
INDICE - 148.206.53.84148.206.53.84/tesiuami/203322770.pdf · precisa en el espacio y de la...
16
1
-
Upload
truongkhuong -
Category
Documents
-
view
217 -
download
0
Transcript of INDICE - 148.206.53.84148.206.53.84/tesiuami/203322770.pdf · precisa en el espacio y de la...

1

2
INDICE
1. INTRODUCCIÓN. 3
2. ANTECEDENTES 5
3. OBJETIVO GENERAL 7
3.1 Objetivos Particulares 7
4. MATERIALES Y MÉTODOS 7
4.1 Ensayos de actividad enzimática 7
4.2 Perfiles de desnaturalización térmica 8
4.3 Espectroscopia de Fluorescencia 8
4.4 Ensayos de apagamiento de la Fluorescencia. 8
5. RESULTADOS Y DISCUSIÓN. 9
5.1 Ensayos de actividad enzimática 9
5.2 Perfiles de desnaturalización térmica 10
5.3 Espectroscopia de Fluorescencia 12
5.4 Ensayos de apagamiento de la Fluorescencia. 13
6. CONCLUSIONES. 14
7. REFERENCIAS 15

3
1. INTRODUCCIÓN
Las proteínas son polímeros largos de aminoácidos unidos por medio de enlaces
peptídicos y dispuestos en una secuencia lineal. Están compuestas básicamente por
carbono, hidrogeno, oxígeno, nitrógeno y azufre aunque pueden contener fosforo,
hierro, magnesio y cobre. Estas macromoléculas presentan una gran diversidad
estructural y funcional y varían en tamaño desde polipéptidos relativamente pequeños
hasta polímeros enormes de masas moleculares del orden de millones de daltones. Las
proteínas constituyen más del 50% del peso seco de las células, y desempeñan una gran
variedad y cantidad de funciones vitales que derivan principalmente de su estructura
precisa en el espacio y de la estabilidad de ésta en el entorno celular. El vocablo
proteína deriva del griego proteos, que significa “primero o fundamental”. Las proteínas
cuentan con cuatro niveles de organización estructural. Estructura primaria, la cual
describe la secuencia de los residuos de aminoácidos unidos de forma covalente por
enlaces peptídicos. Estructura secundaria se refiere a disposiciones particularmente
estables de los aminoácidos que dan lugar a patrones estructurales repetitivos, como las
hélices y las hojas β. Estructura terciaria representa la manera en cómo la cadena
polipeptídica se curva para formar la estructura estrechamente plegada y compacta de
una proteína globular. Estructura cuaternaria es la disposición espacial de dos o más
cadenas polipeptídicas que poseen algunas proteínas llamadas oligoméricas. [1,2]
Otra función importante de las proteínas es el ser los instrumentos moleculares
mediante los cuales se expresa la información genética, para preservar las características
de los miembros de una especie que les hayan permitido adaptarse a entornos
ambientales muy diversos y en algunos casos extremos, como aquéllos con
temperaturas bajas o elevadas, con alta salinidad o bajo pH.
Cada especie de microorganismo o bacteria tiene temperaturas naturales distintas, de
modo que un microorganismo puede presentar una temperatura óptima superior a la
temperatura máxima de otra, o inferior a la temperatura mínima de una tercera, estos
organismos se determinan extremófilos. Según el rango de temperaturas al que pueden
crecer los distintos microorganismos, se pueden establecer tres tipos principales:
psicrófilos, mesófilos y termófilos. [1-3]
Esta clasificación, está basada con respecto a los organismos mesófilos, los cuales
realizan sus funciones óptimas en un intervalo de temperatura de 25 a 45 °C. Los
psicrófilos se localizan en ambientes de baja temperatura (0 a 20 °C). En contraparte se
encuentran los organismos termófilos adaptados a altas temperaturas (de 45 °C a
mayores de 85 °C ). [4]
Los microorganismos extremófilos han despertado el interés de los
investigadores, debido a sus enzimas, catalizadores biológicos que aceleran las
reacciones químicas de la célula. Las enzimas habituales tienen baja estabilidad a
temperaturas moderadamente altas y dejan de funcionar luego de un corto tiempo, sin
embargo las enzimas de estos organismos empiezan a operar justo en el punto donde las
habituales dejan de funcionar. Existen numerosos procesos industriales que requieren el
uso de catalizadores, tal es el caso de las enzimas que participan en la producción de
edulcorantes, papel, síntesis de detergentes, elaboración de alimentos como pan y vino,
tratamiento de residuos, extracción de petróleo, obtención de biochips y el diagnóstico
de enfermedades. [4-6] La mayoría de las enzimas usadas hasta la fecha se originan de
organismos mesófilos y, a pesar de sus muchas ventajas, el uso de estas enzimas está

4
