
FISICOQUÍMICA FARMACÉUTICA (0108) UNIDAD 2....
35
1 FISICOQUÍMICA FARMACÉUTICA (0108) UNIDAD 2. FENÓMENOS SUPERFICIALES EN SISTEMAS FARMACÉUTICOS. Mtra. Josefina Viades Trejo 21 de agosto de 2012 2.1. Definición, tipos de superficies e importancia. 1. Superficie . Una superficie es en realidad una interfase = intercara = interfaz 2. Fase. Es una parte (porción) homogénea del sistema, con propiedades intensivas constantes a lo largo de toda la fase. Por ejemplo: un sistema constituido por un cristal de AgBr en equilibrio con una solución de dicha sal tiene dos fases el sólido (cristal) y la disolución. Propiedades intensivas son aquellas propiedades que no dependen de la cantidad de materia en el sistema como la densidad y la presión (una gota de agua y una piscina tienen la misma densidad). Una fase puede constar de varias partes separadas, en el caso anterior, puede haber más de un cristal de AgBr en equilibrio con la disolución y todos los cristales forman parte de la misma fase, o en el caso de una bebida carbonatada todas las burbujas del gas forman una sola fase. La definición de fase no hace referencia a sólidos, líquidos o gases, un sistema puede ser enteramente líquido o sólido y tener más de una fase; dos líquidos casi inmiscibles o dos sólidos por ejemplo diamante y grafito. 3. Interfase . Aquella región en el espacio en la cual el sistema como un todo sufre una transición de una fase a la otra, o también se puede definir como la región a
Transcript of FISICOQUÍMICA FARMACÉUTICA (0108) UNIDAD 2....

1
FISICOQUÍMICA FARMACÉUTICA (0108)
UNIDAD 2.
FENÓMENOS SUPERFICIALES EN SISTEMAS FARMACÉUTICOS.
Mtra. Josefina Viades Trejo
21 de agosto de 2012
2.1. Definición, tipos de superficies e importancia.
1. Superficie. Una superficie es en realidad una interfase = intercara = interfaz
2. Fase. Es una parte (porción) homogénea del sistema, con propiedades intensivas
constantes a lo largo de toda la fase. Por ejemplo: un sistema constituido por un
cristal de AgBr en equilibrio con una solución de dicha sal tiene dos fases el
sólido (cristal) y la disolución.
Propiedades intensivas son aquellas propiedades que no dependen de la
cantidad de materia en el sistema como la densidad y la presión (una gota de
agua y una piscina tienen la misma densidad).
Una fase puede constar de varias partes separadas, en el caso anterior, puede
haber más de un cristal de AgBr en equilibrio con la disolución y todos los
cristales forman parte de la misma fase, o en el caso de una bebida carbonatada
todas las burbujas del gas forman una sola fase.
La definición de fase no hace referencia a sólidos, líquidos o gases, un sistema
puede ser enteramente líquido o sólido y tener más de una fase; dos líquidos
casi inmiscibles o dos sólidos por ejemplo diamante y grafito.
3. Interfase.
Aquella región en el espacio en la cual el sistema como un todo sufre una
transición de una fase a la otra, o también se puede definir como la región a

2
través de la cual las propiedades intensivas del sistema cambian “desde” aquellas
correspondientes a una fase “hasta” las de la otra fase.
Si la interfase no tiene una energía libre positiva, no puede existir como una
frontera estable entre dos fases. Para que la interfase sea estable, debe tener una
energía libre positiva interfacial tal, que deba realizarse trabajo para aumentar la
interfase; en caso contrario y de no existir otras fuerzas externas como la fuerza
de la gravedad que separa las fases por densidad y otros factores, entonces
factores aleatorios como el movimiento Browniano distorsionan la interfase
hasta que las fases se mezclan.
Existen los siguientes tipos de interfases:
i. Sólido - Sólido
ii. Sólido – Líquido
iii. Sólido – Gas
iv. Líquido – Líquido
v. Líquido - gas
4. Fenómenos Interfaciales.
Un fenómeno interfacial puede definirse como aquel relacionado a la interacción
de por lo menos el seno de una fase (sólida o líquida) con otra fase (sólida,
líquida o gaseosa) o con el vacío, en la región estrecha en que sucede la
transición de una fase a la otra (interfase).
Los fenómenos interfaciales son los siguientes:
i. Tensión superficial.
ii. Adhesión.
iii. Cohesión
iv. Extensión
v. Adsorción

3
2.2. Tensión superficial e interfacial
La energía cohesiva que une a los átomos o moléculas, no es igual para todos sino
que difiere para los de la superficie porque éstos están rodeados de menos vecinos
que los del interior por tanto su energía es mayor que la de las moléculas en el
interior.
Trabajo superficial en sistemas de un componente.
Para crear una superficie es necesario romper enlaces y desplazar átomos
circundantes por tanto se ejerce un trabajo sobre el sistema.
Si el trabajo se realiza en condiciones de T y p constantes, entonces en el equilibrio
el trabajo superficial reversible es:
dAW S
PT γδ =, (2.2.6)
Se usa δ porque no es una diferencial exacta pues depende de las condiciones en
que se realiza el proceso. Este trabajo es comparable al trabajo reversible necesario
para incrementar el volumen de un sistema a p constante (pdV). El término γγγγ tiene
un significado similar pero en dos dimensiones a la p y se puede considerar como
una presión a lo largo del plano superficial que se opone a la creación de más
superficie por tanto es una fuerza por unidad de longitud dina / cm o erg/cm2. dV se
corresponde con dA.
Tensión Superficial.

