
Deficiencias congénitas: mecanismo extrínseco y...
58
Deficiencias cong Deficiencias cong é é nitas: nitas: mecanismo extr mecanismo extr í í nseco y tronco nseco y tronco com com ú ú n n
Transcript of Deficiencias congénitas: mecanismo extrínseco y...

Deficiencias congDeficiencias congéénitas: nitas: mecanismo extrmecanismo extríínseco y tronco nseco y tronco
comcomúúnn
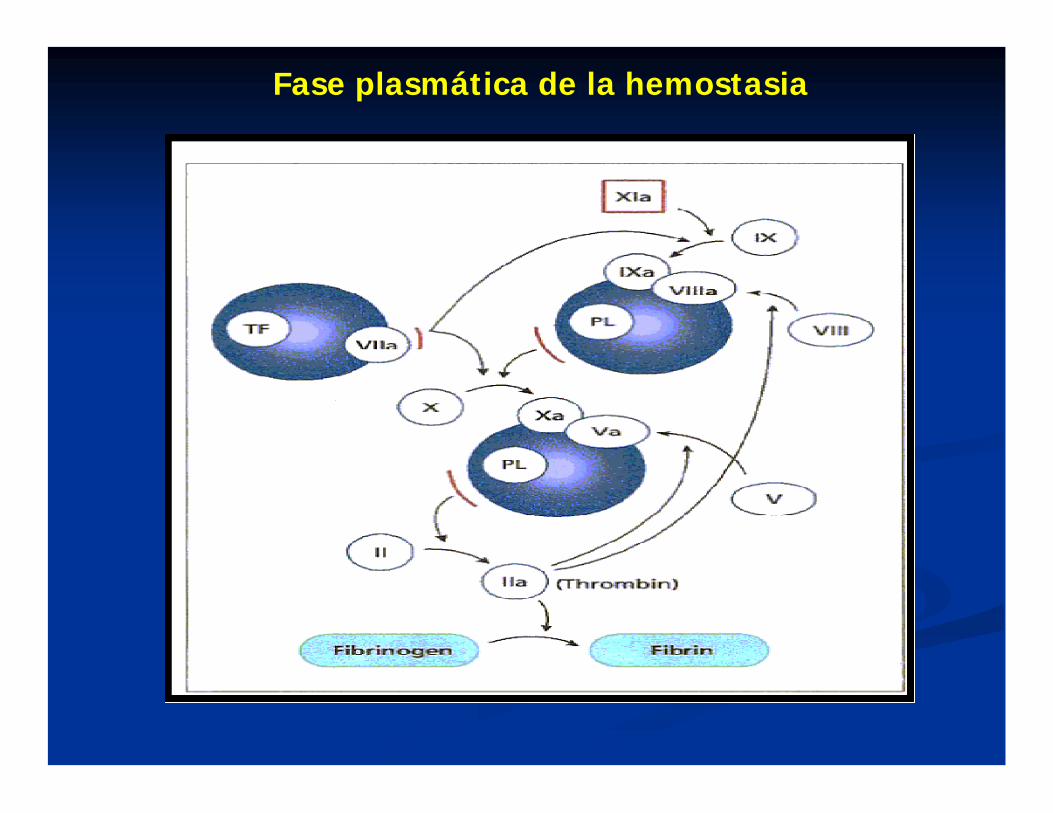
Fase plasmática de la hemostasia

FISIOPATOLOGIACoagulopatías plasmáticas:
1) Alteraciones de la Síntesis:- Déficit congénitos de factores- Hepatopatías agudas o crónicas- Deficiencia de vitamina K- Alteraciones cualitativas
2) Depuración aumentada:- CID- Fibrinolisis primaria- Proteolisis inespecífica
3) Inactivación – inhibición:- “Anticoagulante lúpico”
- Inhibidores de Factor

DIAGNOSTICO
• EXAMEN CLINICO:
- Interrogatorio
- Examen físico
• PRUEBAS DE LABORATORIO