restringido debido a su estabilidad limitada en los extremos de la temperatura. Por otra
parte, los extremófilos son una fuente muy grande de enzimas, que demuestran alta
estabilidad bajo condiciones extremas. La comparación entre estructuras atómicas de
grupos de enzimas termófilas y mesófilas, apoyado con las comparaciones masivas de
secuencias en proyectos genómicos, han permitido dilucidar características que se
presentan de manera frecuente en proteínas térmicamente adaptadas: la optimización de
las interacciones electrostáticas carga-carga en la superficie de las proteínas, el
incremento de dipolos en las hélices, las interacciones entre Lys (catión) y anillos
aromáticos (Tyr,Phe y Trp). [7] Se ha observado un incremento de residuos aromáticos
y una menor presencia de residuos lábiles como: cisteínas, asparaginas y glutaminas, así
como un aumento de argininas. Por otra parte se han encontrado alargamientos en
estructuras regulares, acortamiento de las asas, disminución del número y tamaño de
espacios vacios al interior de la estructura, aumento de interacciones hidrofóbicas y
disminución de residuos lábiles. [1,2,7]
Esto ha llevado a buscar nuevas estrategias de estabilización de proteínas, teniendo
como uno de los resultados el incremento de termoestabilidad. La estabilización de
enzimas de organismos mesófilos se ha logrado utilizando dos metodologías distintas:
conocidas como “diseño racional” y la evolución dirigida.
Diseño racional. Involucra una serie de principios a través de los cuales la
termoestabilización de una proteína puede ser alcanzada o incrementada al aplicar
varios de ellos, otorgando cada uno un efecto pequeño pero aditivo. Tales principios
consisten en mutaciones del tipo X→Pro; incrementar la propensidad de las α-hélices
mediante sustituciones Gly→Ala o por estabilización de los macrodipolos de las
hélices; mejorar las interacciones electrostáticas entre residuos cargados superficiales
por la introducción de puentes salinos o redes de puentes salinos y prediciendo
mutaciones termo estabilizantes en base a cálculos de potenciales electrostáticos. [1,2]
Evolución dirigida. Comprende una serie de técnicas experimentales, mediante las
cuales se trata de imitar en una escala de tiempo acelerada, el proceso de selección
natural de una proteína. Esto se puede lograr a través de la combinación de mutagénesis
al azar seguida de un proceso de selección de fácil aplicación. Con frecuencia se emplea
también la técnica de intercambio de fragmentos de DNA (DNA shuffling). [1,2]
Otra característica importante de resaltar es como la catálisis enzimática de
microorganismos adaptados a climas totalmente opuestos se lleva acabo de manera
eficiente y comparativa en los distintos microorganismos. Para dilucidar este
comportamiento de las enzimas, diferentes investigaciones han encontrado que la
relación entre la actividad, flexibilidad y la estabilidad de una proteína representan un
tema central en la adaptación de estas a diferentes entornos [8]. Tal es el caso de las
proteínas de organismos psicrófilos que el disminuir su estabilidad con una mayor
flexibilidad genera un aumento en su actividad en comparación con sus homólogos
mesófilos y termófilos que cuenta con una mayor estabilidad y rigidez.
El aumento en la flexibilidad de una proteína se puede demostrar experimentalmente,
mediante el seguimiento de las tasa de intercambio de hidrogeno por deuterio, o bien
por el apagamiento de la fluorescencia por efecto de la acrilamida. [8-10]

5
2. ANTECEDENTES
La rizobacteria mesofíla Paenibacillus polymyxa habita en el suelo donde actúa como
facilitador biológico y al asociarse con plantas permite el crecimiento y desarrollo de
estas, protegiéndolas de organismos que provocan enfermedades. En Paenibacillus
polymyxa, existen dos β-glucosidasas homologas que pertenecen a la familia 1 de las
glicosil hidrolasas de la clasificación de Henrissat [11], la β-glucosidasa A (βglA) y la
β-glucosidasa B (βglB), ambas proteínas están conformadas por 448 residuos de
aminoácidos y comparten un 47% de identidad en su secuencia, las dos enzimas
presentan diferentes estructuras cuaternarias; la βglA es un octámero mientras que la
βglB es un monómero. La β-glucosidasa B tiene una masa de 52.5 kDa, su estructura ha
sido determinada por difracción de rayos X con una resolución de 2.10 Å [12],
presentando una estructura de barril (β/α)8 con algunas estructuras secundarias
periféricas adicionales, la cual se muestra en la figura 1, su código PDB es 2O9P.