4
Definición: Fuerza perpendicular a la superficie del líquido dirigida hacia el seno
de éste. Fuerza en dinas que actúa en dirección perpendicular, sobre toda línea de 1
cm de longitud en la superficie. Actualmente N/m
Es la fuerza responsable de la tendencia de la superficie a contraerse y hacer que el
líquido asuma el estado de energía mínima (superficie mínima). Ej. Gotas y
burbujas.
Tensión interfacial.
Tensión interfacial intermedia entre los valores de las tensiones superficiales.
En procesos reversibles δWS es igual a la variación de energía libre total de la
superficie la cual es igual a GS por el área de la interfase
)(, AGdW SS
PT =δ (2.2.7)
La creación de una interfase va acompañada de una G de formación positiva y esta
resistencia del sólido o líquido a formar una superficie define muchas propiedades
de las interfases.
Si la nueva superficie se obtiene incrementando el área, por ejemplo si se
incrementa el área del sólido o cristal por fisura, o se somete el sólido a tensión a
γα/β = γβ/α
α γα
γβ β

5
altas temperaturas en condiciones en que los átomo pueden difundirse hacia la
superficie, en estas circunstancias no se mantiene el estado de tensión por tanto GS
no es proporcional a A y su derivada parcial es cero
dAA
GAGdA
A
AGW
PT
SS
PT
SS
PT
∂∂
+=
∂∂
=,,
,
)(δ (2.2.8)
dAGW SS
PT =,δ (2.2.9)
Igualando (2.2.6 y 2.2.9)
SG=γ (2.2.10)
PT
S
A
GG
,
∂∂
==γ (2.2.11)
La derivada parcial en la ecuación (2.2.11) es el incremento de energía libre total
del sistema/unidad de área superficial incrementada
Tensión superficial como fuerza por unidad de longitud.
Como resultado de la tensión superficial, para aumentar la superficie han de
moverse moléculas desde el seno hacia la superficie contra las fuerzas de atracción
intermolecular, por lo que es necesario efectuar un trabajo, o sea, suministrar
energía.

6
A
B C
D
L
f
dS
Para definir este W se puede pensar en un bastidor de alambre que tiene una varilla
móvil y poniendo en él una película líquida (solución jabonosa) extendida sobre el
área ABCD, la que puede estirarse aplicando una fuerza (peso) en la varilla móvil
de longitud L, la cual actúa contra la γ (si se quita el peso la superficie se vuelve a
contraer) por tanto la γ es la fuerza por unidad de longitud a lo largo de la varilla
existente en la superficie de la película y que se opone al aumento de la superficie.
La longitud total a considerar es 2L (película bidimensional)
L
f
2=γ (2.2.12)
f fuerza necesaria para romper la película, para calcular el trabajo realizado se
despeja f de la ecuación (2.2.12).
Lf 2×= γ (2.2.13)

7
Cuando la varilla está en AD y se cuelga el peso para aumentar el área en una
cantidad dS el dW (fuerza x distancia) será
dSLfdSdW ××== 2γ (2.2.14)
2LxdS =dA es el aumento de área superficial producido por la extensión de la
película de jabón
dAdW γ= (2.2.15)
Para una variación finita:
AW ∆= γ (2.2.16)
W es el trabajo realizado, o sea, el incremento de la energía libre superficial.
2.3. Efecto de Temperatura.
La tensión superficial disminuye al aumentar la temperatura. Por el aumento de la
energía cinética, el movimiento térmico y las tendencias de escape.
S
APAP
S
STT
G−=
∂∂
=
∂∂
,,
γ (2.2.1)
Van der Waalls y Guggenheim

8
n
cT
T
−=
10γγ γγγγ→→→→0; conforme T→→→→Tc (2.3.2)
γγγγ0 es la tensión superficial del líquido en el cero absoluto, Tc es la temperatura
crítica del líquido o sólido. n~1 para metales y n>1 para líquidos orgánicos.
En una gráfica de γ vs T la pendiente es -SS y es negativa, en ocasiones la
superficie se hace más ordenada al aumentar la T y entonces la pendiente es
positiva. Otras veces la superficie se desordena más rápido que el resto de de la
fase condensada y γγγγ→→→→0 antes de lo predicho por la ecuación (2.3.7).
2.4. Tensión superficial en sistemas multi componentes. Isoterma de adsorción
de Gibbs.
El potencial químico del componente “i” en un sistema multi componente está
definido como:
PTni
ii
j
n
GG
,,
∂∂
==−
µ (2.4.1)
Esto es el potencial químico es igual a la energía libre molar parcial del
componente, la derivada parcial (∂G/∂ni) es la variación de la energía libre total del
sistema con respecto a ni. La variación total de G puede expresarse:
i
i i
dnn
GdAVdPSdTdG ∑
∂∂
+++−= γ (2.4.2)

