Documento cinética química
59
CINÉTICA QUÍMICA 1. INTRODUCCIÓN La cinética química trata de la velocidad y el mecanismo de las reacciones químicas. Este tópico es de gran interés en la Química del agua debido a que en muchos casos la velocidad a la cual una reacción procede hacia el equilibrio, y no la condición de equilibrio en sí, es la que determina el diseño y funcionamiento de un proceso de tratamiento y el comportamiento de sistemas de aguas naturales. En el tratamiento de las aguas y aguas residuales, por ejemplo, la oxidación química de compuestos orgánicos por cloro y ozono, la precipitación de fosfatos de calcio, carbonato de calcio, e hidróxido de magnesio, y la oxidación química de hierro (II) y manganeso (II) son reacciones cuyas velocidades controlan al diseño y eficiencia del sistema de tratamiento. En aguas naturales, las velocidades de reacción controlan diversas reacciones tales como la precipitación, disolución, oxidación-reducción y las concentraciones existentes no son aquellas predichas por los cálculos de equilibrio. Estas situaciones de no equilibrio son particularmente comunes para reacciones que involucran oxidación-reducción y precipitación-disolución. Por ejemplo, con sólo cálculos de equilibrio podemos predecir que sería imposible que existieran sulfuros en un agua con oxígeno -1-
-
Upload
steven-moreno -
Category
Documents
-
view
709 -
download
0
Transcript of Documento cinética química

CINÉTICA QUÍMICA
1. INTRODUCCIÓN
La cinética química trata de la velocidad y el mecanismo de las reacciones químicas. Este
tópico es de gran interés en la Química del agua debido a que en muchos casos la velocidad
a la cual una reacción procede hacia el equilibrio, y no la condición de equilibrio en sí, es la
que determina el diseño y funcionamiento de un proceso de tratamiento y el
comportamiento de sistemas de aguas naturales.
En el tratamiento de las aguas y aguas residuales, por ejemplo, la oxidación química de
compuestos orgánicos por cloro y ozono, la precipitación de fosfatos de calcio, carbonato
de calcio, e hidróxido de magnesio, y la oxidación química de hierro (II) y manganeso (II)
son reacciones cuyas velocidades controlan al diseño y eficiencia del sistema de
tratamiento. En aguas naturales, las velocidades de reacción controlan diversas reacciones
tales como la precipitación, disolución, oxidación-reducción y las concentraciones
existentes no son aquellas predichas por los cálculos de equilibrio. Estas situaciones de no
equilibrio son particularmente comunes para reacciones que involucran oxidación-
reducción y precipitación-disolución. Por ejemplo, con sólo cálculos de equilibrio podemos
predecir que sería imposible que existieran sulfuros en un agua con oxígeno disuelto. Sin
embargo datos de la Bahía de San Francisco en 1962 mostraron que el sulfuro puede ser
detectado en aguas que contiene hasta 3 mg/L de oxígeno disuelto, debido a que la
reacción entre el oxígeno y el sulfuro en soluciones acuosas diluidas no es rápida.
La información derivada de la cinética química nos ayuda a entender por qué, cuando
mezclamos los gases hidrógeno y oxígeno, no hay reacción, mientras que en presencia de
un poco de platino o una chispa, una mezcla de estos dos gases producirá agua con
violencia explosiva. De nuestra investigación de cinética química debemos entender por
qué algunas reacciones de oxidación-reducción ocurren lentamente en ausencia de
microorganismos mientras en su presencia ocurren rápidamente. Por ejemplo, las
predicciones termodinámicas o de equilibrio nos llevan a concluir que el amoniaco se oxida
a nitratos en aguas oxigenadas y que una solución estéril y aireada de cloruro de amonio es
-1-

estable indefinidamente; sin embargo, si introducimos microorganismo del género de
nitrosomas y nitrobacterias se llevará a cabo una rápida conversión del ión amonio en
nitritos y luego a nitratos.
2. COLISIONES DE LAS ESPECIES REACCIONANTES
Con pocas excepciones, las colisiones entre las especies reaccionantes son necesarias para
que ocurran las reacciones químicas. Aunque las colisiones facilitan que las especies
reaccionantes se hallen lo suficiente cercanas para que la reacción ocurra, no todas las
colisiones satisfactorias producen una reacción química.
Las colisiones entre dos especies (colisión bimolecular) son más frecuentes que la colisión
simultánea de tres moléculas (trimolecular) o más especies. Por ejemplo, en el aire a
condiciones de laboratorio ordinarias donde 0.1% del volumen del gas está ocupado por
moléculas gaseosas, una molécula choca con otra (colisión bimolecular) aproximadamente
109 veces por segundo. Tres moléculas colisionan simultáneamente (colisión trimolecular) a
una velocidad cerca de 105 veces por segundo. Podemos entonces razonar sobre la base de
mayor frecuencia de colisión bimolecular, que ésta forma es la responsable de las
reacciones químicas.
Una línea similar de razonamiento puede seguirse para las sustancias que reaccionan en
solución. El número de colisiones entre las especies diferentes al solvente en una solución
es solamente un poco mayor que en la fase gaseosa. Luego, para las mismas
concentraciones y temperaturas, las reacciones en solución deben ocurrir aproximadamente
a la misma velocidad que en la fase gaseosa. Sin embargo, las especies reaccionantes,
especialmente sustancias iónicas, frecuentemente interaccionan con el solvente, y esto
puede afectar considerablemente la velocidad a la cual ocurren las colisiones.
-2-