La βglB cataliza la hidrólisis de celobiosa, celodextrinas de mayor grado de
polimerización y otros glicósidos relacionados. El mecanismo de reacción de la enzima
con su sustrato se muestra en la figura 2. Los residuos catalíticos de βglB son E167
que
actúa como donador de protones y E356
que actúa como nucleófilo. [1, 2, 12]
Figura 1. Estructura tridimensional de la enzima β-glucosidasa B de Paenibacillus
polymyxa. En rojo se representan las hojas β, en turquesa las hélices α y en violeta las
estructuras irregulares. Los residuos catalíticos se encuentran en el centro. Código PDB:
2O9P.

6
Figura 2. Representación del mecanismo de reacción de la βglB sobre su sustrato.
Para el presente trabajo es preciso hablar de las mutantes V323R y
H62R/N223Y/M319I de la β-glucosidasa B, la primera muestra una baja en su
estabilidad térmica, mientras que la segunda adquirió un aumento en su estabilidad
térmica respecto a la enzima nativa βglB. [7,13]
La mutante V323R se obtuvo por una estrategia de diseño racional, donde se
buscó optimizar la energía electrostática en la superficie de la proteína (Figura 3A).
[1,7]
La triple mutante H62R/N223Y/M319I se obtuvo a partir de un proceso de
intercambio de DNA a partir de un grupo de mutantes termorresistentes sencillas (con
un solo residuo cambiado) obtenidas por evolución dirigida. Figura 3B. [1,13]
En nuestro estudio seguiremos el apagamiento de fluorescencia para dos mutantes
de la enzima β-glucosidasa B, una de las cuales muestra una mejor termorresistencia
que la enzima nativa, mientras que la otra muestra una pobre resistencia a la
temperatura.
A B
Figura 3. (A) Posición del residuo mutado en la βglB, para obtener la mutante de carga.
(B) Posición de los residuos mutados en βglB, para obtener la mutante por evolución
dirigida. Se resaltan en esferas a los residuos de aminoácidos involucrados.

7
3. OBJETIVO GENERAL
Estudiar la movilidad de las mutantes H62R/N223Y/M319I y V323R de la β-
glucosidasa B y su relación con la estabilidad térmica.
3.1 Objetivos Particulares
- Determinar los parámetros cinéticos de las enzimas mutantes
H62R/N223Y/M319I y V323R.
- Obtener los perfiles de desnaturalización térmica por Dicroísmo Circular de las
enzimas mutantes H62R/N223Y/M319I y V323R.
- Determinar la movilidad de ambas mutantes midiendo el apagamiento de su
fluorescencia intrínseca por efecto de la acrilamida.
4. MATERIALES Y MÉTODOS
4.1 Ensayos de actividad enzimática
Los parámetros cinéticos se determinaron a través de mediciones de las velocidades
iníciales de la reacción de hidrólisis catalizada con las enzimas mutantes de la β–
glucosidasa B, usando diferentes concentraciones de p-nitrofenil-β-D-glucopiranósido,
PNPG, como sustrato; la hidrólisis de este libera nitrofenilo de color amarillo que
seguimos a través del aumento de absorbancia a 400nm. El PNPG se utilizó en un
intervalo de concentraciones de 0.50 mM a 44 mM en buffer de fosfatos 50 mM a pH
7.0. Los ensayos de actividad se realizaron en un espectrofotómetro UV-Visible Hewlett
Packard 8453, que tiene un controlador de temperatura HP 89090A. En los ensayos de
actividad se utilizaron celdas de 1.0 cm de paso óptico, donde se incubó el PNPG en el
amortiguador a 25 ºC. Una vez equilibrada la temperatura, se adicionó el volumen
necesario de enzima concentrada para tener una concentración final de 2.0µg mL-1
. El
volumen final de reacción fue de 1.0 mL. Después se mezcló por inversión el contenido
de la celda y se inició la medición de absorbancia en función del tiempo.
Las velocidades iníciales (v) se expresaron en micromoles de sustrato transformado por
minuto por miligramo de enzima (µmol min-1
mg-1
). Los resultados obtenidos al variar
la concentración de sustrato (S) se ajustaron a la ecuación de Michaelis-Menten (1).