9
Sustituyendo (2.4.1 en 2.4.2)
i
i
idndAVdPSdTdG ∑+++−= µγ (2.4.3)
Considerando el cambio de G al variar el área en una cantidad dA transfiriendo
desde el interior dni moles hacia la superficie a T y P constantes:
i
i
iPT dndAdG ∑−= µγ, (2.4.4)
dG a T y P constantes corresponde a (∂G/∂A) a T y P constantes. El signo negativo
se debe a que la concentración de “i” en el bulto (seno) disminuye por lo que en la
superficie hay un incremento correspondiente.
dACdn S
ii =− (2.4.5)
CSi es la concentración de “i” en la superficie en mol/cm2. en un sistema
multicomponente SG≠γ (ver ecuación 11 al inicio este tema) ahora dAGA
G S=∂∂
y
entonces la ecuación (2.4.4) queda:
( ) dACdAdAG S
i
i
iPT
S ∑+= µγ, (2.4.6)
dividiendo entre dA y despejando:
S
i
i
i
S CG ∑−= µγ (2.4.7)

10
i
i
S
i
S
i
i
i
S dCdCdGd µµγ ∑∑ −−= (2.4.8)
dGS es función de T, P, A y CSi. En una superficie plana P es constante por tanto su
derivada parcial es cero, si el A es constante también su derivada parcial es cero,
entonces:
S
i
i
i
SSdCdTSdG ∑+−= µ (2.4.9)
Sustituyendo (2.4.9) en (2.4.8) se obtiene la ecuación de Gibbs:
i
i
S
i
S dCdTSd µγ ∑−−= (2.4.10)
A T constante el primer término desaparece, definiendo AnC i
S
i /= entonces:
i
i
i dA
nd µγ ∑
−= (2.4.11)
Para un sistema de dos componentes:
22
11 µµγ d
A
nd
A
nd
−
−= (2.4.12)
Si el componente 1 es el disolvente y su concentración en la superficie es cero:

11
22 µγ dA
nd
−= (2.4.13)
Definiendo (n2/A) = Γ como el exceso de concentración en la superficie
22 µγ dd Γ−= (2.4.14)
2022 lnaRT+= µµ y 22 ln aRTdd =µ
Despejando Γ y sustituyendo dµ2
−=
−=Γ
ada
d
RT
a
ad
d
RT
γγ 2
22 ln
1 (2.4.15)
Si la solución es ideal (infinitamente diluida), la actividad se puede sustituir por la
concentración:
−=ΓdC
d
RT
C γ2 (2.4.16)
Se pueden tener dos situaciones:
i) El soluto tiende a ir a la superficie, entonces Γ (+)
ii) El soluto tiende a permanecer en el bulto, entonces Γ (-).
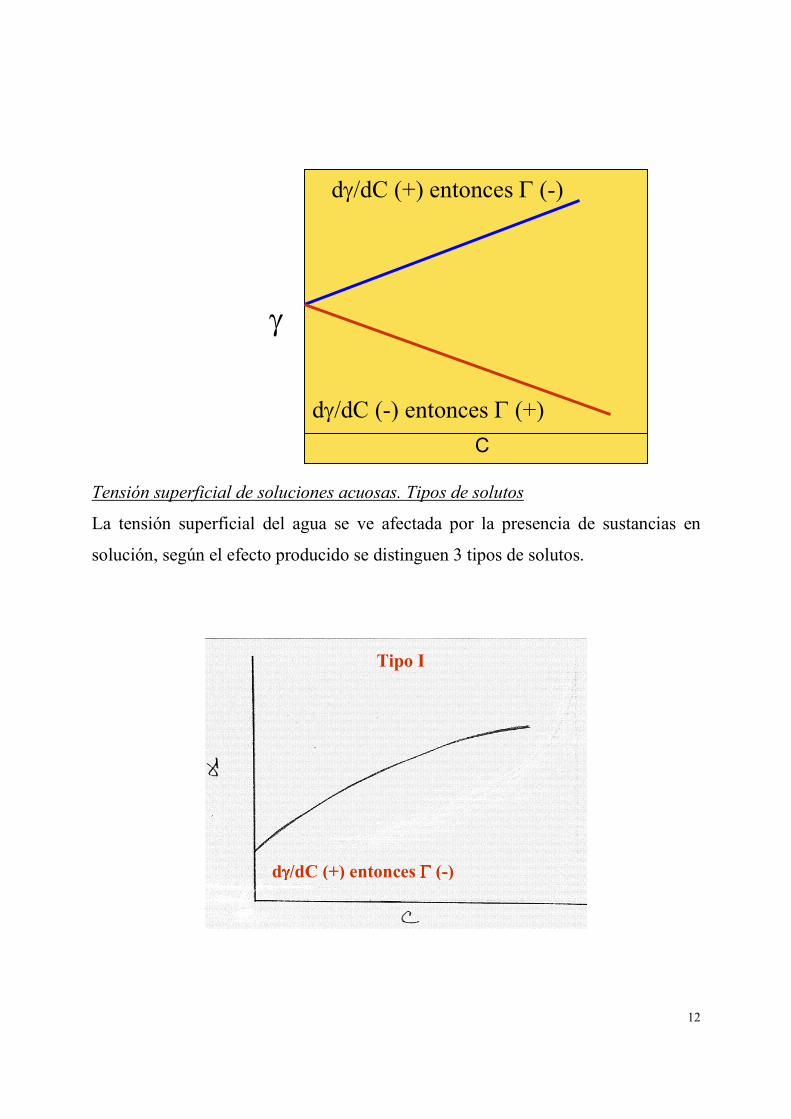
12
Tensión superficial de soluciones acuosas. Tipos de solutos
La tensión superficial del agua se ve afectada por la presencia de sustancias en
solución, según el efecto producido se distinguen 3 tipos de solutos.
γ
C
dγ/dC (+) entonces Γ (-)
dγ/dC (-) entonces Γ (+)
Tipo I
dγγγγ/dC (+) entonces ΓΓΓΓ (-)

