
genetica fenilcetonuria
20
GENETICA REPASO ;)
-
Upload
josselyn-roxana-uribe-matamoros -
Category
Education
-
view
80 -
download
2
Transcript of genetica fenilcetonuria

GENETICA REPASO ;)

Errores innatos del metabolismoCromosómicos: error en un cariotipoMultifactoriales: ambiente + multiples genes.Genéticos: duplicación de un gen

Fenilcetonuria (PKU) La fenilcetonuria es un error innato del metabolismo
producido por la deficiencia de fenilalanina hidroxilasa, se debe a una anomalía génica localizada en el cromosoma 12.
Es un enfermedad genética hereditaria autosómica recesiva. Se trata de una enfermedad infantil metabólica progresiva
severa, que puede producir retraso mental. La fenilalanina es un teratógeno.

Enfermedad genética autosómica recesiva, es decir, los padres son portadores de los genes defectuosos y al ser traspasados de ambos progenitores, la enfermedad se expresa en los descendientes.
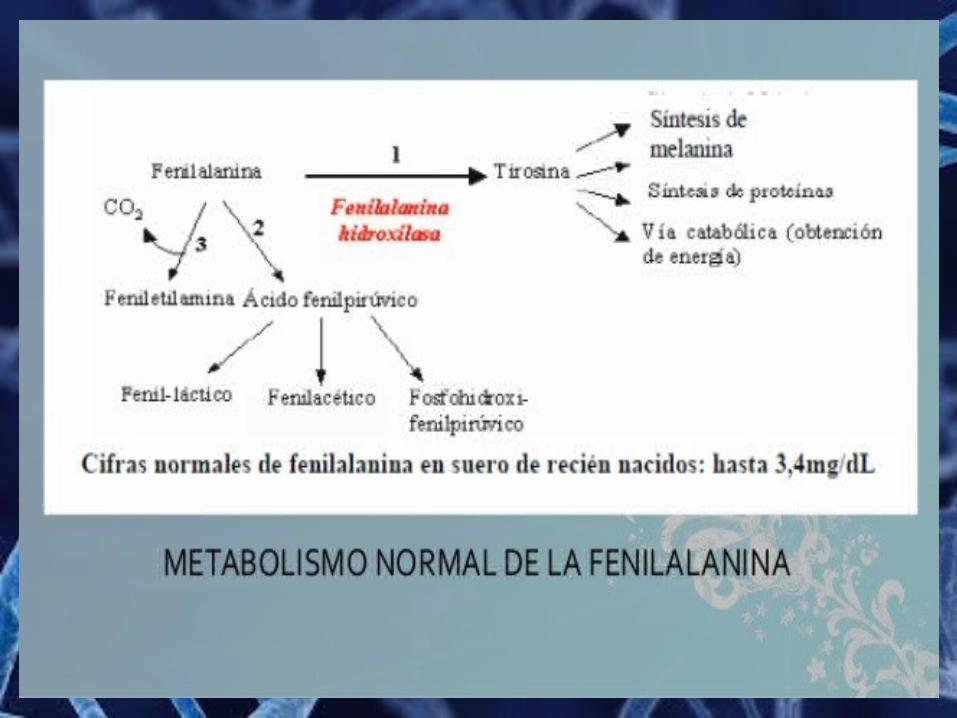
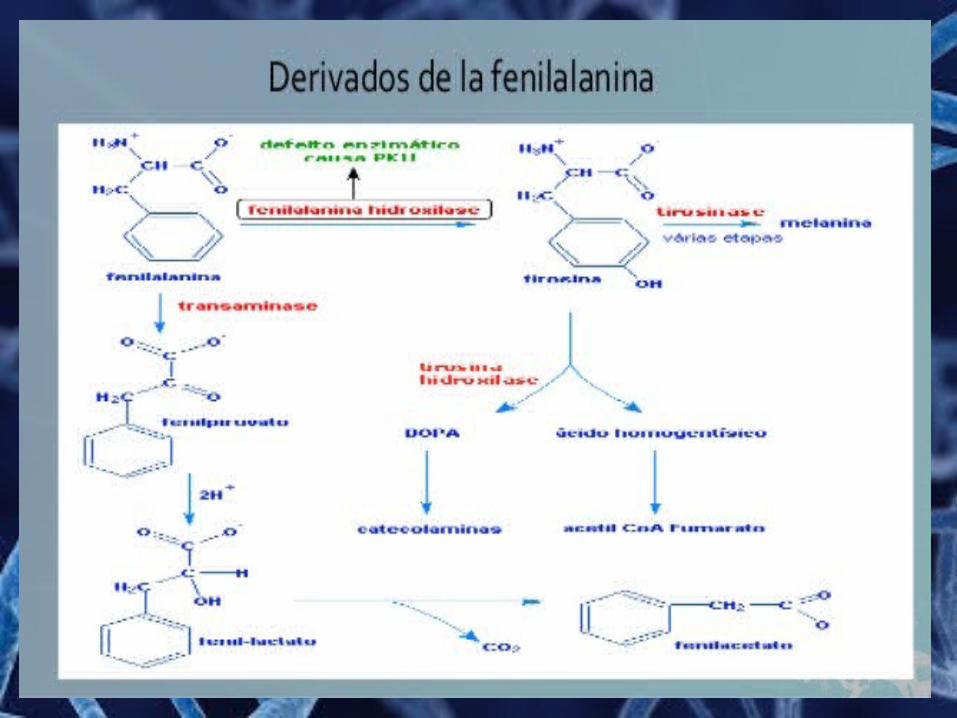
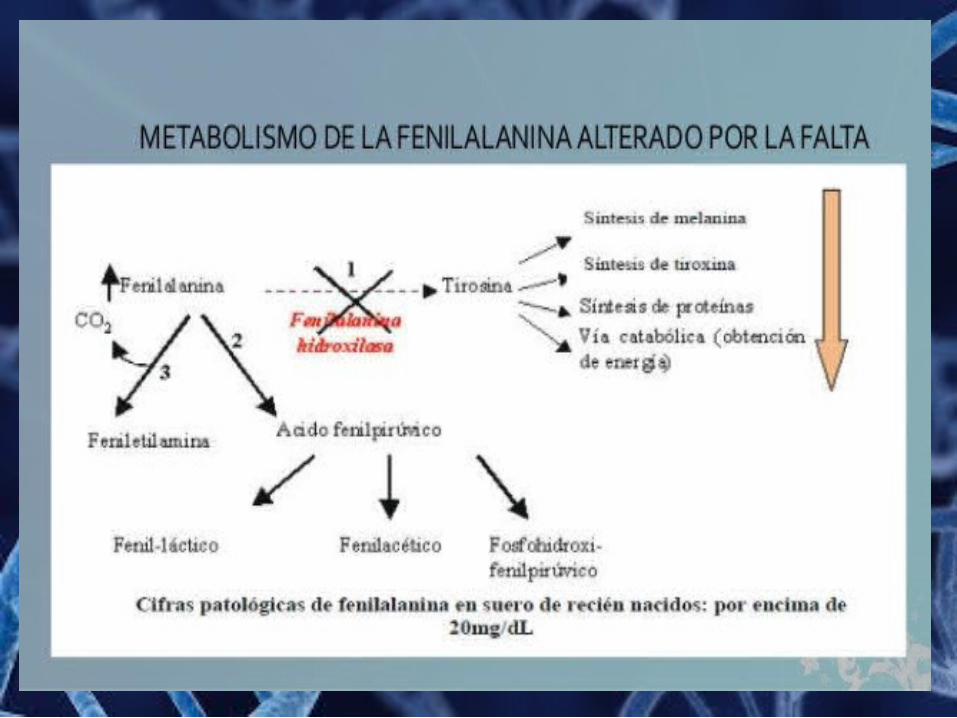

