil derivado está en una dis-posición adecuada para abririntramolecularmente el oxirano(intermedio 13.6) truncando la reac-ción deseada. Sin embargo, loscambios de grupo protector ensaya-dos tampoco produjeron ningúnresultado positivo (Esquema 13). Lasíntesis de la (-)-chaparrinona selogró, finalmente, mediante unaestrategia totalmente diferente. Los ejemplos anteriores indican quecuanto mayor es la complejidadestructural y/o funcional de un inter-medio sintético, el numero de va-riables de las que dependen inclusolas transformaciones mas sencillasaumenta exponencialmente. Estohace que cualquier predicción seamuy arriesgada. El hecho de sercapaces de minimizar dichas va-riables es un indicio de la capacidadpredictiva de la síntesis orgánicamoderna.
¿POR QUÉ NO CONSEGUIMOSCERRAR EL ANILLO?.
Para terminar este artículo discutire-mos uno de los aspectos mas atrac-tivos de la síntesis total: la forma-ción de anillos. Una buena parte delos objetivos sintéticos son produc-tos naturales que tienen en suestructura al menos un ciclo. Porello, el desarrollo de metodologíapara la formación de anillos ha sidoy sigue siendo un aspecto prioritarioen química. Continuando con elespíritu de este artículo hemosseleccionado algunos ejemplos sen-cillos que muestran lo lejos que
estamos de controlar eficientementelos procesos de ciclación. Los prob-lemas que surgen de la imposibili-dad de cerrar un anillo en unasecuencia sintética, causan cam-bios más drásticos en la estrategiaoriginal que cualquiera de los prob-lemas que hemos discutido hastaahora.En la síntesis de la makaluvamina D(14.1) de White, la construcción delnúcleo tricíclico se intentó via unadoble ciclación de la diaminaaromática 14.2.30 Este proceso for-maría simultáneamente los anillos By C del alcaloide (Esquema 14). Eldiseño de la síntesis fue correcto encuanto a la formación del anillo B.Así, la diamina 14.2, en presenciade acetoxiyodosobenceno (PIFA)en 2,2,2-trifluoroetanol, experimentala ciclación oxidativa esperada yforma el biciclo 14.3. Sin embargo,el cierre del anillo C resultó ser másproblemático. El tratamiento de 14.3 con nitrato de cerio y amonio (CAN)condujo a la iminoquinona inestable
14.4. Esta quinona no produjo el tri-ciclo deseado con reactivos básicoscomo NaH, sino que condujo a laquinolina 14.6. Este compuestopodría proceder de la aromatizacióndel enolato intermedio 14.5. Seintentó provocar un aumento de laacidez del protón amídico de lacadena lateral, utilizando una sul-fonamida en lugar de un grupoacetato, pero tampoco se produjo laesperada ciclación (Esquema 14). Ante la imposibilidad de cerrar elanillo C desde el intermedio 14.2, sereconsideró la estrategia originalbasada en la construcción secuen-cial de los anillos A-B-C. La estrate-gia alternativa se basó en la con-strucción inicial del núcleo indólico,a partir de la hidrazona 15.1, paracerrar posteriormente el anillo B. Laelaboración del indol 15.2 se efec-tuó sin novedad y en esta ocasión sise pudo cerrar el anillo B, en condi-ciones básicas. En estas condi-ciones se obtuvo el compuesto 15.3que tiene el núcleo tricíclico de la
23
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:05 PÆgina 23

makaluvamina D (14.1). El interme-dio 15.3 se convirtió en varias eta-pas en el producto final (Esquema15).El ejemplo anterior muestra clara-mente el cambio radical de estrate-gia que requiere el fracaso del cierrede un anillo. La situación puedeverse exacerbada cuando lamolécula objetivo es un macrociclo.En estos casos, el cierre del ciclosuele ser, normalmente, la últimaetapa sintética. Esto puede hacerseen principio desde diferentes pre-cursores dependiendo del tipo dereacción que se utilice para cerrar elanillo. Sin embargo, prever cual esel sustrato mas idóneo para laciclación no es en absoluto evi-dente, ya que incluso el orden en elque se unen los fragmentos puededeterminar el éxito o el fracaso de lasíntesis.Esto ocurre, por ejemplo, en la sín-tesis de la (-)-papuamina deWeinreb, (16.1).31 La primera a-proximación sintética se basaba enla formación del macrociclo a partirdel intermedio 16.2, en el que se hainstalado previamente el sistemadiénico (Esquema 16). Se probarondiferentes condiciones para efectuar