13
Son sustancias polares que se solvatan fácilmente, por lo que tienden a permanecer
en el seno de la solución, atrayendo hacia ellas a las moléculas de agua y
sumándose a las fuerzas de cohesión entre estas últimas, produciendo un
incremento en la tensión superficial. Ejemplos son las sales y algunas sustancias
orgánicas polares como azúcares.
Son sustancias orgánicas de bajo peso molecular como alcoholes, aminas, ácidos
carboxílicos, cuyos grupos polares son afines al agua pero sus pequeñas cadenas de
hidrocarburo son hidrófobas, de modo que migran a la interfase dejando hacia fuera
del agua la cadena hidrófoba y hacia adentro los grupos polares, estos últimos
atraen hacia ellos a las moléculas del agua venciendo parcialmente las fuerzas de
cohesión y disminuyendo la tensión superficial.
Tipo II
dγγγγ/dC (-) entonces ΓΓΓΓ (+)

14
Presentan igual comportamiento que los solutos Tipo II, pero su efecto sobre la
tensión superficial del agua es mucho más marcado, porque sus cadenas hidrófobas
son mucho mayores lo que incrementa su tendencia a emigrar a la superficie,
igualmente su parte polar o hidrófila presenta mayor número de grupos polares
(OH, COOH, NH2). Sus pesos moleculares son mucho mayores. Son los
verdaderos tensoactivos
2.5. Agentes tensoactivos.
Son sustancias que cuando se dispersan en el seno de un líquido tienden a emigrar y
concentrarse en la superficie o interfase, se llaman también agentes
superficialmente activos. Winsor los llama agentes anfífilos o anfifílicos
Se caracterizan por tener un grupo polar (hidrófilo, lipófobo, cabeza) y un grupo no
polar (hidrófobo, lipófilo, cola) lo que les permite formar una especie de puente
entre ambas fases, debe estar equilibrado en cuanto a poseer la cantidad de grupos
Tipo III
dγγγγ/dC (-) entonces ΓΓΓΓ (+)

15
hidrosolubles y liposolubles para que se concentre en la interfase y por lo tanta
produzca un descenso en la tensión superficial. Si es muy hidrófilo permanece en el
interior de la fase acuosa y no ejerce efecto sobre la interfase, lo mismo si es muy
lipófilo, por tanto deben estar equilibrados de tal modo que cuando se dispersen
inicialmente en cualquiera de las fases emigre a la interfase y se oriente.
Propiedades físicas de soluciones de tensoactivos.
Formación de micelas: Los tensoactivos en solución diluida se comportan como
electrolitos normales, pero a una concentración dada y bien definida ocurren
cambios bruscos en su presión osmótica, conductividad eléctrica y tensión
superficial.
Mc. Bain explicó estos cambios como debidos a la formación de micelas que son
agregados de los iones del tensoactivo con las cadenas del hidrocarburo hacia el
interior y los grupos hidrófilos hacia el exterior en contacto con el medio acuoso.
La concentración que da lugar a la formación apreciable de micelas se llama
concentración micelar crítica (cmc), como la cmc es la concentración a la que
ocurren los cambios bruscos mencionados, tales propiedades pueden utilizarse para
determinar la cmc.