Las diferencias entre las reacciones en fase líquida y en fase gaseosa ocurren debido al
llamado “efecto de jaula”. En una fase gaseosa cuando dos especies reaccionantes chocan y
después rebotan ellas llegan a estar separadas por una gran distancia en un período muy
corto. Sin embargo en solución, las moléculas de solvente que rodean a las especies
colisionantes sirven para atraparlas luego de la colisión e inmediatamente colisionar con las
especies que rebotan. Además en muchos casos, los reactantes son devueltos así que otra
colisión entre ellos puede ocurrir. Debido a esto, pueden ocurrir colisiones sucesivas de las
mismas moléculas de un solvente.
Podemos además, aumentar el número de colisiones entre las especies reaccionantes
aumentando la temperatura y si el medio reaccionante es un gas comprimido, aumentando
la presión.
3. ORIENTACIÓN DE LOS REACCIONANTES
Las reacciones entre especies que no requieren especial orientación durante la colisión para
llevarse a cabo tienden a ser más rápidas que las reacciones entre especies que deben estar
adecuadamente alineadas para que la colisión resulte en una reacción. El llamado “efecto de
orientación” explica particularmente por qué muchas colisiones entre especies
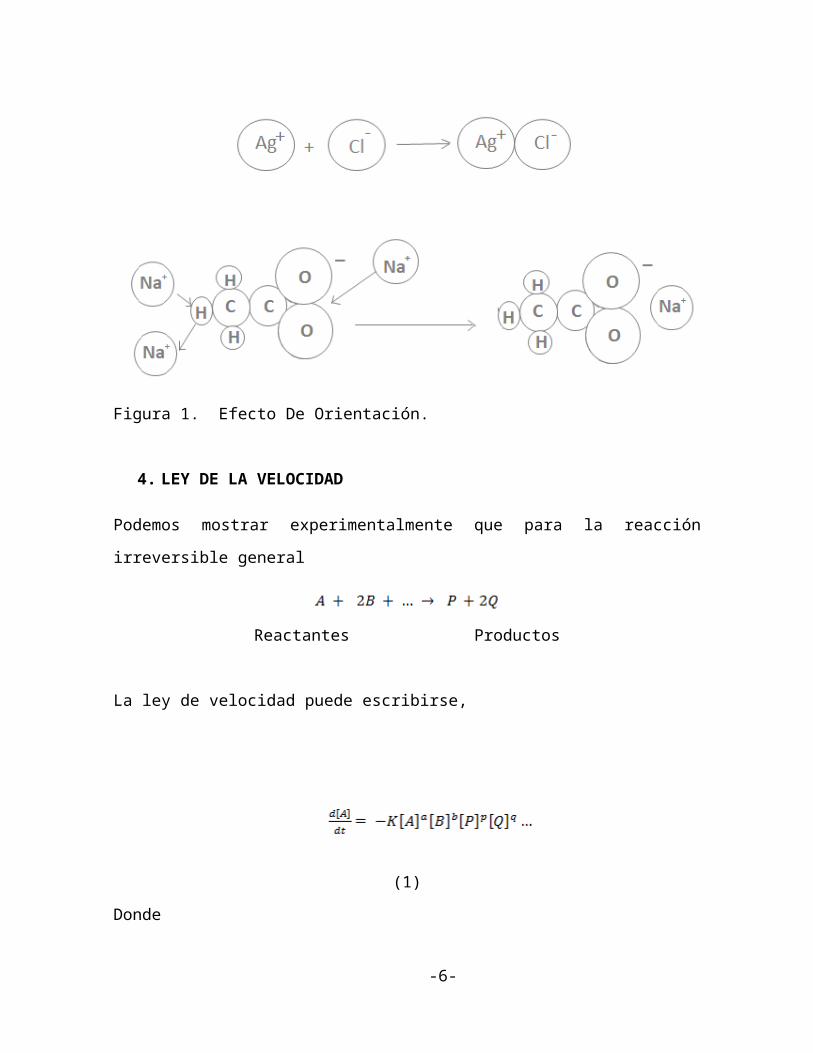
reaccionantes no son satisfactorias en producir una reacción. Por ejemplo el ión Ag+ y el
ion esférico Cl- se combinan rápidamente para formar el precipitado AgCl(s) cuando están
presentes en una solución sobresaturada.
Un gran porcentaje de colisiones entre estas especies es exitoso, en dar lugar a reacciones
debido a que su forma esférica no requiere orientación (figura 1).
La formación de acetato de sodio sólido de soluciones de acetato de sodio sobresaturadas es
lenta en comparación y, las soluciones sobresaturadas pueden existir por largos periodos sin
formación de precipitado. Muchas colisiones entre el ión esférico Na+ y el ión linear
CH3COO- (acetato) ocurren pero no dan lugar a reacción porque solamente la colisión entre
el Na+ y el oxígeno final de CH3COO- (figura 1) da lugar a reacción. Una colisión entre el
Na+ cualquiera y otra parte del ión acetato no puede formar precipitado de acetato de sodio.
-3-

Figura 1. Efecto De Orientación.
4. LEY DE LA VELOCIDAD
Podemos mostrar experimentalmente que para la reacción irreversible general
Reactantes Productos
La ley de velocidad puede escribirse,
(1)
Donde
-4-

El término [ ] significa concentración en moles/L. Se pueden utilizar otras unidades de
concentración en la ley de la velocidad pero utilizando éstas tendremos las mismas
unidades de concentración para cada especie y lograremos que tanto los valores numéricos
y las unidades de las constantes de la velocidad de reacción difieran de las encontradas
cuando se usan concentraciones moleculares.
De acuerdo con la estequiometria de la reacción, esto es, el número relativo de moles de las
especies reaccionantes y el número relativo de moles de los productos que se están
formando a medida que la reacción se lleva a cabo, podemos establecer que,
(2)
Porque 1 mol de A reacciona con dos (2) moles de B y así sucesivamente, y 1 mol de P es
formado por cada mol de A que reacciona y así sucesivamente. Podemos determinar el
orden de reacción de la ley de velocidad.
El orden de cualquier reaccion es
(3)
Mientras que el orden con respecto a A es a, el orden con respecto a B es b, y así
sucesivamente. Si la reacción es irreversible, entonces p, q, … los exponente de los
productos de la reacción, son generalmente cero. Por ejemplo, si
-5-
Entonces, se puede decir que la reacción es de primer orden respecto a A, de segundo orden
respecto a B y de tercer orden total. Es importante tener en cuenta que el orden de la
reacción generalmente no se determina por la estequiometria de la reacción total; para
determinar el orden se requiere experimentación a nivel de laboratorio.
El siguiente ejemplo ilustra varios puntos que son importantes para el buen entendimiento
de la ley de velocidad:
EJEMPLO 1
El amoniaco, NH3 es un constituyente común en muchas aguas naturales y residuales. El
reacciona con el ácido hipocloroso (HOCl), desinfectante, en solución, para formar
monocloramina, NH2Cl, como sigue,
1.
La constante de velocidad, K, se encontró experimentalmente 5.1 x106 (litros /mol segundo)
a 25 °C. La ley de velocidad determinada fue
2.
(a) ¿Cuál es el orden total de la reacción?
(b) ¿Qué porcentaje de descenso en la velocidad de reacción ocurre si la concentración
de cada reaccionante es reducida a un 50%?
(c) Determine el valor de la constante de velocidad cuando las concentraciones son
expresadas en unidades de mg/L en lugar de moles/L.
SOLUCIÓN
(a) La ecuación 2 puede escribirse más explícitamente como
-6-

Donde a=1 y b= 1, El orden total de la reacción es a + b = 2
Determinando las unidades de la constante de velocidad para esta reacción de
segundo orden,
(b) Si asumimos que las concentraciones de NH3 y HOCl son X y Y respectivamente, la
velocidad de reacción es entonces,
Cuando X y Y son reducidas cada una un 50% la velocidad de reacción llega a ser,
La nueva velocidad de reacción es entonces el 25% de la velocidad de reacción
inicial.
(c) Cuando las concentraciones HOCl y NH3 son expresadas en moles/ Litro, la
contantes de velocidad puede escribirse,
(1)
-7-

Llamando K ´= constante de velocidad cuando las concentraciones son expresadas
en mg/L, podemos establecer
(2)
Puesto que 1 mol NH3/L = 17.000 mg NH3/L.
(3) mg NH3/L = [NH3] x 17.000
Combinando (a), (b) y (c) tenemos:
Puesto que K= 5.1 x 106 L/mol. Segundo
Las formas integradas de las leyes de velocidad son muy útiles para analizar los datos de
velocidad para determinar las constantes de velocidad y el orden de reacción. Vamos a
considerar primero la reacción irreversible,
-8-