DIAGNOSTICO
Alteraciones plasmáticas
• Manifestaciones características:- Hematomas - Hemartrosis- Hemorragias mucosas
• Diagnósticos diferenciales:- Congénitas : antecedentes familiares y
comienzo en edad precoz
- Adquiridas: aparición tardía, ausencia deantecedentes familiares y enfermedad co-existente

DIAGNOSTICO
- Prolongación exclusiva del tiempo de Quick:
- Déficit de factor VII
- Pruebas adicionales:- corrección con plasma normal- dosaje de factor VII

DIAGNOSTICO
- Alteraciones combinadas del APTT y tiempo de Quick:- Déficit de factores de tronco común- Inhibidores de la coagulación- Hepatopatías- Déficit de vitamina K- CID- Fibrinolisis- Proteolisis inespecífica
- Pruebas adicionales:- corrección con plasma normal- dosaje de factores- PDF y dímero- D

DIAGNOSTICO
- Alteración aislada del tiempo de trombina:
- Hipo- afibrinogenemias- Disfibrinogenemias- Heparina
- Pruebas adicionales:- dosaje de fibrinógeno funcional e inmunológico

DEFICIENCIA DE VIIDEFICIENCIA DE VII
HERENCIA: autosHERENCIA: autosóómica recesiva.mica recesiva. HeterocigotasHeterocigotas asintomasintomááticos ticos Se ha asociado a Se ha asociado a hiperhiper BilBil. cong. congéénita nita
enfermedad de Gilbert y enfermedad de Gilbert y SmeSme de de DubinDubinJohnson)Johnson)
ClClíínica variable no siempre asociada a nica variable no siempre asociada a los valores del factor. los valores del factor.

DEFICIENCIA DE VIIDEFICIENCIA DE VII
Alteraciones moleculares: Alteraciones moleculares: mutaciones mutaciones sin sentido y sin sentido y delecionesdeleciones cortascortas
En 1997 se revisaron 35 mutaciones En 1997 se revisaron 35 mutaciones puntuales y hay muchas mpuntuales y hay muchas máás publicadas. s publicadas. (Cooper y col. (Cooper y col. ThromboThrombo. . HaemostaHaemosta.78:151. 1997).78:151. 1997)

Manifestaciones clManifestaciones clíínicasnicas
Semejante al hemofSemejante al hemofíílicolico hemartrosishemartrosis hemorragias digestivashemorragias digestivas hematomas subcuthematomas subcutááneosneos hemorragias genitourinariashemorragias genitourinarias RN pueden presentar hemorragias RN pueden presentar hemorragias
cerebrales.cerebrales.

LaboratorioLaboratorio
TP alargadoTP alargado
Resto de las pruebas normalesResto de las pruebas normales
Test de Test de StypvenStypven normal (activa al X normal (activa al X directamente)directamente)

TratamientoTratamiento
Plasma fresco congeladoPlasma fresco congelado
FracciFraccióón que contenga a dicho factorn que contenga a dicho factor
Suero conservadoSuero conservadoVida media aprox 6 hs, lo que obliga a perfusiones continuas. Utilización de VIIr
Vida media aprox 6 hs, lo que obliga a perfusiones continuas. Utilización de VIIr

Deficiencia congDeficiencia congéénita de factor Xnita de factor X
HerenciaHerencia: : autosautosóómica recesivamica recesiva
Es una de las deficiencias de mEs una de las deficiencias de máás rara s rara apariciaparicióónn
Manifestaciones clManifestaciones clíínicasnicas: caso leves : caso leves son asintomson asintomááticos, los mticos, los máás graves s graves similares a la deficiencia de factor VII: similares a la deficiencia de factor VII: equimosis, epistaxis, hemorragias equimosis, epistaxis, hemorragias gastrointestinalesgastrointestinales

Deficiencia congDeficiencia congéénita de factor Xnita de factor X
Alteraciones molecularesAlteraciones moleculares
Grandes Grandes delecionesdeleciones..
PequePequeññas as delecionesdeleciones que modifican el que modifican el marco de lectura y mutaciones sin marco de lectura y mutaciones sin sentido.sentido.