16
La micelización es un mecanismo diferente a la adsorción por medio del cual
disminuye la tensión superficial. Lo deseable es tener una baja cmc. La agitación
térmica y la repulsión eléctrica entre las cabezas se oponen a la formación de
micelas, por lo que una baja cmc estaría favorecida por:
Aumento de la longitud de la cadena de hidrocarburo, en una serie homóloga
cada CH2 reduce la cmc a la mitad de su valor.
Disminución de temperatura, movimiento térmico que se opone a la formación
de cmc disminuye al disminuir T.
Adición de sales simples (KCl) que reducen la repulsión entre cabezas por
efecto de pantalla.
Estructuras de las micelas. Hay varios modelos propuestos:
Hartley micela esférica, la mayor parte de los conocimientos experimentales
apoyan este modelo.
Mc. Bain micela laminar que propone que las moléculas se organizan en una
doble capa.
Harkins micela cilíndrica.

17
Clasificación de tensoactivos:
� Iónicos
1. Aniónicos
2. Catiónicos
3. Zwitter iónicos
� No iónicos
Balance Hidrófilo-Lipófilo (BHL).
Griffin ideó una escala arbitraria de valores que sirviera como medida del balance
hidrófilo-lipófilo, escala que nos permite establecer un intervalo de la máxima
eficacia para cada tipo de agente. 10-20 hidrófilos, de 0-10 lipófilos
1 – 3 La mayoría de los agentes antiespumantes.
3 – 8 Agentes emulgentes w/o
6 – 9 Agentes humectantes y de extensión
8 – 16 Agentes emulgentes o/w
12 – 16 Agentes detergentes
16 – 20 Agentes solubilizantes
Moore y Bell idearon una clasificación similar asignando a los anfífilos del tipo del
polioxietileno los llamados índices H/L
100×
=n
E
LH
(2.5.1)
E es el número de unidades de óxido de etileno y n es el número de C en la cadena.
Cálculos de HLB
I. Para un tensoactivo.

18
1.- Para algunos ésteres de ácidos grasos y alcoholes poli hidroxilados
−=A
SBHL 120 (2.5.2)
Donde S es el índice de saponificación y A es el índice de acidez
2.- Cuando no es posible obtener buenos índices de saponificación (derivados de la
cera de abeja)
5
PEBHL
+= (2.5.3)
Donde E % en peso de las cadena oxietilénica y P el % en peso de los grupos
alcohólicos polihidroxilados,
3.- Si la molécula está formada solamente por grupos oxietilénicos, de aquí surge
que el valor máximo de HLB sea 20.
5
EBHL = (2.5.4)
4.- Greenwald, Brown y Finnegan proponen un método en el cual la disolución del
agente en dioxano-benceno se valora con agua hasta un punto final de turbidez
y los resultados se expresan como el Num de mililitros de agua necesarios para
producir una turbidez pronunciada.
II. Para mezclas de tensoactivos.
1.- Para mezclas de tensoactivos. Método de la cruz, solo para dos tensoactivos, no
se necesita conocer el porcentaje de cada uno dado un HLB óptimo o deseado

19
BA
bD
HLBHLB
HLBHLBA
−
−=
)(100% (2.5.5)
AB %100% −= (2.5.6)
2.- Método de la suma, para “n” tensoactivos dado el % de cada uno, da el HLB
para esas cantidades específicas
100
)%...()%( nBHLABHLBHL nA
T
×+×= (2.5.7)
3.- Método gráfico, solo para dos tensoactivos dado un HLB deseado
2.6. Medición de tensión superficial. Tensiómetros.
2.6.1. Método del Ascenso Capilar.
Cuando fuerzas de adhesión superan a fuerzas de cohesión se da el ascenso capilar
HLBD HLBD
HLBA
HLBB
%TB 0 100
0 0
20 20

20
21 FF = (2.6.1.1)
θγπ cos21 rF = (2.6.1.2)
ghrF ρπ 22 = (2.6.1.3)
θρ
γcos2
grh= (2.6.1.4)
Si θθθθ = 00 entonces cos θθθθ = 1
grhργ2
1= (2.6.1.5)
h
2r

21
2.6.2. Tensiómetro del Anillo (Du Nouy)
Se basa en la medición de la fuerza necesaria para separar un anillo de platino-
iridio de la superficie del líquido. La fuerza se suministra por medio de un hilo de
torsión y se registra en dinas/cm en un disco calibrado
γπ )2(2 RF = (2.6.2.1)
R
F
πγ
4= (2.6.2.2)
f es un factor de corrección empírico en función del R y de r (radio del anillo en su
grosor) se encuentra en tablas
Rf
F
πγ
4= (2.6.2.3)
2.6.3. Método de la placa (Wilhelmy)
El principio es el mismo del tensiómetro del anillo. Del brazo de una balanza se
cuelga una placa de mica o un cubreobjetos de microscopio, que se sumerge
parcialmente en un líquido, el depósito donde está el líquido se baja gradualmente y
se anota el tirón sobre la balanza en el punto de despegue. Para una lámina de
longitud “x” y anchura “y” y peso “w” suponiendo un ángulo de contacto igual a
cero:
γ)(2 yxWWdesp +=− (2.6.3.1)