La cual tiene la ley de velocidad
Para determinar el comportamiento de [A] como función del tiempo, debemos integrar la
expresión de velocidad con respecto al tiempo. Haremos esto para varios valores del orden
de reacción, n. Cuando n=0, el orden de reacción es cero, y
(4)
Luego de integrar, tenemos,
(5)
Donde [A]0 = concentración de A al t=0, esto es, la concentración inicial de A. El tiempo de
“vida media”, t1/2, o el tiempo al cual el 50% de la concentración ha reaccionado puede
obtenerse de la ecuación (5) estableciendo [A] = 0.5 [A]0 cuando t= t1/2. Entonces
Cuando n= 1, la reacción es de primer orden, con respecto a A y a la reacción total, y podemos escribir,
(6)
Reorganizando la ecuación (6) y resolviendo la integral,
-9-

Encontramos
(7)
O
(8)
Examinando la ecuación (7) sugiere que la constante de velocidad K puede determinarse
experimentalmente de una gráfica de la [A] versus t, la cual tiene una pendiente de –K.
También, de la ecuación (8), cuando [A] = 0.5 [A]0, encontramos que la vida media será
Si la reacción es de orden mayor a primer orden, podremos entonces escribir,
(9)
Integrando, obtenemos
O = -kt (10)
-10-

Si n=2, por ejemplo, la reacción es de segundo orden, tanto con respecto a A y
globalmente, y podemos escribir,
(11)
Y la vida media es
Para una reacción de segundo orden involucrando un solo reactante, podríamos determinar
la constante de velocidad K graficando versus t para dar lugar a una línea recta con
pendiente K de acuerdo con la ecuación (11).
Vamos ahora a examinar un caso más complejo de reacción irreversible en el cual dos
reactantes dan productos. Podemos escribir.
(12)
La cual tiene la ley de velocidad
(13)
Cuando a= 1 y b=1, la reacción es de segundo orden en total y de primer orden con respecto
tanto a A como a B.
Existen dos situaciones generales que debemos examinar. Estas son cuando [A]0 = [B]0 y
cuando [A]0 ≠ [B]0. Para el primer caso,
-11-

(14)
De la estequiometria de la reacción en ecuación (12), podemos ver que por cada molécula
de A que reacciona, una molécula de B reaccionará. Las concentraciones de A y B
decrecerán a la misma velocidad y puesto que ellas inicialmente eran iguales significa que
ellas serán siempre iguales. Podemos además sustituir [A] para [B] y dar
La cual, luego de integrada, da la misma expresión de la ecuación (11).
Cuando a=b y [A]0 ≈ [B]0 es útil utilizar la sustitución
(16)
(17)
Donde X es la concentración en moles/L de cada especie que ha reaccionado.
Diferenciando la ecuación (16) se obtiene.
(18)
De la ecuación (14)
-12-

(19)
Y sustituyendo para [A] y [B] de las ecuaciones (16) y (17) en la ecuación (19) obtenemos,
(20)
Integrando, reorganizando y sustituyendo para X de las ecuaciones (16) y (17) obtenemos
(21)
Una gráfica de datos experimentales en la forma Ln ([B] / [A]) versus tiempo da lugar a
una línea recta con una pendiente de ([B]0 – [A]0) K y puesto que [A]0 y [B]0 son conocidos,
K puede determinarse como K = pendiente / ([B]0 – [A]0). Es importante observar que la
ecuación (21) es válida solamente para la reacción estequiométrica dada en la ecuación
(12). Si la estequiometria es diferente, la forma integrada de la ley de velocidad también
será diferente. Por ejemplo, para la reacción
(22)
Para la condición inicial de [A]0 = 2[B]0 y la ley de velocidad de
(23)
La forma integrada de la expresión de velocidad es
-13-

(24)
Para obtener K de los datos experimentales para una reacción que sigue la ley de velocidad,
debemos graficar versus tiempo y obtener la pendiente la cual es 8K. Luego
Otro orden de reacción puede ser aquí considerado, la llamada reacción de seudo-primer
orden. La reacción,
(25)
La cual tiene la ley de velocidad
(26)
Puede ser tratada como una reacción de primer orden si uno de los reactantes está presente
en tal exceso que su concentración es virtualmente invariable durante el curso de la
reacción. Po ejemplo, si el reactante B está presente en gran exceso sobre el reactante A,
entonces
(27)
Donde K´= K [B] y K´ es una constante de seudo-primer orden. Esta ecuación puede ser
integrada para dar,
-14-

(28)
Cuando las reacciones se llevan a cabo en soluciones acuosas diluidas teniendo el agua
como uno de los reactantes, es a menudo posible asumir que el agua está presente en gran
exceso. Por ejemplo, la hidrólisis de una molécula de sacarosa (disacárido) para dar dos
moléculas de monosacárido (una molécula de glucosa y fructosa) puede ser escrita como
Sacarosa Glucosa Fructosa
Experimentos muestran que esta reacción es de segundo orden en total, pero debido a que la
concentración del agua permanece constante en solución acuosa diluida, puede considerarse
como una reacción de seudo-primer orden.
Una técnica experimental común usada para el estudio de las velocidades de reacción es
hacer cada uno de los reactantes presentes en gran exceso. La dependencia de la velocidad
de reacción sobre la concentración de otros reactantes puede entonces ser estudiada.
El siguiente ejemplo ilustra un procedimiento para determinar el orden de reacción y la
constante de velocidad.
EJEMPLO 2.
El peróxido de hidrógeno, H2O2 es un agente oxidante que a menudo se usa en el proceso
de purificación del agua. El se descompone rápidamente en el agua en presencia de un
catalizador el dióxido de manganeso (MnO2). Dado que es irreversible, se encontró que
Determine la constante de velocidad y el orden de esta reacción.
-15-