Síntomas
o Retardo mentalo Hiperactividado Convulsioneso Erupción cutáneao Tembloreso Microcefalia
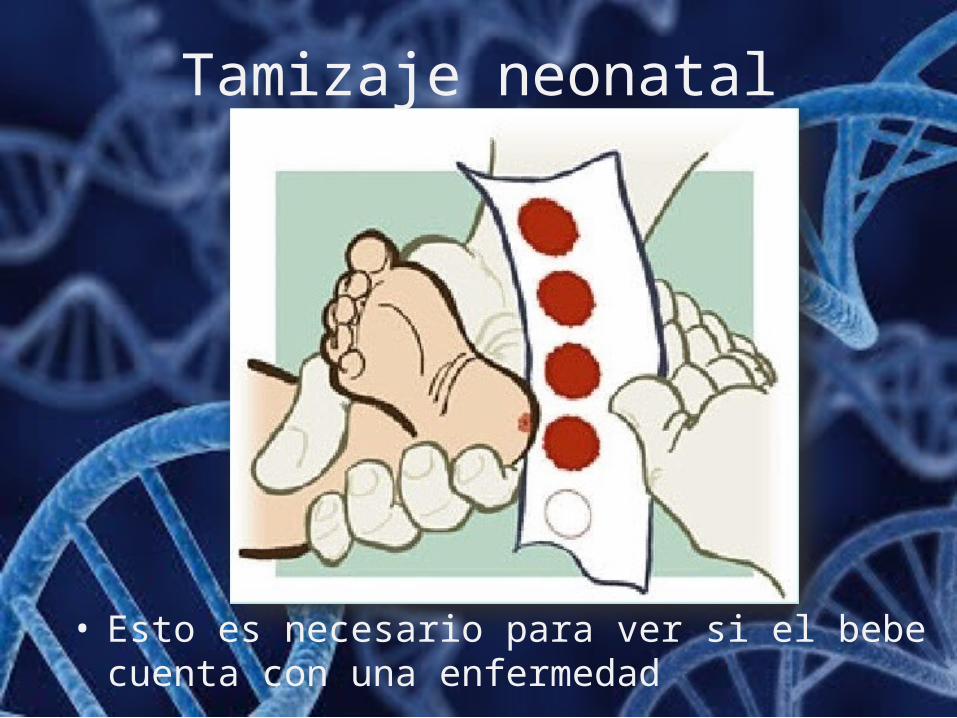
Tamizaje neonatal
• Esto es necesario para ver si el bebe cuenta con una enfermedad