22
)(2 yx
WWdesp
+
−=γ (2.6.3.2)
Otra forma de calcular la tensión superficial es:
PFF a γ+= (2.6.3.a)
Donde F es la fuerza hacia abajo cuando la placa toca la superficie (fuerza total),
Fa es también una fuerza hacia abajo pero cuando la placa está suspendida
libremente en el aire y P es el perímetro de la placa.
P
FF a−=γ (2.6.3.b)
2.7. Interfases curvas. Ecuación de Young-La Place.
Establecen las condiciones de equilibrio mecánico en una superficie curva entre dos
fases. Consideremos una superficie esférica de radio r que separa dos fases α y β;
en la superficie hay una tensión γγγγ, por lo que el equilibrio mecánico entre las fases
se mantiene a diferentes presiones Pαααα > Pββββ en tanto que en las superficies planas
ambas presiones son iguales.
r
Pα
Pβ

23
La ecuación para la superficie esférica es:
βα PPP −=∆ (2.7.1)
El volumen de la fase αααα aumenta en una expansión reversible en una cantidad dV,
el W necesario es:
dAdVPPW γδ βα =−= )( (2.7.2)
3
3
4rV π= drrdV 24π=
24 rA π= rdrdA π8=
)8()4)(( 2 rdrdrrPP πγπβα =− (2.7.3)
rP
γ2=∆ (2.7.4)
Capilaridad: La elevación capilar se debe a un balance entre adhesión y cohesión
el cual a su vez determina el ángulo de contacto entre el líquido y las paredes del
tubo.
Fuerzas de cohesión son las que se ejercen entre moléculas de igual naturaleza
química.

24
θ
R
r
Fuerzas de adhesión son las que se ejercen entre moléculas de distintas especies
químicas.
El ángulo de contacto se define como el ángulo en el líquido entre la pared del
tubo y la tangente a la superficie del líquido en la pared.
R es el radio de curvatura del casquete esférico (menisco), r es el radio del tubo, θθθθ
es el ángulo de contacto
r
R=θcos (2.7.5)
Si θθθθ < 900 moja (menisco cóncavo)
θθθθ = 900 superficie plana
θθθθ > 900 no moja (menisco convexo)

25
L
L
L
γL
Rr
θcos1= (2.7.6)
RP
θγ cos2=∆ (2.7.7)
γθ
2cos
PR∆= (2.7.8)
2.8. Trabajos de adhesión y de cohesión. Coeficiente de extensión.
Trabajo de cohesión.
Trabajo de cohesión: es la energía necesaria para separar en contra de fuerzas de
cohesión las moléculas del líquido que se extiende para que pueda fluir sobre le
sustrato (sólido o líquido)
LcW γ2= (2.7.9)

26
Trabajo de adhesión
Trabajo de adhesión: es la diferencia en la energía libre entre dos estados, es decir,
cuando el líquido que está extendido es separado en contra de fuerzas de adhesión
de la superficie sobre la que se extendió (sólida o líquida)
LSSLAW /γγγ −+= (2.7.10)
Coeficiente de extensión.
Harkins: explica la extensión de aceite sobre una superficie acuosa como sigue: Si
el aceite gusta más de sí mismo que de el agua no se extiende sobre de ella, pero si
lo hará si gusta más del agua que de sí mismo.
Coeficiente de extensión o esparcimiento es la diferencia entre los trabajos de
adhesión y cohesión
cA WWS −= (2.7.11)
S
L
γL
S
L
γS γS/L

27
Sustituyendo las expresiones para los trabajo:
LLSSLS γγγγ 2)( / −−+= (2.7.12)
)( / LSLSS γγγ +−= (2.7.13)
Se pueden presentar dos situaciones:
i) S (+) si WA>WC HAY EXTENSION (MOJA)
ii) S (-) si WA<WC NO HAY EXTENSIÓN (NO MOJA )
El balance de fuerzas superficiales en el equilibrio si θ es pequeño
gSLSgL /// cos γγθγ =+ (2.7.14)
gL
LSgS
/
//cos
γ
γγθ
−= (2.7.15)