SOLUCIÓN
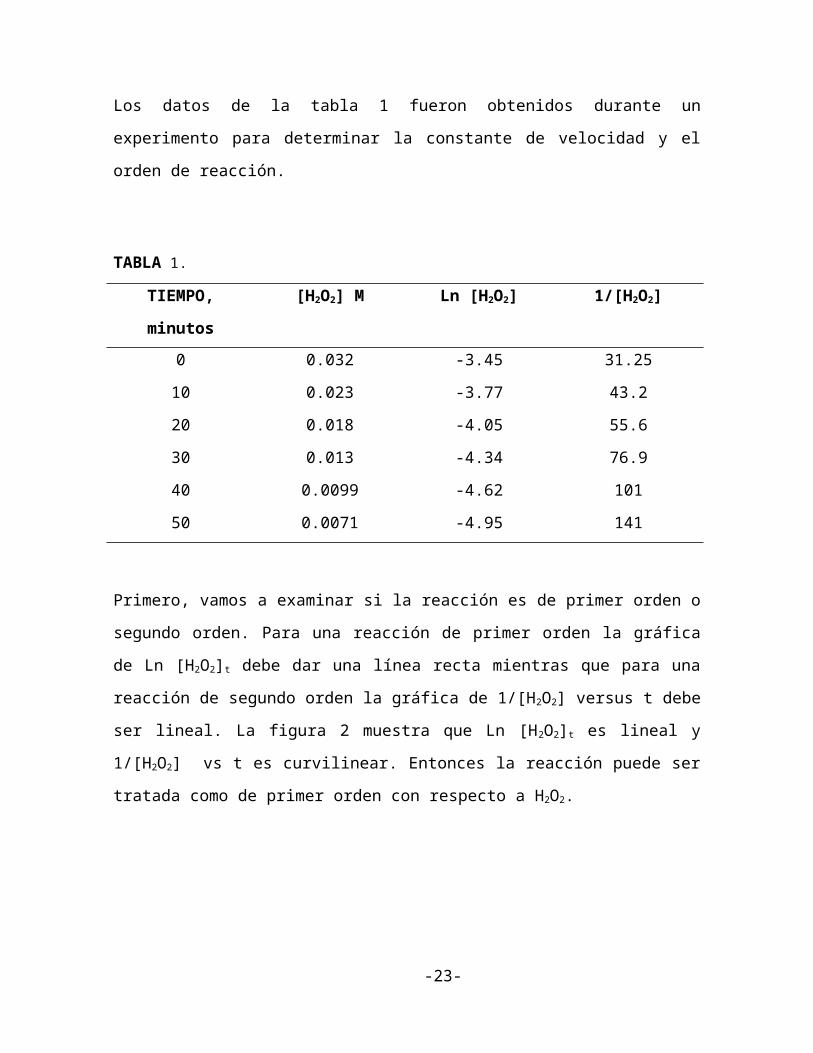
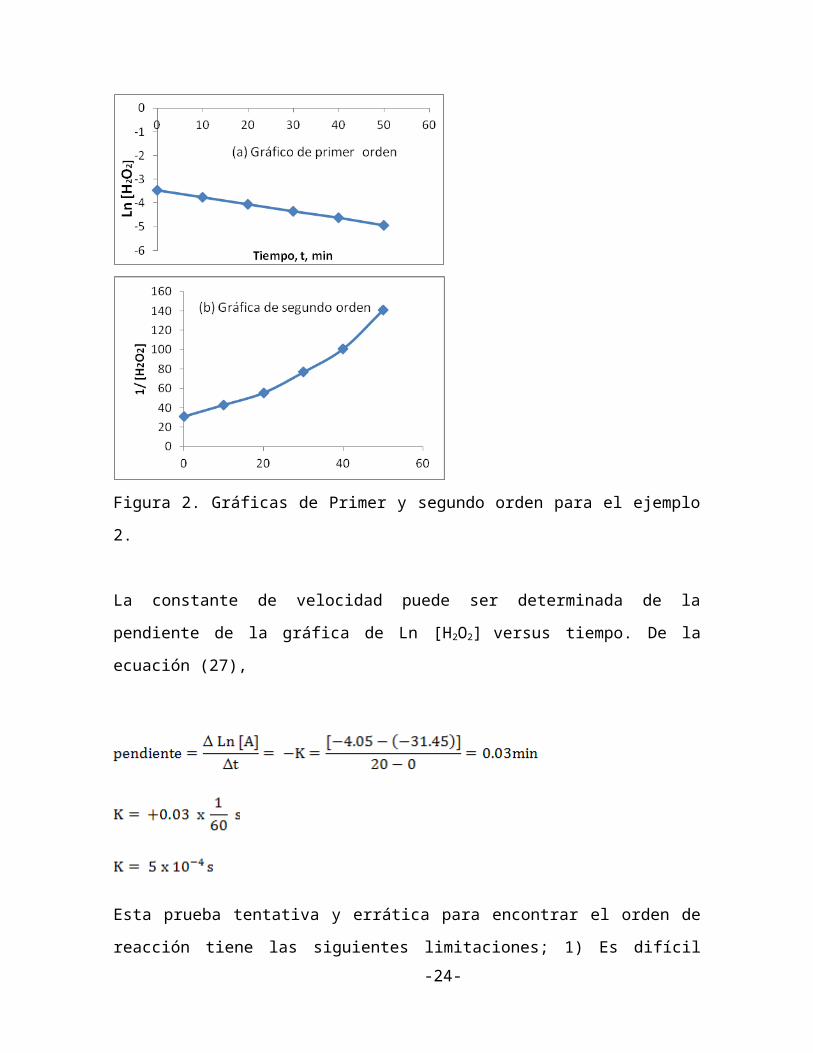
Los datos de la tabla 1 fueron obtenidos durante un experimento para determinar la
constante de velocidad y el orden de reacción.
TABLA 1.
TIEMPO, minutos [H2O2] M Ln [H2O2] 1/[H2O2]
0
10
20
30
40
50
0.032
0.023
0.018
0.013
0.0099
0.0071
-3.45
-3.77
-4.05
-4.34
-4.62
-4.95
31.25
43.2
55.6
76.9
101
141
Primero, vamos a examinar si la reacción es de primer orden o segundo orden. Para una
reacción de primer orden la gráfica de Ln [H2O2]t debe dar una línea recta mientras que para
una reacción de segundo orden la gráfica de 1/[H2O2] versus t debe ser lineal. La figura 2
muestra que Ln [H2O2]t es lineal y 1/[H2O2] vs t es curvilinear. Entonces la reacción puede
ser tratada como de primer orden con respecto a H2O2.
-16-

Figura 2. Gráficas de Primer y segundo orden para el ejemplo 2.
La constante de velocidad puede ser determinada de la pendiente de la gráfica de Ln [H 2O2]
versus tiempo. De la ecuación (27),
Esta prueba tentativa y errática para encontrar el orden de reacción tiene las siguientes
limitaciones; 1) Es difícil aplicarlo si el orden de la reacción no es un entero, y 2) los datos
experimentales dispersos conllevan a un error que es difícil de juzgar ahí la línea recta se
ajusta a un punto determinado asumiendo que el orden es mayor que otro. Muchos órdenes
de reacción son enteros, sin embargo si los datos son colectados cuidadosamente y el
experimento es diseñado adecuadamente el procedimiento da resultados bastante
confiables.
5. MECANÍSMOS DE REACCIÓN
-17-

Hemos observado previamente que el orden de una reacción no puede necesariamente ser
determinado de la estequiometria de la reacción. Además podemos definir un tipo particular
de reacción, la reacción elemental la cual tiene la propiedad especial de que su orden de
reacción puede ser determinado de su estequiometria. Las reacciones elementales pueden
ser monomoleculares, donde una especie simple reacciona y su sola concentración
determina el orden de la reacción.
Generalmente, se establece que si la reacción A → productos, es elemental y
monomolecular, la ley de velocidad será
La descomposición radioactiva es un ejemplo de tal reacción como lo es la descomposición
del gas ciclopropano a propileno.
Las reacciones elementales que son bimoleculares involucran la interacción de dos
moléculas las cuales pueden ser de la misma especie o moléculas de especies diferentes.
Luego,
o
Las leyes de velocidad son, respectivamente,
-18-

Ejemplos de tales reacciones son la descomposición del dióxido de nitrógeno a óxido
nítrico y oxígeno en fase gaseosa.
Y la formación de yodometano de bromometano.
Reacciones mucho más complejas son la regla general en lugar de la excepción. Las
reacciones totales, cuando se escriben estequiométricamente, pueden conducir a creer que
tiene un mecanismo sencillo. Sin embargo la experimentación revela que leyes de
velocidad complicadas indican mecanismos complejos.
Por ejemplo, el contraste de las dos reacciones siguientes: La formación del yoduro de
hidrógeno del hidrógeno gaseoso y el yodo gaseoso es representada estequiométricamente
como:
La reacción es elemental, bimolecular y de primer orden con respecto al [ y al
. La ley de velocidad es
-19-