LisosomasEnfermedades lisosomales:• Esfingolipidosis• Mucopolisacaridosis• Oligosacaridosis• Sialidosis• Glicogenosis • Lipofuccinosis

Tay-sachs (Autosómica Recesiva)• Es una de las Gangliosidosis GM2
causada por una deficiencia de Hexosaminidasa A, debida a mutaciones en la subunidad α de dicha enzima.
• Esta enfermedad se desarrolla cuando se acumula grasa en el cerebro.

¿QUÉ SON LAS GANGLIOSIDOSIS GM2? Las Gangliosidosis GM2 son un grupo de enfermedades lisosomales en las que se produce un acúmulo de Gangliósidos GM2 que no se metabolizan bien debido a la deficiencia de unos enzimas llamados Hexosaminidasa A y Hexosaminidasa B o debido a la deficiencia de la proteína activadora de GM2.

Mutaciones en cualquiera de estos tres genes pueden alterar la actividad enzimática de Hexosaminidasa A: • Las que afectan al gen HEXA, alteran a la subunidad α y con ello, la actividad de Hexosaminidasa A, causando la enfermedad de Tay-Sachs. • Las que afectan al gen HEXB, alteran a la subunidad β y, con ello, la actividad de Hexosaminidasa B, causando la enfermedad de Sandhoff. • Las que afectan al gen GM2A, alteran la actividad del activador de GM2, impidiendo así su degradación

Manifestaciones clínicas
• Apatía• Ceguera• ligero retraso mental • irritabilidad convulsiones• disminución del tono
muscular• Sordera• Demencia• Parálisis• degeneración progresiva del
sistema nervioso central

Gauchear ( Autosómica Recesiva) La enfermedad de Gauchear es una enfermedad genética hereditaria que causa la acumulación de depósitos grasos en ciertos órganos y en los huesos. Esta enfermedad puede causar una gran variedad de síntomas.Células de Gauchear • Nuestros cuerpos contienen miles de sustancias activas
llamadas enzimas. En individuos sanos, la enzima glucocerebrosidasa ayuda al organismo a degradar un cierto tipo de molécula grasa (glucocerebrósido). Las personas con la enfermedad de Gauchear no tienen cantidades suficientes de esta enzima. Como resultado, las células se llenan de esta grasa no digerida. Estas células se conocen como células de Gauchear.
Manifestaciones clínicas• La acumulación de las células de Gauchear puede causar
el agrandamiento del bazo y el hígado, anemia, y un número diverso de signos y síntomas. En ciertos casos el cerebro y el sistema nervioso central resultan afectados.

Enfermedad de Fabry(recesiva ligada al X)
La enfermedad de Fabry (EF) es una patología progresiva, hereditaria y multisistémica de almacenamiento lisosómico, caracterizada por manifestaciones neurológicas, cutáneas, renales, cardiovasculares, cocleovestibulares y cerebrovasculares específicas.La enfermedad de Fabry es un trastorno del metabolismo de los glicoesfingolípidos causado por el déficit o ausencia de actividad de la enzima lisosomal alfa-galactosidasa A ligada a mutaciones en el gen GLA (Xq21.3-q22) que codifica la enzima. El déficit de actividad da lugar a la acumulación de globotriaosilceramida (Gb3) en los lisosomas, lo que se cree que desencadena una cascada de eventos celulares.