28
La ecuación (2.7.15) define el punto de contacto o línea de contacto en su posición
de equilibrio, excepto en dos casos extremos: θ = 00 (moja) y θ>900 (no moja)
θ = 00 γS/g > γS/L + γL/g
θ > 00 γS/L > γS/g + γL/g
El gas desplaza al líquido sobre la superficie sólida.
2.8. Adsorción. La adsorción puede definirse como la tendencia de un componente del sistema a concentrarse en la interfase, donde la composición interfacial es diferente a las composiciones correspondientes al seno de las fases. Hay una clara diferencia entre el fenómeno de adsorción y el de absorción, en el segundo existe una penetración física de una fase en la otra; sin embargo es factible que ambos sucedan simultáneamente, y en este caso puede ser muy difícil separar los efectos de ambos fenómenos, inclusive un fenómeno puede afectar al otro.
Adsorción Absorción
Sorción
29
El fenómeno de adsorción es de particular relevancia en la ciencia de los coloides y superficies. El proceso de adsorción de átomos y moléculas en las interfases, es una de las principales formas en que las interfases de alta energía pueden modificarse para disminuir la energía total del sistema. La adsorción puede ocurrir en cualquier tipo de interfase (L-G, S-G, L-S), sin embrago las diferentes características de las interfases sólidas y líquidas hace necesario un análisis particular de cada caso. En los procesos de adsorción hay dos aspectos que deben ser considerados; 1) El efecto de la adsorción sobre la energía interfacial del sistema en el equilibrio
(termodinámica) 2) La rapidez del proceso de adsorción (cinética) Definición de términos:
� Adsorbato: Sustancia que se pega (adsorbe) en la superficie. � Adsorbente: Superficie sobre la que sucede la adsorción. � x Cantidad de adsorbato adsorbida en una cantidad dada de adsorbente. � m Cantidad de adsorbente sobre la que sucede la adsorción. � x/m = y Cantidad adsorbida por gramo de adsorbente. � ym Cantidad adsorbida por gramo de adsorbente en la monocapa. � ΣΣΣΣ Área superficial específica del adsorbente (m2/g). � σσσσ Área molecular del adsorbato (m2/molécula). � θθθθ Fracción de superficie cubierta por el adsorbato. � (1 – θθθθ) Fracción de superficie no cubierta por el adsorbato. Isoterma de adsorción
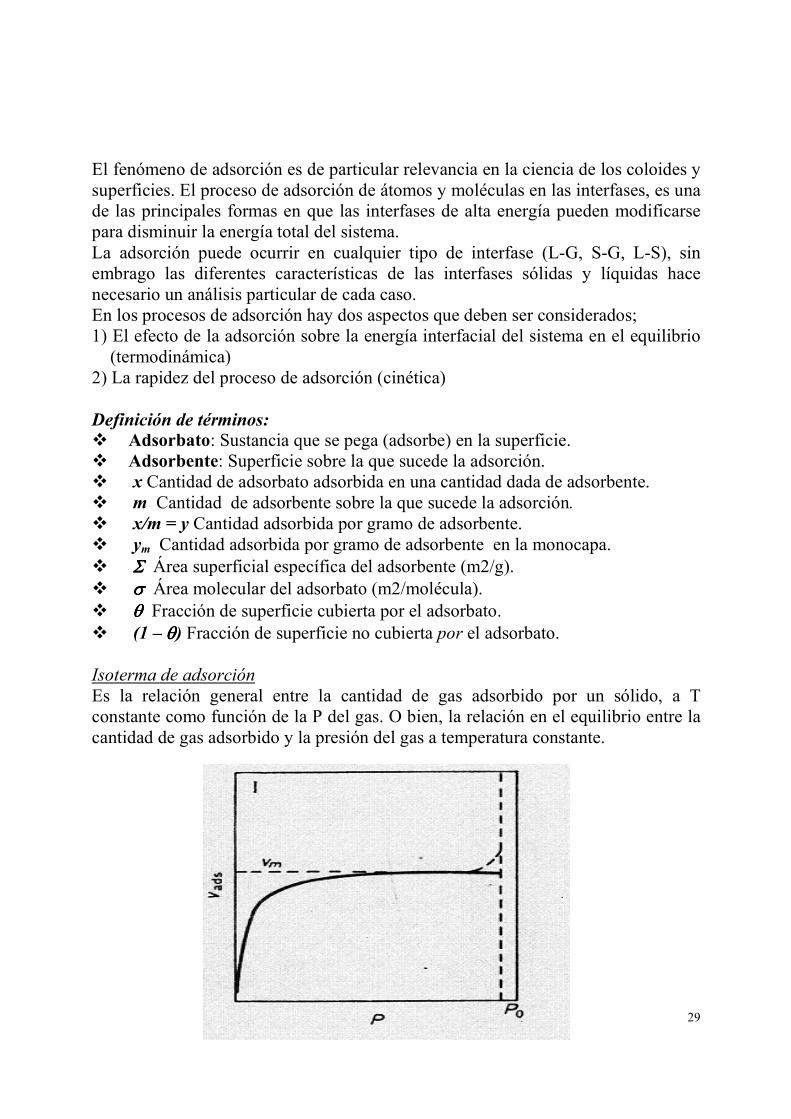
Es la relación general entre la cantidad de gas adsorbido por un sólido, a T constante como función de la P del gas. O bien, la relación en el equilibrio entre la cantidad de gas adsorbido y la presión del gas a temperatura constante.