La formación del bromuro de hidrógeno gaseoso del hidrógeno gaseoso y bromo gaseoso
puede ser estequiométricamente establecida como,
La ley de velocidad determinada experimentalmente es,
La reacción evidentemente no es elemental. Tales reacciones complejas, como esta, sin
embargo, están compuestas de una serie o una secuencia de reacciones elementales.
Podemos determinar el orden de la reacción con respecto a cada uno de los reactantes (y
productos) y la influencia de la concentración de todas las especies sobre la velocidad de
toda la reacción si conocemos la secuencia y las velocidades de las reacciones elementales
que constituyen la reacción total.
Para ilustrar más estos puntos, vamos a examinar los resultados de un experimento
bioquímico en el cual la velocidad de hidrólisis de la sacarosa por la enzima sacarasa es
investigada. Esta enzima, es un catalizador biológico (sobre lo cual hablaremos en mayor
detalle posteriormente) que cataliza la reacción:
Sacarosa Glucosa Fructosa
-20-

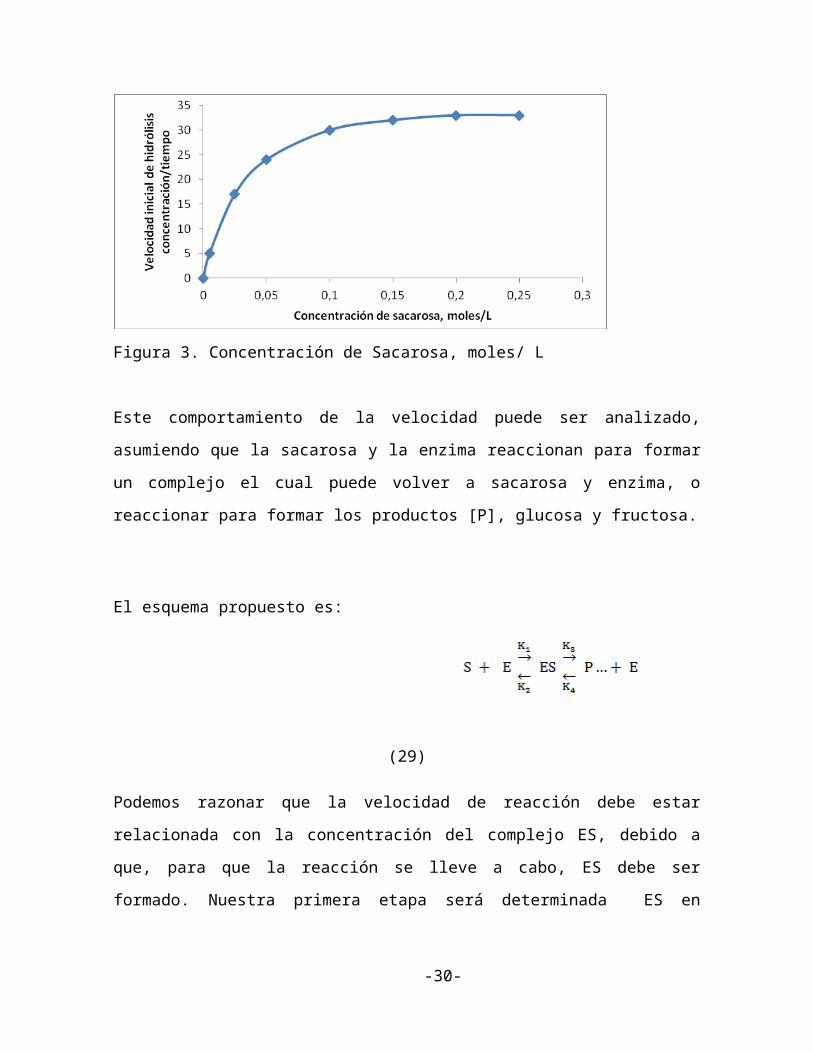
Datos experimentales muestran que la velocidad inicial de hidrólisis del sustrato sacarosa (-
d[s]/dt) es una función de la concentración del sustrato [s] donde la concentración de la
enzima sacarasa [E] era constante. (Figura 3)
A bajas concentraciones de sustrato la velocidad de hidrólisis era proporcional a la
concentración de sacarosa, esto es
Mientras a concentraciones de sacarosa más altas, la velocidad de hidrólisis alcanzaba una
velocidad máxima que era aparentemente independiente de la concentración de sacarosa,
esto es,
Figura 3. Concentración de Sacarosa, moles/ L
Este comportamiento de la velocidad puede ser analizado, asumiendo que la sacarosa y la
enzima reaccionan para formar un complejo el cual puede volver a sacarosa y enzima, o
reaccionar para formar los productos [P], glucosa y fructosa.
-21-

El esquema propuesto es:
(29)
Podemos razonar que la velocidad de reacción debe estar relacionada con la concentración
del complejo ES, debido a que, para que la reacción se lleve a cabo, ES debe ser formado.
Nuestra primera etapa será determinada ES en términos de la cantidad total de especies que
contengan E y de la concentración de sustrato.
Cuando la reacción descrita en la ecuación (29) está en el estado estacionario,
Velocidad de formación de ES= velocidad de remoción de ES.
Luego, debido a que las reacciones de la ecuación (29) son elementales,
Dividiendo por [E] y reorganizando da,
Si ahora asumimos, que la velocidad de reacción, V, es proporcional a [ES], entonces la
velocidad máxima de reacción Vmax ocurrirá cuando toda la enzima esté presente como
[ES], cuando [ES] = [E]t, así establecemos Km = (K2 + K3) / K1, obtenemos,
-22-
(30)
Una gráfica de V versus [S] usando esta expresión, la ecuación de Michaelis-Menten, da
una hipérbola rectangular, una curva de la forma de la figura 3. Esto da soporte a la
aseveración de que nuestro modelo cinético es descriptivo del mecanismo de la reacción.
Como hemos visto a lo largo de este estudio, esta expresión hiperbólica rectangular da un
modelo útil para la cinética de crecimiento microbial. Examinemos, las dos constantes, que
determinan la forma de la curva V versus [S]. El valor de Vmax es la máxima velocidad de
reacción, esto es, la velocidad alcanzada a valores altos de [S]. Cuando Vmax es alcanzada,
un aumento adicional en [S] no afecta la velocidad de reacción.
Cuando [S]>> Km, la reacción (30) muestra que V= Vmax, la reacción entonces es cero
respecto a [S], una propiedad que está de acuerdo con una de nuestras observaciones
experimentales iniciales. El valor de Km es igual al valor de [S] cuando V= Vmax/2 y el
valor del sustrato será [S]1/2 (ver figura 3) esto se muestra usando la ecuación (3) si, V=
Vmax/2 y [S] =[S]1/2 por ejemplo,
O
La constante, Km, es llamada la constante de velocidad media, o constante de Michaelis-
Menten y es un indicativo de la fuerza del enlace entre la enzima y el sustrato. A menor
valor de Km, mayor es la afinidad entre la enzima y el sustrato. Para reacciones enzima-
sustrato simples los valores de Km están ligeramente entre 10-2 y 10-5 M. El significado de
este rango de valores es que solamente se requiere 10-5 a 10-2 M de sustrato para permitir
que la enzima opere a la mitad de su velocidad máxima.
-23-
En el rango de concentración de sustrato donde Km >> [S] la ecuación (3) predice que,
Esta es la expresión para una reacción la cual es de primer orden con respecto a [S], la cual
está de acuerdo con el comportamiento observado en la figura 3. Así vemos que la misma
reacción puede ser cero o de primer orden dependiendo de la concentración de los
reactantes.
6. EFECTO DE LA TEMPERATURA SOBRE LA VELOCIDAD DE LA
REACCIÓN
Ensayos experimentales han demostrado que la velocidad de las reacciones químicas
aumenta con el aumento de la temperatura. En muchos casos, el efecto de la temperatura
sobre la velocidad de la reacción está relacionado con su efecto sobre la constante de
velocidad.
Arrhenius formuló la ley de velocidad empírica,
(31)
La cual puede expresarse también,
(32)
-24-