LaboratorioLaboratorio
TPR alargado TPR alargado
TP alargadoTP alargado
APTT alargadoAPTT alargado
Test de Test de StypvenStypven alargadoalargado

TratamientoTratamiento
Plasma.Plasma.
Concentrado de plasma rico en XConcentrado de plasma rico en X
Vida media 30-50 hs. Experiencia escasa, poco frecuente.
Vida media 30-50 hs. Experiencia escasa, poco frecuente.

DEFICIENCIA DE VDEFICIENCIA DE V
HERENCIA: autosHERENCIA: autosóómica recesivamica recesiva Ligeramente mLigeramente máás frecuente que la s frecuente que la
deficiencia de Xdeficiencia de X Afecta a los dos mecanismosAfecta a los dos mecanismos CLCLÍÍNICANICA: s: sóólo se presenta cuando el lo se presenta cuando el
individuo es individuo es homocigotahomocigota: hemorragias : hemorragias en piel y mucosas, sangrados en piel y mucosas, sangrados postquirpostquirúúrgicos y postraumrgicos y postraumááticos ticos

LaboratorioLaboratorio
TPR alargado TPR alargado TP alargadoTP alargado APTT alargadoAPTT alargado Test de Test de StypvenStypven alargadoalargado TS alargado en 1/3 de los pacientesTS alargado en 1/3 de los pacientes

TratamientoTratamiento
Plasma fresco. Transfusiones Plasma fresco. Transfusiones frecuentes, vida media corta.frecuentes, vida media corta.
Vida media 16-24 hs.Vida media 16-24 hs.

DEFICIENCIA DE IIDEFICIENCIA DE II
HERENCIA: autosHERENCIA: autosóómica recesivamica recesivaEs la mEs la máás rara de las deficiencias s rara de las deficiencias congcongéénitas de la coagulacinitas de la coagulacióónn
Cuando es severa es Cuando es severa es incompatible con incompatible con la vida y puede causar muerte la vida y puede causar muerte intrauterinaintrauterina

Manifestaciones clManifestaciones clíínicasnicas
Pueden clasificarse en dos grupos: Pueden clasificarse en dos grupos:
HIPOPROTROMBINEMIASHIPOPROTROMBINEMIASGeneralmente con valores de Generalmente con valores de II del 10%II del 10%
DISPROTROMBINEMIASDISPROTROMBINEMIASCon valores de Con valores de II normalesII normales(inmunol(inmunolóógicos) y gicos) y actividad coagulante actividad coagulante variables.variables.

DEFICIENCIA DE IIDEFICIENCIA DE II
Los defectos moleculares responsables de la deficiencia hereditaria de protrombina son su mayoría mutaciones sin sentido.
Producen activación anormal de la protrombina o la trombina formada tiene escasa capacidad para formar el coágulo de fibrina

LaboratorioLaboratorio
TPR alargado TPR alargado TP alargadoTP alargado APTT alargadoAPTT alargado Test de Test de StypvenStypven alargadoalargado TT normal.TT normal.

TratamientoTratamiento
La profilaxis y el tratamiento se realiza con CCP (Prothromplex® , Baxter): 20-30 U/kg seguidos de un mantenimiento de 5 U/kg/24 horas)
Plasma frescoPlasma fresco
Vida media 60-80 hs.Vida media 60-80 hs.

DEFICIENCIA DE IDEFICIENCIA DE I
AFIBRINOGENEMIAS: dosaje de I menor al AFIBRINOGENEMIAS: dosaje de I menor al 5% (10mg/5% (10mg/dldl))
Herencia: autosHerencia: autosóómica recesivamica recesiva Sumamente infrecuenteSumamente infrecuente CosanguinidadCosanguinidad en los ascendientes en men los ascendientes en máás s
de la mitad de las familias estudiadas.de la mitad de las familias estudiadas.