4.2 Perfiles de desnaturalización térmica
Los perfiles de desnaturalización térmica para las enzimas mutantes
H62R/N223Y/M319I y V323R se obtuvieron siguiendo su señal de dicroísmo circular.
Los datos de elipticidad se tomaron a 220 nm con una velocidad de barrido de 2 ºC por
minuto en un intervalo de temperaturas de 25 ºC a 75 ºC, con agitación constante. La
celda que contenía las soluciones contaba con 1.0 cm de paso óptico, la concentración
de enzima utilizada fue de 20µg mL-1
en regulador de fosfatos 50mM a pH 7.0, el cual

8
fue desgasificado previamente para cada ensayo. Para medir el cambio de elipticidad se
utilizó un espectropolarímetro JASCO J-715 que cuenta con un controlador de
temperatura tipo Peltier PTC-348WI.
La fracción desnaturalizada (f D) de proteína se calculó por medio de los valores
de elipticidad (θobs) obtenidos a cada temperatura en el barrido, con ayuda de la
ecuación (2).
Donde θN y θD representa a la elipticidad de la forma nativa y desnaturalizada
respectivamente.
4.3 Espectroscopia de Fluorescencia
Se determinaron los espectros de fluorescencia de las enzimas, H62R/N223Y/M319I y
V323R a 25 ºC, variando la concentración de enzima de 100 µg/mL a 20 µg/mL en
amortiguador de fosfatos 50mM, pH 7.0, excitando a una longitud de onda de 295 nm y
registrando la emisión de fluorescencia en un intervalo de 300 a 450 nm. Se utilizó una
celda de 1.0 cm de paso óptico con agitación continua, los espectros se corrigieron con
la señal del amortiguador de fosfatos, 50 mM, pH 7.0. Se utilizó un espectrofluorómetro
ISS K2 (Champaign, IL, USA), que cuenta con un controlador de temperatura tipo
Peltier.
4.4 Ensayos de apagamiento de la Fluorescencia.
Para los ensayos de apagamiento de la fluorescencia intrínseca por efecto de la
acrilamida, se adicionaron alícuotas de 7.0 µL de acrilamida 2.30M variando su
concentración en la celda de 0 a 0.114M. Nuestras enzimas se incubaron en un volumen
inicial de 2.0 mL de amortiguador de fosfatos50 mM a pH 7.0, a una concentración de
50 µg/mL. La concentración de enzima se corrigió por la dilución generada por la
adición de acrilamida. La longitud de onda de excitación fue de 280 nm, mientras que la
emisión de fluorescencia fue registrada a 350 nm. Se utilizó una celda de 1.0 cm de paso
óptico con agitación constante. Los ensayos se realizaron a las temperaturas de 12 ºC,
25 ºC y 38 ºC. Los datos obtenidos del apagamiento de la fluorescencia fueron tratados
de acuerdo a la ecuación de Stern-Volmer.
Donde F0 es la fluorescencia intrínseca en ausencia de acrilamida, F es la intensidad de
fluorescencia a una concentración dada de acrilamida [Q] y KSV es la constante de
Stern-Volmer relacionada con el apagamiento de la fluorescencia por efecto de las
colisiones.

9
Figura 4. Ubicación de los residuos de triptófano en la estructura cristalográfica de la
β-glucosidasa B. Se resaltan los 16 residuos de triptófano en color azul.
5. RESULTADOS Y DISCUSIÓN.
5.1 Ensayos de actividad enzimática
En la tabla 1 se muestran los valores obtenidos de los para metros cinéticos kcat y KM así
como el valor de Vmáx y la eficiencia catalítica para las enzimas H62R/N223Y/M319I y
V323R a 25 ºC. Mientras que en la figura 5 se puede apreciar la tendencia promedio de
las velocidades iníciales con las cuales se determinaron las constantes catalíticas para
las enzimas de estudio. La tendencia de las determinaciones describe adecuadamente el
comportamiento del modelo de Michaelis-Menten.
ENZIMA Vmáx (µmol min-1 mg-1)
KM
(mM)
kcat
(s-1
)
kcat/KM
(s-1
M-1
)
H62R/N223Y/M319I 16,6 ± 0,2 0,2 ± 0,0 14,5 ± 0,2 55657 ± 798
V323R 11,1 ± 0,1 4,1 ± 0,2 9,7 ± 0,1 2403 ± 120
Tabla 1. Constantes catalíticas de ambas enzimas, H62R/N223Y/M319I y V323R a 25
ºC. La desviación estándar (± DE) se determinó con las dos repeticiones de cada
experimento.