la reacción de N-alquilación con 1,3-diyodo- y 1,3-dibromopropano, peroen ningún caso se obtuvo N,N-diacetilpapuamina 16.3. Se recon-sideró una nueva ruta sintética con-sistente en instalar en primer lugarla cadena de propano de la mitadnorte, y cerrar posteriormente elanillo en la parte sur. En este casose sintetizó el bis-estannano 16.4 y,después de intentar condiciones dereacción muy diversas, la ciclaciónse produjo por reacción conPdCl2(PPh3)2/DMF en presencia deoxígeno.No es fácil racionalizar el fallo de laprimera ciclación por el cierre delpuente alifático de la mitad norte dela molécula. La N,N-diacetilpapua-mina (16.2) existe como isómero s-trans. Se podría pensar que el cam-bio conformacional del precursor16.2 de la forma s-trans a s-cisdebería producirse antes de laciclación. La energía asociada condicho cambio es de 6.5 Kcal mol-1.No obstante, la distancia N-N en elconformero s-trans-16.2 es de 5.1Å,menor que en el conformero s-cis-16.2 en el que es de 6.3Å. Más aún,la s-trans-papuamina es 1.6 Kcalmol-1 mas estable que la s-cis-
papuamina. Teóricamente laciclación debería producirse peroen la práctica no se da.
CONCLUSIONES.
En la literatura se pueden encontrarotros muchos ejemplos que comen-tan los problemas que han surgidodurante la síntesis de una moléculaobjetivo.32 Los ejemplos que hemosdiscutido son casos particulares deun problema bastante más amplio.La complejidad estructural, los efec-tos electrónicos, estéricos y confor-macionales son los motivos últimosque se aducen como causa de lastransformaciones fallidas duranteuna síntesis. Evidentemente, estodemuestra que el conocimiento quetenemos sobre el comportamientoquímico de moléculas densamentefuncionalizadas, conformacional-mente flexibles es bastante escaso.La capacidad del cálculo computa-cional llega al límite en estos casos,y aún queda mucho por aprendersobre cómo ligeros cambios a nivelmolecular afectan al resultado finalde una reacción. Por otro lado, reac-tivos altamente selectivos que seensayan en transformaciones sim-ples, pueden y suelen fallar cuandohay grupos funcionales en el sustra-to. El síndrome metilo, etilo, propiloe inutilo es especialmente aplicablea estos casos.33
Sin lugar a dudas, puede decirseque cualquier molécula se puedesintetizar con la tecnología y elconocimiento existente, pero esigualmente cierto que el esfuerzonecesario es enorme.Evidentemente el reto es hacerlomás simple, mejor y de una maneramás eficaz. Ciertamente y a pesarde opiniones en contra que pecande un optimismo injustificado, la sín-tesis orgánica aún tiene un largo yapasionante camino para alcanzarla plena madurez.
AGRADECIMIENTOS
Queremos agradecer al MCYT laconcesión del proyecto BQU2001-1283 y a la Comunidad Autonomade Madrid la financiación de losproyectos 07M/0044/2002 (MCT) y07M/0043/2002 (MAS).
24
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:06 PÆgina 24

1. Algunas de las síntesis másbellas de la historia se recogen y secomentan magistralmente en ellibro: K. C. Nicolaou, E. J.Sorensen, Classics in TotalSynthesis, VCH, Weinheim, 1996;Veáse, adicionalmente, la revision:K. C. Nicolaou, D. Vourloumis, N.Winssinger, P. S. Baran, Angew.Chem. Int. Ed. 2000, 39, 44-122.
2. Un ejemplo representativo deesta forma de pensar se recoge eneste artículo de revision: D.Seebach, Angew. Chem. Int. Ed.Engl. 1990, 28, 1320-1367.
3. Los dos artículos siguientesescritos por Heathcok reflejan laforma de pensar alternativa: (a) C.H. Heathcock, Angew. Chem. Int.Ed. Engl. 1992, 31, 665-681. (b) C.H. Heathcock, Proc. Natl. Acad. Sci.USA 1996, 93, 14323-14327.