Así que una gráfica de Ln K versus 1/T puede dar una línea recta con una pendiente -Ea/RT
y un intercepto sobre el eje 1/T de Ln (A)/ Ea/RT,
Donde,
A = Factor pre-experimental, o factor de frecuencia, y es usualmente tratado
como una constante que es independiente de la temperatura para una
reacción particular.
Ea = Energía de activación y es tratada también como una constante para una
reacción particular.
R = Constante de los gases ideales.
T = Temperatura en °K
Es posible relacionar el efecto de la temperatura sobre la velocidad de reacción observado
experimentalmente y el establecido empíricamente para dar una descripción teórica del
efecto de la temperatura sobre la distribución de niveles energéticos de las especies
reaccionantes. Para hacer esto, asumimos, que la reacción,
Procede a través de un intermediario inestable, de alta energía conocido como un complejo
de transición o complejo activado.
-25-

Podemos ilustrar los niveles energéticos promedio relativos de los reactantes, productos y
complejo activado por un diagrama como el que se presenta en la figura 4.
Figura 4. Energía De Activación
La figura 4 muestra que la reacción compleja procede, en este ejemplo con la liberación de
energía ΔH. Sin embargo la formación intermedia del complejo activado requiere
suministro de una cantidad promedia de energía Ea, la energía de activación de la reacción
en progreso. Esta energía, también llamada ΔH, calor de reacción, es liberada cuando el
complejo activado se descompone en los productos de la reacción. Para que esta reacción
ocurra reversiblemente, se requiere una energía adicional (ΔH + Ea) para formar el
complejo activado del cual solamente Ea es liberada cuando A y B se forman de nuevo. La
gran cantidad (ΔH + Ea) es la energía de activación de la reacción inversa.
La figura 4 está basada sobre niveles energéticos promedio de los reactantes, productos y el
complejo activado. Para entender como la idea de un complejo de alta energía está
relacionada al efecto de la temperatura sobre la velocidad de reacción, debemos examinar la
distribución de los niveles energéticos en un reactante a una temperatura dada. (Figura 5)
-26-

FIGURA 5. DISTRIBUCIÓN DE LA ENERGÍA DE LOS REACTANTES
El efecto de la temperatura sobre la distribución de los niveles energéticos para una especie
dada, puede ser descrita por la teoría de Maxwell-Boltzman, la cual relaciona la variación
del número de moléculas con una energía igual o mayor que un nivel de energía dado a la
temperatura absoluta. Luego,
(33)
Donde,
E = Nivel energético estipulado.
No = Número total de moléculas.
N = Numero de moléculas con energía igual o mayor que E
T = Temperatura absoluta, en °K
D = Constante.
De esta ecuación y de la figura 5 podemos ver que, a una temperatura dada, el valor de N
decrecerá al aumentar el valor de E. Simplemente se establece que cuando un nivel
energético estipulado se aumenta, una pequeña fracción de las moléculas totales están
incluidas en N.
-27-

Un análisis adicional de la ecuación (33) revela que para un nivel de energía dado, al
aumentar la temperatura debe aumentarse exponencialmente la población de moléculas
presentes a este nivel o un nivel superior.
Podemos ahora postular que solamente las moléculas con un nivel de energía mayor o igual
que E son capaces de formar un complejo de transición de alta energía.
El procedimiento para determinar los valores de Ea y A para una reacción es ilustrado en el
siguiente ejemplo:
EJEMPLO 3.
La constante de velocidad K para la reacción entre el peróxido de hidrógeno y el yoduro de
potasio para formar yodo y agua.
Se encontró que variaba con la temperatura, cuando las concentraciones de los reactantes se
mantenían constantes a [H2O2] = 5.56 x 10-1 M y [KI] = 1.2 x 102 M.
TEMPERATURA CONSTANTE DE VELOCIDAD, K
44.5 1.66 x 10-3
35.0 1.02 x 10-3
25.7 6.63 x10-4
15.1 2.98 x10-4
4.5 1.17 x10-4
Determine la energía de activación para esta reacción y el valor del factor exponencial.
-28-

Graficando los datos de acuerdo con la ecuación (32)
Ln K TEMPERATURA °K (T) 1/T x 10-3
-6.401 317.7 3.15
-6.888 308.2 3.25
-7.319 298.9 3.35
-8.118 288.3 3.47
-9.053 277.7 3.60
Figura 6. grafica de Arrhenius para ejemplo 3
De la figura 6,
Puesto que R= 1.99 calorías/ mol °K
Eactivación = 11.400 cal /mol
= 11.4 Kcal/ mol
Usando la ecuación y observando que cuando Ln K = -7,
-29-
= 0.00327 °K-1
Las unidades de A son las mismas de K (ver ecuación 31) puesto que no tiene
dimensiones.
Figura 7. efecto de un catalizador sobre la energía de activación.
Por ejemplo, podemos mostrar que si la velocidad de una reacción (a 25 °C) es aumentada
por un factor de dos luego de la adición de un catalizador, la energía de activación es
reducida 409 cal/mol. Sin catalizador,
1.
-30-

Luego Ea ha disminuido en 0.409 Kcal/mol
Aunque existe un cambio en Ea atribuible a la presencia de un catalizador, el calor de
reacción, ΔH, no se ha alterado, de lo cual deducimos que el catalizador tendrá influencia
sólo sobre la velocidad de la reacción y no de su extensión. Un catalizador puede participar
en la reacción por ejemplo, llegando a formar parte del complejo activado y su
concentración no varía durante el tiempo de reacción, esto es
Los catalizadores pueden ser clasificados como homogéneos cuando son uniformemente
distribuidos sobre un nivel molecular a través del medio de reacción, o heterogéneos
cuando el catalizador está presente como una fase separada distinguible. Estos catalizadores
químicos como también los catalizadores biológicos específicos conocidos como enzimas
son importantes en la química acuática.
El ión hidrógeno (H+) y el ión hidróxido (OH-) son catalizadores comunes en los sistemas
acuáticos. El efecto de su catálisis se manifiesta por cambios en la velocidad de reacción
que ocurre con cambios en pH. Por ejemplo, uno de los principales constituyentes de los
detergentes sintéticos caseros es la sal del ácido fosfórico condensado, tal como el ácido
pirofosfórico, H4P2O7 y el ácido tripoliforfótico H5P3O10. Estos compuestos reaccionan con
agua en una reacción conocida como hidrólisis para formar ácido ortofosfórico, entonces:
-31-