Manifestaciones clinicas• Dolor• Opacidad de la cornea• Problemas de sudoración• Intolerancia al calor y al ejercicio• Manifestaciones: digestivas, del corazón, de los
riñones , del SNC , psicosociales.
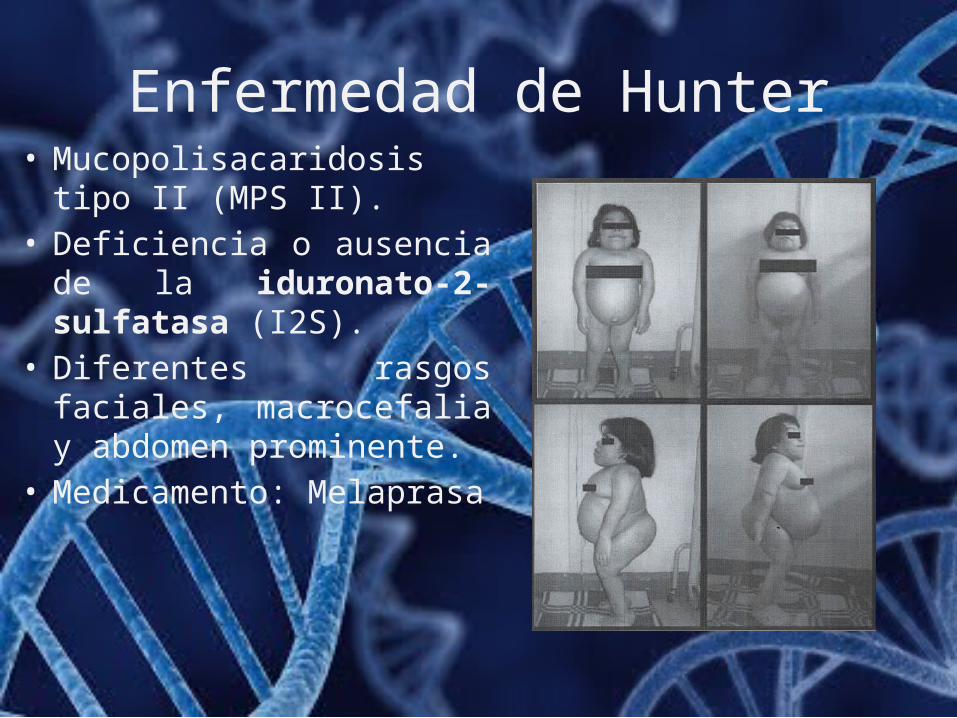
Enfermedad de Hunter• Mucopolisacaridosis tipo II
(MPS II).• Deficiencia o ausencia de la
iduronato-2-sulfatasa (I2S).• Diferentes rasgos faciales,
macrocefalia y abdomen prominente.
• Medicamento: Melaprasa
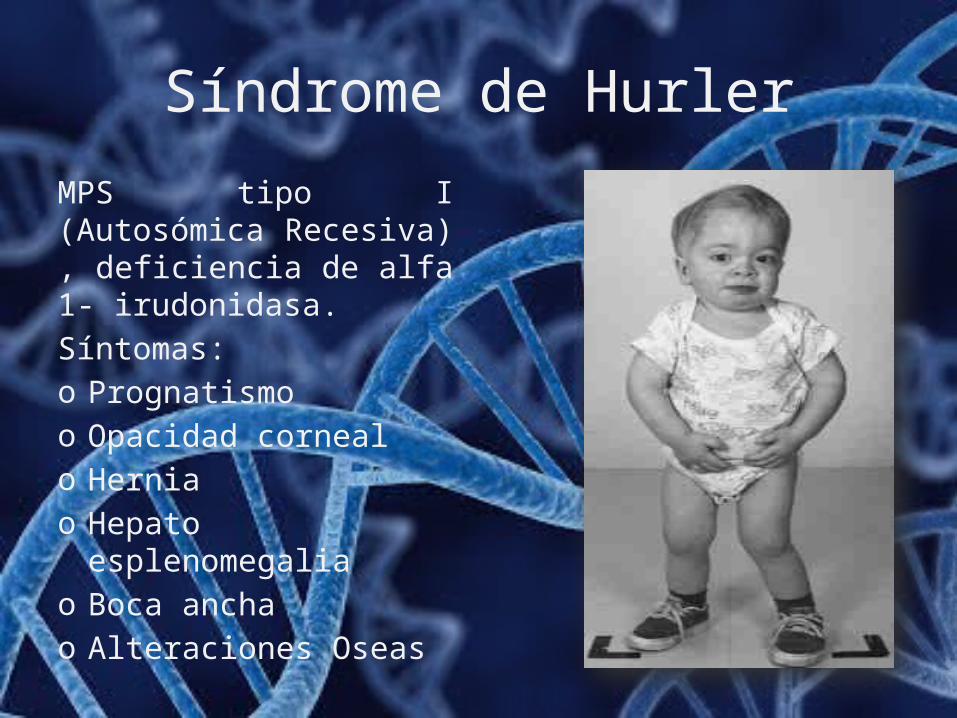
Síndrome de Hurler
MPS tipo I (Autosómica Recesiva) , deficiencia de alfa 1- irudonidasa.Síntomas:o Prognatismoo Opacidad cornealo Herniao Hepato esplenomegaliao Boca anchao Alteraciones Oseas


