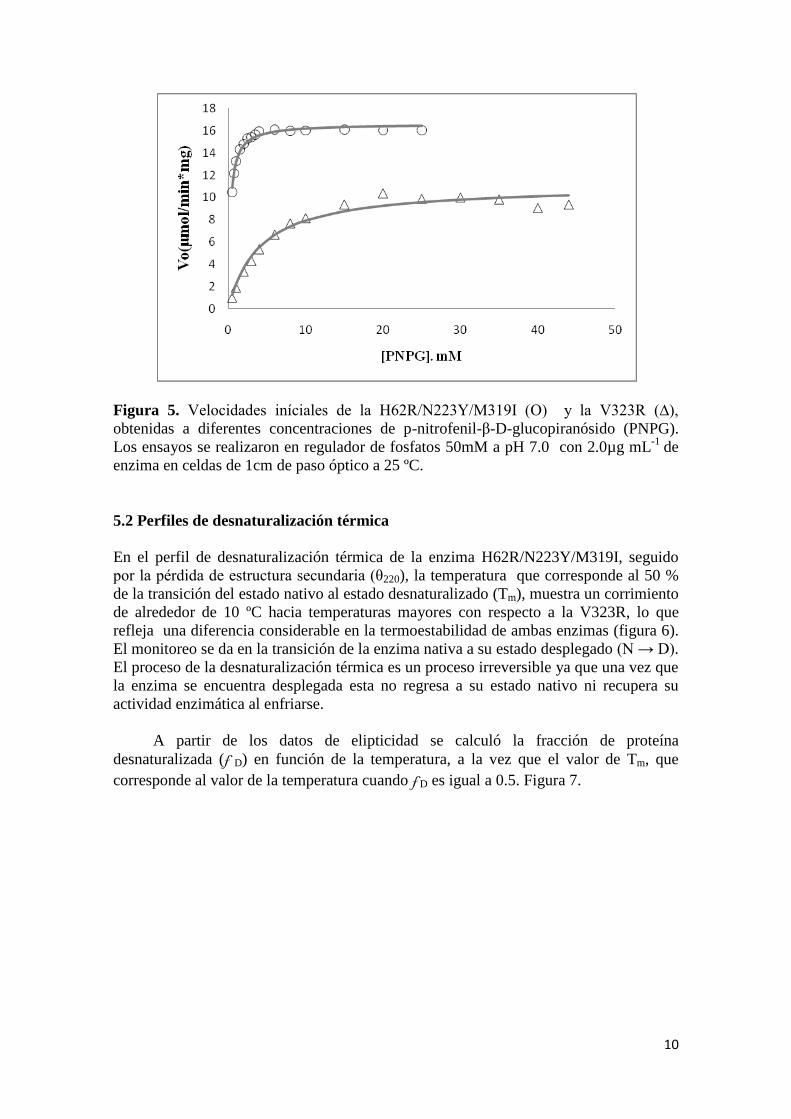
10
Figura 5. Velocidades iníciales de la H62R/N223Y/M319I (O) y la V323R (∆),
obtenidas a diferentes concentraciones de p-nitrofenil-β-D-glucopiranósido (PNPG).
Los ensayos se realizaron en regulador de fosfatos 50mM a pH 7.0 con 2.0µg mL-1
de
enzima en celdas de 1cm de paso óptico a 25 ºC.
5.2 Perfiles de desnaturalización térmica
En el perfil de desnaturalización térmica de la enzima H62R/N223Y/M319I, seguido
por la pérdida de estructura secundaria (θ220), la temperatura que corresponde al 50 %
de la transición del estado nativo al estado desnaturalizado (Tm), muestra un corrimiento
de alrededor de 10 ºC hacia temperaturas mayores con respecto a la V323R, lo que
refleja una diferencia considerable en la termoestabilidad de ambas enzimas (figura 6).
El monitoreo se da en la transición de la enzima nativa a su estado desplegado (N → D).
El proceso de la desnaturalización térmica es un proceso irreversible ya que una vez que
la enzima se encuentra desplegada esta no regresa a su estado nativo ni recupera su
actividad enzimática al enfriarse.
A partir de los datos de elipticidad se calculó la fracción de proteína
desnaturalizada (f D) en función de la temperatura, a la vez que el valor de Tm, que
corresponde al valor de la temperatura cuando f D es igual a 0.5. Figura 7.

11
Figura 6. Desnaturalización térmica de la enzima H62R/N223Y/M319I (∆) y de la
enzima V323R (O). Los dos barridos térmicos se hicieron con una concentración de
20µg mL-1
de enzima y a una velocidad de calentamiento de 2 ºC por minuto en
regulador de fosfatos 50mM pH 7.0, en una celda de 1 cm de paso óptico con agitación
constante.