(34)
Ácido pirofosfórico Ácido fosfórico
(35)
Pentasodio tripolifosfato Fosfato trisódico
Los fosfatos condensados contribuyen aproximadamente con un 50 a 60% de los fosfatos
totales en las aguas residuales domésticas. Además, son usados en la formulación de
detergentes sintéticos comerciales para acomplejar los iones Ca+2 y Mg+2 y prevenir su
interacción con el surfactante. Los ortofosfatos no llevan a cabo su función acomplejante
tan bien como los fosfatos condensados, así que es importante anotar que los fosfatos
condensados son estables (no realizan hidrólisis como, ecuaciones (34) y (35)) durante el
proceso de limpieza. La hidrólisis de los fosfatos condensados es catalizada por H+. La
figura 8 muestra, que el tiempo para que se lleve a cabo el 5% de una solución de
pirofosfato a 10°C es cerca de 1 año a pH 4, muchos años más a pH 7, y por encima de un
siglo a pH 10.
-32-

FIGURA 8. HIDRÓLISIS DEL 5% DE PIROFOSFATO EN UNA SOL. APROX. 1%
La ecuación de la velocidad para la hidrólisis de pirofosfato es
La constante de velocidad depende altamente de la [H+] y varía desde 0.534/hora a pH = 0
a 0.0318/hora, a pH 1.1 a 0.00272/hora, a pH = 33.
Muchas aguas residuales domésticas tienen valores de pH en el rango de 6.5 a 8.0 y
temperaturas entre 10 a 20 °C
La figura 8 nos muestra que los pirofosfatos son bastante estables bajo estas condiciones.
Sin embargo, observemos que mucho de los fosfatos condensados en aguas residuales
-33-

domésticas han formado ortofosfatos en el tiempo que el agua residual permanece en la
planta de tratamiento, generalmente un período significativamente menos a un día.
Además, los fosfatos no condensados podrán sobrevivir en un tratamiento biológico de
desechos, máximo un periodo de un día. La razón de nuevo es la catálisis pero en este caso
enzimática. Muchos microorganismos poseen una enzima (catalizador biológico-específico)
que podrá controlar la hidrólisis de fosfatos condensados: Un ejemplo de tal enzima es la
pirofosfato hidrogenasa, la cual cataliza la hidrólisis de pirofosfato inorgánico a dos moles
de ortofosfato.
La eficiencia con la cual un aparato de aireación transfiere oxígeno al agua o agua residual
es medida, determinando la velocidad a la cual el equipo aumenta la concentración de
oxígeno disuelto líquido. Antes de la prueba, el líquido a investigar debe ser desoxigenado.
El método recomendado para llevar a cabo la desoxigenación es la adición de sulfito
El primero se refiere al tratamiento de los datos de la prueba de demanda química de
oxígeno (DBO) la cual es usada para determinar la potencia de un agua residual, el grado
de polución orgánica en aguas de abasto y la velocidad a la cual la materia orgánica en las
aguas residuales puede ser degradada por microorganismos bajo condiciones aeróbicas (y
cuando el oxígeno disuelto (OD) está presente).
En la prueba de la DBO, una muestra diluida de agua residual es incubada por un período
(5 días a 20 °C) y la cantidad de oxígeno disuelto consumido en este período es medido. La
DBO5 es entonces calculada como,
Donde
-34-

La medición de la DBO5 determina solamente un punto de la curva que relaciona el
oxígeno disuelto consumido en un tramo de tiempo. La reacción monitoreada realizando
mediciones de oxígeno consumido se ha expresado como:
Y la velocidad de esta reacción en presencia de un exceso de oxígeno disuelto se ha
establecido a menudo como de primer orden con respecto a la materia orgánica. Se debe
reconocer que éste es un estamento totalmente empírico porque la naturaleza de la materia
orgánica degradable en el agua residual no está bien definida y por cierto las velocidades de
degradación microbial individuales de todos los compuestos orgánicos no son conocidas.
No obstante, el curso total de tiempo del consumo de oxígeno para la degradación de la
materia orgánica carbonácea en la prueba de la DBO tiene muchas características de una
reacción de primer orden. Puesto que la velocidad de la DBO (diagrama de la materia
orgánica) es de importancia en el diseño de plantas de tratamiento de desechos y en el
manejo de aguas de abasto, podemos usualmente emplear una relación empírica de primer
orden para formular “la curva de DBO”. (Figura 9)
FIGURA 9. CURVA DE DBO
-35-

Si L = concentración de la materia orgánica degradable a cualquier tiempo t (días),
entonces para una reacción de primer orden podemos escribir,
(36)
Integrando, tenemos
(37)
Donde
L0 = concentración original de materia orgánica biodegradable
K = constante de velocidad/día
Debido a que L no puede ser medida directamente, la ecuación debe ser modificada para
reemplazar L con un parámetro que pueda ser medido como una función del tiempo.
Podemos lograr esta modificación estableciendo,
(38)
Donde
Y = cantidad de material que ha sido degradado a un tiempo t.
El valor de Y puede ser asignado determinando el consumo de oxígeno disuelto a cualquier
tiempo, a través de mediciones de la concentración de oxígeno disuelto de la muestra.
(39)
-36-
Sustituyendo ecuación (38) en ecuación (37), obtenemos
(40)
La cual es la ecuación de velocidad empírica clásica de primer orden para la DBO.
Existen varios procedimientos para analizar Y versus t y obtener las constantes K y L0.
Uno de estos, el método de la pendiente Thomas, involucra el desarrollo de la ecuación de
una línea recta que aproxima la relación de Y y T.
Si expandimos la porción (1- e-Kt) de la ecuación (40) obtenemos,
La cantidad [ Kt(1 + Kt/6)-3] tiene una expansión similar
(42)
Comparando estas dos expansiones, vemos que sólo hay una pequeña diferencia en el
cuarto término, sobre esta base podemos decir
(43)
-37-

(44)
Reorganizando ecuación (44) obtenemos la ecuación de una línea recta.
(45)
Si llamamos
(46)
(47)
Obtenemos que
(48)
Y
(49)
-38-