4. Danishefsky da razones adicio-nales para no ser demasiado expli-cito en detallar todas las dificultadesencontradas durante el desarrollode una síntesis: … Hence, the sea-soned chemist will appreciate thatalong with these "magic moments"of success, one could have reporteda litany of setbacks and reversals.However, for younger and moreoptimistic enthusiasts, it is appro-priate to underscore the uncertain-ties, the detours and, yes, the frus-trations associated with OrganicSynthesis… S. J. Danishefsky,Tetrahedron 1997, 53, 8689-8730.
5. P. Wipf, Y. Kim, D. M.Goldstein, J. Am. Chem. Soc. 1995,117, 11106-11112.
6. a) O. Mitsunobu, Synthesis1981, 1-28; b) D. L. Hughes, Org.React. 1992, 42, 335-665.
7. K. C. Nicolaou, C.-K. Hwang,M. E. Duggan, D. A. Nugiel, Y. Abe,K. B. Reddy, S. A. DeFrees, D. R.Reddy, R. A. Awartani, S. R. Conley,F. P. J. T. Rutjes, E. A. Theodorakis,J. Am. Chem. Soc.1995, 117,10227-10238. Para una discusióndetallada de ésta aproximación a lasíntesis de Brevetoxina B ver refe-rencia 1 pag. 750-752.
8. a) Y.-Y. Lin, M. Risk, S. M. Ray,D. Van Engen, J. Clardy, J. Golik, J.C. James, K. Nakanisi J. Am. Chem.Soc. 1981, 103, 6773. b) M. S. Lee,D. J. Repeta, K. Nakanishi ibid.1986, 108, 7855.
9. a) D. M. Andersn Sci. Am.
1994, 271(8), 62 y referencias cita-das, b, V. L. Trainer, D. G. Baden, W.A. Catterall J. Biol. Chem. 1994,269, 19904.
10. a) K. C. Nicolaou, C.-K.Hwang, M. E. Duggan, K. B. Reddy,B. E. Marron, D. G. McGarry, J. Am.Chem. Soc.1986, 108, 6800-6802;b) K. C. Nicolaou, C.-K. Hwang, B.E. Marron, S. A. DeFrees, E.Couladouros, Y. Abe, P. J. Carroll, J.P. Snyder, J. Am. Chem. Soc.1990,112, 3040-3054.
11. a) T. W. Greene, P. G. M.Wuts, Protective Groups in OrganicSynthesis, 2nd. ed., Wiley, NewYork, 1991; b) M. Schelhaas, H.Waldmann, Angew. Chem. Int. Ed.Eng. 1996, 35, 2056-2083.
12. J. C. McWilliams, J. Clardy, J.Am. Chem. Soc. 1994, 116, 8378-8379.
13. J. Zhu, T. Laïb, J. Chastanet,R. Beugelmans, Angew. Chem. Int.Ed. Eng. 1996, 34, 2517-2519.
14. a) T. F. Jamison, S.Shambayati, W. E. Crowe, S. L.Schreiber, J. Am. Chem. Soc. 1994,116, 5505-5506; b) T. F. Jamison, S.Shambayati, W. E. Crowe, S. L.Schreiber, J. Am. Chem. Soc. 1997,119, 4353-4363.
15. a) N. E. Schore inComprehensive OrganometallicChemistry II, vol 12 (Eds.: E. W.Abel, F. G. A. Stone, G. Wilkinson),Pergamon. Oxford, 1995, pp.703-739; b) N. E. Schore, Org, Reac.1991, 40, 1-90; c) P.L. Pauson,Tetrahedron 1985, 41, 5855-5860.
16. A. J. M. Caffyn, K. M. Nicholasin Comprehensive OrganometallicChemistry II, vol 12 (Eds.: E. W.Abel, F. G. A. Stone, G. Wilkinson),Pergamon. Oxford, 1995, p. 685-702.
17. (a) M. Y. Chu-Moyer, S. J.Danishefsky, G. K. Schulte, J. Am.Chem. Soc. 1994, 116, 11213-11228. (b) M. Y. Chu-Moyer, S. J.Danishefsky, J. Am. Chem. Soc.1992, 114, 8333-8334.