30
Según sus características la adsorción puede clasificarse en dos tipos:
a) Adsorción física (fisisorción): fuerzas de van der Waalls, ∆H~ 20 KJ/mol, reversible ⇒ desorción ⇐ si T ↑ y P↓, no es de naturaleza específica (sitios activos), formación de multicapas, el adsorbato conserva su identidad.
b) Adsorción química (quimisorción): fuerzas análogas al enlace químico (atracción entre iones opuestos o coulombicas, coordinadas o covalentes), ∆H~ 200 KJ/mol, irreversible ⇒ no hay desorción ⇐ si T ↑ y P↓ a menos que sean cambios muy drásticos, es de naturaleza específica (sitios activos), formación de monocapas, el adsorbato puede perder su identidad,
Factores que afectan a la cantidad adsorbida
1. Cantidad del adsorbente (m): Superficie disponible 2. P o C del adsorbato: Cantidad de material disponible 3. Temperatura: Generalmente la adsorción es exotérmica
Si T, P ó C y m dadas, entonces Y (x/m) se ve afectada por:
1. Naturaleza del adsorbato. Solubilidad, la cantidad adsorbida generalmente es inversamente proporcional a la solubilidad, esto se conoce como la Regla de Lundelius. pH la adsorción aumenta conforme el grado de ionización disminuye y alcanza su máximo cuando la ionización es cero.
monocapa
Pérdida de identidad
desorción
multicapa

31
2. Naturaleza del adsorbente. Área superficial específica que depende a su vez del tamaño de partícula (grado de subdivisión) y de la porosidad.
3. Naturaleza del disolvente. Su afinidad con el adsorbato. Tipos de isotermas.
Existen 5 tipos o perfiles de isotermas I-V. Para explicarlos se han propuesto diferentes ecuaciones. Isoterma de Langmuir. 1. Langmuir intenta explicar la isoterma Tipo I, como una adsorción en monocapa
(monomolecular) en la cual gracias al equilibrio adsorción-desorción no hay formación de monocapas.
2. El límite de capacidad del adsorbente es cuando la monocapa está completa (región asintótica en la gráfica).
3. La velocidad de adsorción es proporcional a la P y a la fracción de superficie NO cubierta
)1( θ−= Pkv aa 4. En tanto que la velocidad de desorción solo depende de la fracción de superficie
cubierta.
θdd kv =

32
5. En el equilibrio de adsorción – desorción se tiene que (va = vd)
θθ daa kPkPk =−
6. Si despejamos la fracción de superficie cubierta (θ); dividimos entre kd y definiendo b = ka / kd
bP
bP
Pkk
Pk
ad
a
+=
+=
1θ
+=
pbyyy mm
1111
1/y
1/p
pendiente
my
y=θ
bp
bpyy m
+=1
pybyy
p
mm
11+=
p/y
p
pendiente
7. A presiones bajas la cantidad adsorbida es directamente proporcional a la presión

33
bPy
y
m
==θ bPyy m=
8. A presiones altas en cambio se alcanza el límite de la capacidad del adsorbente (monocapa completa)
myy = 9. A presiones intermedias la cantidad adsorbida depende del coeficiente de
adsorción o constante de equilibrio (b) y por tanto también de la temperatura. 10. La limitante más importante de la isoterma de Langmuir es que supone que
el calor de adsorción es independiente del recubrimiento de la superficie. Isoterma de Freundlich.
1. Aunque esta isoterma tiene un origen empírico, puede demostrarse teóricamente
considerando que la magnitud del calor de adsorción varía exponencialmente con el recubrimiento de la superficie.
2. En esta isoterma no hay un recubrimiento límite ni se propone una adsorción monomolecular, sino multimolecular.
3. A presiones moderadamente bajas la dependencia de la cantidad adsorbida con la presión presenta un comportamiento del tipo:
nkPy1
= K y 1/n son constantes. El exponente 1/n varía entre 1 y 0.1.
pn
ky log1
loglog +=
Log p
pendientelog yordenada

34
Isoterma de Brunauer, Emmett y Teller (BET).
Brunauer, Emmett y Teller extienden el tratamiento de Langmuir a la adsorción multimolecular. Debido a la similitud entre las fuerzas responsables de la adsorción física y las de licuefacción (vander Waals), la adsorción en las superficies planas y convexas continua hasta que la superficie queda cubierta con una capa multi molecular del líquido. Igualando las velocidades de condensación y evaporación en las diversas capas se obtiene la ecuación BET. Suponiendo que la energía característica de adsorción del vapor corresponde a la primera capa, en tanto que la energía de licuefacción del vapor sirve para las siguientes capas. p0 presión de vapor de saturación, c diferencia de energía entre las moléculas adsorbidas en la primera capa y las de las capas siguientes, Vm capacidad de adsorción en la primera capa
pendiente
ordenada)( 0 ppv
p
−
0pp
( )( )
00
11
p
p
cV
c
cVppV
p
mm
−+=
−
( )( )
00
11
p
p
cV
c
cVppV
p
mm
−+=
−
−≅
RT
EEc L1exp

35
pendienteordenadayV mm +
==1
myordenadac
∗=
1
Determinación del área superficial específica del adsorbente
Σ es función de grado de subdivisión, porosidad (número y tamaño de poros).
Método BET para adsorbatos gaseosos
Para adsorbatos en solución:
σ00 NRT
yp m
=Σ
σ0Nym=Σ


