Con datos experimentales relacionando Y y tiempo, podemos graficar (t/Y)1/3 versus
tiempo. Sin embargo, valores experimentales de Y> 0.9 L0 no deben ser usados para la línea
recta porque la ecuación (42) es significativamente diferente a la ecuación (41) en este
rango. Llevando la gráfica a una línea recta y determinando el intercepto a y la pendiente,
b, las ecuaciones (47) y (48) pueden ser usadas para calcular K y L0
EJEMPLO 4.
Los métodos estándar, establecen que la constante de la velocidad (base 10) para el
consumo de oxígeno de una mezcla de glucosa (150 mg/L) y ácido glutámico (150 mg/L)
debe estar entre 0.16 a 0.19/ día cuando se utiliza una semilla de microorganismo adecuada.
Los datos de la Tabla 2 fueron obtenidos s 20°C de un posible material a utilizar como
semilla por un estudiante graduado. Satisface la semilla las especificaciones anteriores?
Tabla 2.
TIEMPO, t días DBO, Y mg/L t/Y (t/Y )1/3
1 122 0.0082 0.202
2 117 0.017 0.25
3 184 0.016 0.25
4 193 0.021 0.276
5 203 0.025 0.292
6 205 0.029 0.307
7 207 0.034 0.324
-39-
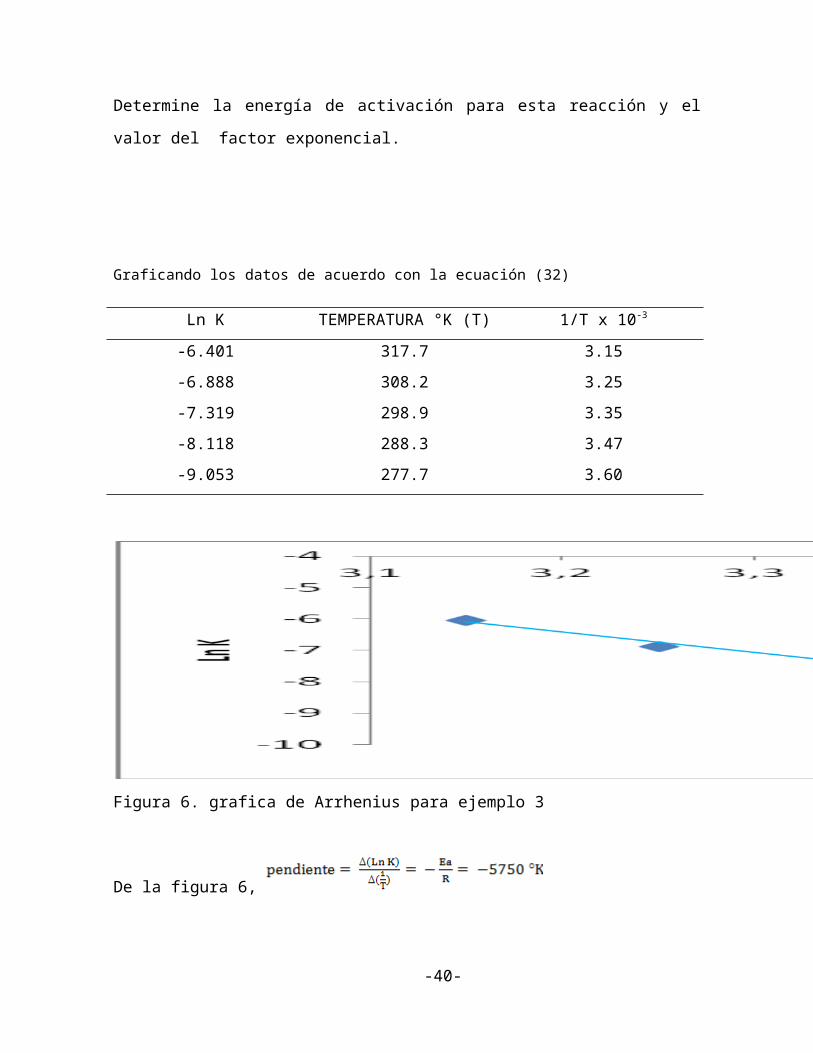
FIGURA 10. GRÁFICA DE (t/Y)1/3
De la figura 10, una gráfica de (t/Y)1/3 versus tiempo de acuerdo con la ecuación (25)
encontramos,
a= 0.182 y b= 0.0216
De la ecuación (48)
6 x 0.0216/0.182 = 0.71/ día
O
De la ecuación (49)
-40-

Puesto que K es mucho mayor que 0.16 a 0.19/día la semilla no es aceptable.
El segundo uso de las expresiones cinéticas empíricas en las aguas residuales y en el campo
del control de la contaminación del agua es la cinética del crecimiento de cultivos
microbianos mixtos sobre sustratos simples o múltiples. Ejemplos típicos son el
crecimiento de sólidos de lodos activados sobre la DBO del agua residual y el crecimiento
de algas sobre nutrientes como nitrato y fosfato.
El microbiólogo Monod derivó una relación entre la velocidad de crecimiento del sustrato
que limita el crecimiento de las bacterias.
(50)
Donde
M= velocidad de crecimiento = (1/x) (dx/dt)
Mmax = velocidad de crecimiento máxima
X= masa microbial o número de concentración
Ks = concentración del sustrato cuando M= Mmáx/2
La relación expresada en la ecuación (50) es similar a la expresión de Michaelis –Menten
desarrollada previamente para la interacción enzima-sustrato. Además, algunos
investigadores creen que la analogía no es coincidencial, estableciendo que los
microorganismos son 2 sacos llenos de enzimas”, así que no es sorprendente que su
velocidad de crecimiento esté relacionada con el comportamiento de catalizadores que
interviene en muchas reacciones que contribuyen al crecimiento.
-41-

La relación velocidad crecimiento del sustrato es expresada empíricamente en aplicaciones
tales como el tratamiento biológico de aguas residuales. En estos procesos, crece una
población de microorganismos mezclada y variable (Ej. Desecho doméstico). Ahora,
tenemos el mismo problema que cuando intentábamos explicar la cinética de la DBO. No
tenemos una medida definitiva de la concentración del sustrato limitante del crecimiento,
como tampoco una medida bien definida de la concentración de microorganismos. Sin ir en
el razonamiento de su selección, el parámetro usado para los microorganismos es
usualmente los sólidos suspendidos o sólidos suspendidos volátiles en la suspensión de
lodos activados. Para el sustrato limitante del crecimiento la concentración de DBO o
DQO.
Los datos de la figura 11 muestran la relación entre la velocidad de crecimiento y la
concentración de sustrato, expresada como DQO soluble, para una unidad de lodos
activados operados con un sustrato de proteína de res. La velocidad de crecimiento es una
función de primer orden de la concentración de materia orgánica degradable. Podemos ver
que para el rango de condiciones estudiado, fue una relación linear entre M y [S] indicando
que estamos en la región donde Ks>> [S] y M = K´[S]. Tal observación es además común
en sistemas de lodos activados tratando aguas residuales prácticamente a la velocidad del
tratamiento. La curva en la figura 11 nos permite que si se desea producir un efluente con
una DQO soluble de menos de 20 mg/L en un tratamiento de lodos activados de este
desecho, debemos diseñar la planta de tratamiento de tal forma que se adopte a una
velocidad de crecimiento de lodos activados de menos que 1.2/día.
-42-

Figura 11. Relación de primer orden entre la velocidad de crecimiento de lodos activados y
la concentración de DQO soluble del efluente degradable.
-43-





![Cinética QuímicaCinética Química - uprh.edu · 1/26/2011 1 Cinética QuímicaCinética Química Química 4042 [C] [P] Ileana Nieves Martínez t [R] Definición de Cinética Química](https://static.fdocuments.mx/doc/165x107/5b5ecbc87f8b9a8b4a8cf716/cinetica-quimicacinetica-quimica-uprh-1262011-1-cinetica-quimicacinetica.jpg)