Figura 7. Fracción de proteína desnaturalizada de la V323R (O) y H62R/N223Y/M319I
(∆), obtenidos a partir de los datos de elipticidad obtenidos en la figura 6. La figura del
recuadro muestra la Tm de ambas mutantes.
12
5.3 Espectroscopia de Fluorescencia
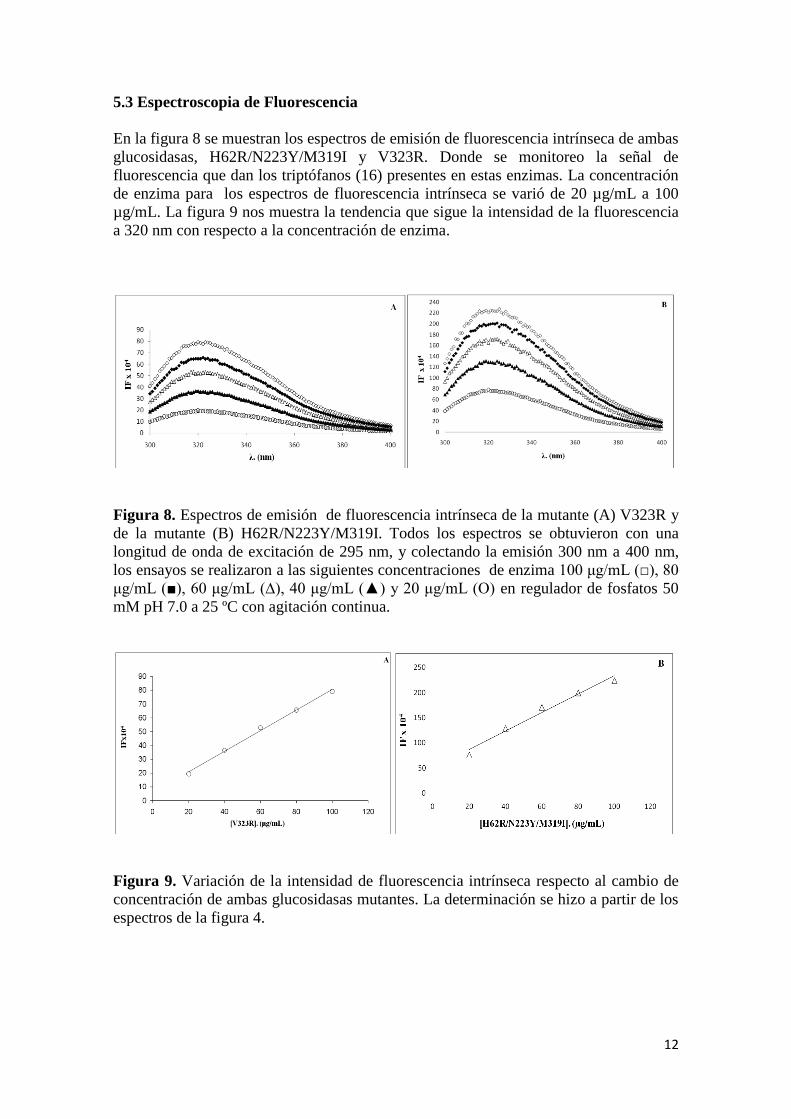
En la figura 8 se muestran los espectros de emisión de fluorescencia intrínseca de ambas
glucosidasas, H62R/N223Y/M319I y V323R. Donde se monitoreo la señal de
fluorescencia que dan los triptófanos (16) presentes en estas enzimas. La concentración
de enzima para los espectros de fluorescencia intrínseca se varió de 20 µg/mL a 100
µg/mL. La figura 9 nos muestra la tendencia que sigue la intensidad de la fluorescencia
a 320 nm con respecto a la concentración de enzima.
Figura 8. Espectros de emisión de fluorescencia intrínseca de la mutante (A) V323R y
de la mutante (B) H62R/N223Y/M319I. Todos los espectros se obtuvieron con una
longitud de onda de excitación de 295 nm, y colectando la emisión 300 nm a 400 nm,
los ensayos se realizaron a las siguientes concentraciones de enzima 100 μg/mL (□), 80
μg/mL (■), 60 μg/mL (∆), 40 μg/mL (▲) y 20 μg/mL (O) en regulador de fosfatos 50
mM pH 7.0 a 25 ºC con agitación continua.