18. A. B. Smith III, J. R. Empfield,R. A. Rivero, H. A. Vaccaro, J. Am.Chem. Soc. 1991, 113, 4037-4038.
19. L. A. Paquette, L.-Q. Sun, D.Friederich, P. B. Savage, J. Am.Chem. Soc. 1997, 119, 8438-8450.
20. S. C. Sinha, A. Sinha, S. C.Sinha, E. Keinan, J. Am. Chem.Soc. 1997, 119, 12014-12015.
21. S. Masamune, W. Choy, J. S.Petersen, L. R. Sita, Angew. Chem.Int. Ed. Eng. 1985, 24, 1-30.
22. D. A. Evans, S. W. Kaldor, T.K. Jones, J. Clardy, T. J. Stout, J.Am. Chem. Soc. 1990, 112, 7001-7031.
23. a) D. A. Evans, J. V. Nelson, T.R. Taber, Top. Stereochem. 1982,13, 1-115; b) D. A. Evans, Aldrichim.Acta 1982, 15, 23-32.
24. N. T. Ahn, O. Eisenstein, Nov.J. Chem. 1977, 1, 61.
25. a) A. J. Duplantier, M. H.Nantz, J. C. Roberts, R. P. Short, P.Somfai, S. Masamune, TetrahedronLett. 1987, 30, 7357-7360; b) W. R.Roush, ChemTracts-OrganicChemistry 1990, 3, 201-203.
26. a) M. T. Crimmins, Z. Wang, L.A. McKerlie, J. Am. Chem. Soc.1998, 120, 1747-1756; b) M. T.Crimmins, Z. Wang, L. A. McKerlie,Tetrahedron Lett. 1996, 37, 8703-8706.
27. P. Dowd, W. Zhang, Chem.Rev. 1993, 93, 2091-2115.
28. P. A. Grieco, J. L. Collins, E.D. Moher, T. J. Flek, R. S. Gross, J.Am. Chem. Soc. 1993, 115, 6078-6093.
29. a) P. A. Grieco, D. T. Parker,R. P. Nargund, J. Am. Chem. Soc.1988, 110, 5568-5569; b) P. A.Grieco, R. P. Nargund, D. T. Parker,J. Am. Chem. Soc. 1989, 111, 6287-6294.
30. J. D. White, K. M. Yager, T.Yakura, J. Am. Chem. Soc. 1994,116, 1831-1838.
31. a) R. M. Borzilleri, S. M.Weinreb, M. Parvez, J. Am. Chem.Soc. 1995, 117, 10905-10913; b) R.M. Borzilleri, S. M. Weinreb, M.Parvez, J. Am. Chem. Soc. 1994,116, 9789-9790.
32. Para una discusión generalde los aspectos tratados en ésteartículo así como otros problemasadicionales de la Síntesis Orgánicamoderna, veáse: M. A. Sierra, M. C.de la Torre, Angew. Chem. Intern.Ed. 2000, 39, 1538-1559.
33. Este síndrome es un mal queafecta a una buena parte de laQuímica Moderna y fue descrito porprimera vez por Hegedus hace 15años. Veáse la introducción de surevisión para una definición: L. S.Hegedus, Angew. Chem. Int. Ed.1988, 27, 1113-1126.
BIBLIOGRAFIA
25
TOPICOS Y REALIDADES EN SINTESIS ORGANICA
topicos y realidades.qxd 11/10/2007 14:06 PÆgina 25






![TEMA 1 [Modo de compatibilidad] - ::WEB DEL …webdelprofesor.ula.ve/ingenieria/josealvarado/TEMA 1 [Modo de... · Características generales: ... combtiblbustible para condi idiciones](https://static.fdocuments.mx/doc/165x107/5baa549f09d3f2c9618b9e59/tema-1-modo-de-compatibilidad-web-del-1-modo-de-caracteristicas-generales.jpg)






![UNIVERSITAT DE BARCELONA DEPARTAMENT DE …diposit.ub.edu/dspace/bitstream/2445/41587/1/01.CEM_1de2.pdf · Síntesis de 2-azabiciclo[3.3.1]nonano V mediante un proceso de ciclación](https://static.fdocuments.mx/doc/165x107/5f4671f650594c1fb40e1e24/universitat-de-barcelona-departament-de-sntesis-de-2-azabiciclo331nonano-v.jpg)