Figura 9. Variación de la intensidad de fluorescencia intrínseca respecto al cambio de
concentración de ambas glucosidasas mutantes. La determinación se hizo a partir de los
espectros de la figura 4.
13
5.4 Ensayos de apagamiento de la fluorescencia.
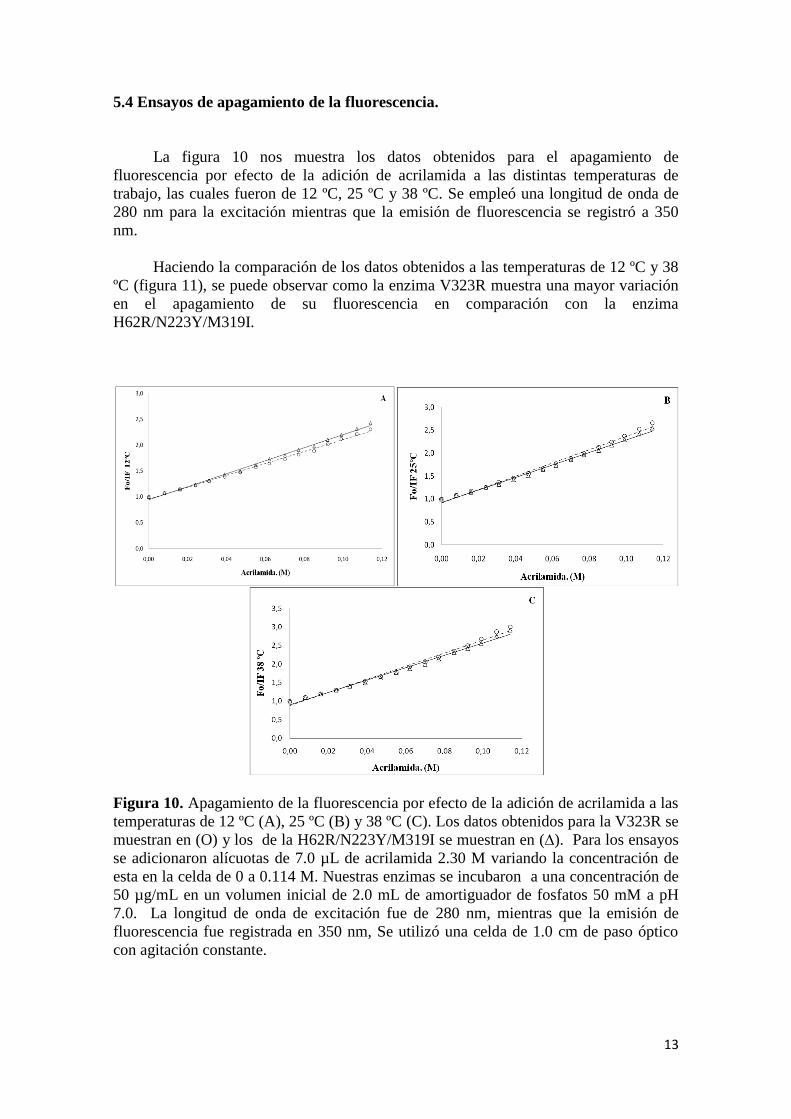
La figura 10 nos muestra los datos obtenidos para el apagamiento de
fluorescencia por efecto de la adición de acrilamida a las distintas temperaturas de
trabajo, las cuales fueron de 12 ºC, 25 ºC y 38 ºC. Se empleó una longitud de onda de
280 nm para la excitación mientras que la emisión de fluorescencia se registró a 350
nm.
Haciendo la comparación de los datos obtenidos a las temperaturas de 12 ºC y 38
ºC (figura 11), se puede observar como la enzima V323R muestra una mayor variación
en el apagamiento de su fluorescencia en comparación con la enzima
H62R/N223Y/M319I.
Figura 10. Apagamiento de la fluorescencia por efecto de la adición de acrilamida a las
temperaturas de 12 ºC (A), 25 ºC (B) y 38 ºC (C). Los datos obtenidos para la V323R se
muestran en (O) y los de la H62R/N223Y/M319I se muestran en (∆). Para los ensayos
se adicionaron alícuotas de 7.0 µL de acrilamida 2.30 M variando la concentración de
esta en la celda de 0 a 0.114 M. Nuestras enzimas se incubaron a una concentración de
50 µg/mL en un volumen inicial de 2.0 mL de amortiguador de fosfatos 50 mM a pH
7.0. La longitud de onda de excitación fue de 280 nm, mientras que la emisión de
fluorescencia fue registrada en 350 nm, Se utilizó una celda de 1.0 cm de paso óptico
con agitación constante.

14
Figura 11. Variación del apagamiento de la emisión de fluorescencia para las enzimas
V323R (o, ●) y H62R/N223Y/M319I (∆, ▲) al agregar alícuotas de acrilamida a 12 ºC
y 38 ºC. Se observa una mayor afectación para la enzima V323R.
6. CONCLUSIONES.
Los parámetros cinéticos (KM; kcat) de las enzimas H62R/N223Y/M319I y V323R
fueron: (0.2 ± 0.0 mM; 14.5 ± 0.2 s-1
) y (4.1 ± 0.2 mM; 9.7 ± 0,1 s-1
) respectivamente.
La enzima H62R/N223Y/M319I muestra un aumento de 10 °C en la Tm respecto a la
V323R, lo que nos habla de su mayor resistencia a la temperatura.
La triple mutante H62R/N223Y/M319I muestra una menor movilidad que la mutante
V323R.
La enzima más termoestable (H62R/N223Y/M319I) muestra entonces una menor
movilidad a temperaturas cercanas a la ambiente (12-38 °C).

15
7. REFERENCIAS
[1] Tesis de Doctorado., Menandro Camarillo Cadena. Nov-2011. “Evaluación
Fisicoquímica de Mutantes Termorresistentes de la β-glucosidasa B de
Paenibacilluspolymyxa”
[2] Tesis de Maestría., Miriam Ordoñez Diaz. (2008). “Diseño de Mutantes
Termorresistentes de la β-Glucosidasa B de Paenibacilluspolymyxa, Empleando
Herramientas de Bioinformática”.
[3] Zubillaga, R. A., García-Hernández, E., Camarillo-Cadena, M., León, M. y
Polaina, J.(2006). Effect of a new ionic pair on the unfolding activation barrier of
β-glucosidase B.Prot Pept Lett. 13:113-118.
[4] Ninfa Ramírez D., José Antonio Serrano R., Horacio Sandoval T. (2006).
Microorganismos extremófilos. Actinomicetos halófilos en México. Revista
Mexicana de Ciencias Farmacéuticas. Vol. 37, numero 003.
[5] Armando Gómez Puyou. (2003). LA COMPLEJIDAD DE LAS PROTEÍNAS:
RELACIÓN ENTRE ESTABILIDAD, FLEXIBILIDAD Y CATÁLISIS. Mensaje
Bioquímico Vol. XXVII.
[6] Jason K. Yano. Y Thomas L. Poulos. (2003). New understandings of
thermostable and peizostable enzymes. CurrOpinBiotechnol. 14:360-365.
[7] Zubillaga, R. A. y Pérez-Hernández, G. (2008). Optimización de interacciones
electrostáticas superficiales como estrategia de termoestabilidad en proteínas., pp.
95-106, eds., El COLEGIO NACIONAL. México.
[8] Tony Collins, Marie-Alice Meuwis, Charles Gerday and Georges Feller. (2003).
Activity, Stability and Flexibility in GlycosidasesFrom Adapted to Extreme
Thermal Environments. J. Mol. Biol. (2003) 328, 419-428.
[9] Murice R. Eftink and Camilo A. Ghiron.Fluorescence Quenching Studies With
Proteins. AnalyticalBiochemistry. 114, 199-227 (1981).
[10] José A. Rodríguez-Martínez, Ricardo J. Solá, Betzaida Castillo, Héctor R.
Cintrón-Colón, Izarys Rivera-Rivera, Gabriel Barletta, KaiGriebenow. Stabilization
of a-Chymotrypsin UponPEGylation Correlates With Reduced Structural
Dynamics.Biotechnol. Bioeng. 2008;101: 1142–1149.
[11] Henrissat, B. y Bairoch, A. (1996). Updating the sequence-based classification
of glycosyl hydrolases. Biochem. J. 316:695-696.
[12] Isorna, P., Polaina, J., Latorre, G, L. Cañada, J. F., González, B., y Sanz, A. J.
(2007).Crystal structures of Paenibacillus polymyxa beta-glucosidase B complexes
reveal the molecular basis of substrate specificity and give new insights into the
catalytic machinery of family I glycosidases. J. Mol. Biol. 371:1204-18.

16
[13] Arrizubieta, M. J. y Polaina, J. (2000). Increased thermal resistance and
modification ofthe catalytic properties of a beta-glucosidase by random mutagenesis
and in vitrorecombination. J. Biol. Chem. 275:28843- 28848.


