Manifestaciones clManifestaciones clíínicasnicas
Manifestaciones hemorrManifestaciones hemorráágicas desde el gicas desde el nacimiento. nacimiento.
CicatrizaciCicatrizacióón anormal de las heridas.n anormal de las heridas.

LaboratorioLaboratorio
Sangre Sangre incoagulableincoagulable
DisfunciDisfuncióón plaquetaria. Disminucin plaquetaria. Disminucióón n del fibrindel fibrinóógeno plaquetariogeno plaquetario

TratamientoTratamiento
Plasma fresco congelado Plasma fresco congelado CrioprecipitadosCrioprecipitados InfusiInfusióón cada 3n cada 3--4 d4 díías. Vida media largaas. Vida media larga
Vida media 100-150 hs.
Vida media 100-150 hs.

DEFICIENCIA DE IDEFICIENCIA DE I HIPOFIBRINOGENEMIAS: dosaje de I entre 20HIPOFIBRINOGENEMIAS: dosaje de I entre 20--80 80
mgmg//dldl
Herencia: Herencia: podrpodríía tratarse de la forma a tratarse de la forma heterocigotaheterocigotadel trastorno. Mdel trastorno. Máás frecuente que la anteriors frecuente que la anterior..
Laboratorio: Laboratorio: pruebas menos alteradas que en la pruebas menos alteradas que en la afibrinogenemiaafibrinogenemia
ClClíínica: nica: Puede detectarse desde el nacimiento. Puede detectarse desde el nacimiento. Hemorragias Hemorragias infrecinfrec. y poco graves.. y poco graves.

DEFICIENCIA DE IDEFICIENCIA DE I DISFIBRINOGENEMIAS: 300 casos DISFIBRINOGENEMIAS: 300 casos
descriptos con fibrindescriptos con fibrinóógeno normal geno normal pero con pero con alteraciones cualitativasalteraciones cualitativas
Herencia: autosHerencia: autosóómica dominantemica dominante
ClClíínica: la mayornica: la mayoríía de los a de los pacpac. no presentan . no presentan signos de sangrado o signos de sangrado o transtornostranstornoshemorrhemorráágicos leves. Puede detectarse desde gicos leves. Puede detectarse desde el nacimiento o en la vida adultael nacimiento o en la vida adulta

LaboratorioLaboratorio
TP y APTT variables TP y APTT variables óó normal.normal. TT casi siempre alargado. TT casi siempre alargado.
Generalmente no corrige con plasma Generalmente no corrige con plasma normal, debido a que el I anormal no normal, debido a que el I anormal no permite la polimerizacipermite la polimerizacióón de la fibrina n de la fibrina
Se puede sospechar Se puede sospechar cdocdo el I da normal el I da normal por precipitacipor precipitacióón pero por tn pero por téécnicas cnicas coagulomcoaguloméétricastricas da anormal.da anormal.
DisfunciDisfuncióón plaquetaria. Disminucin plaquetaria. Disminucióón n del fibrindel fibrinóógeno plaquetariogeno plaquetario

TratamientoTratamiento
Plasma fresco o congelado, Plasma fresco o congelado, concentrados de I. concentrados de I.
InfusiInfusióón cada 3n cada 3--4 d4 díías. Vida media as. Vida media larga.larga.
Vida media 100-150 hs.Vida media 100-150 hs.

DEFICIENCIA DE XIIIDEFICIENCIA DE XIII
HERENCIA: autosHERENCIA: autosóómica recesivamica recesivaMuchas veces los padres son Muchas veces los padres son cosangucosanguííneosneos
Manifestaciones clManifestaciones clíínicas:nicas: Hemorragias al Hemorragias al caer el cordcaer el cordóón, hemorragias n, hemorragias intracraneales. Las hemorragias no se intracraneales. Las hemorragias no se producen inmediatamente. Cicatrizaciproducen inmediatamente. Cicatrizacióón n anormal de las heridas.anormal de las heridas.

LaboratorioLaboratorio
Todas las pruebas de coagulaciTodas las pruebas de coagulacióón son n son normalesnormales
Prueba de lisis del coPrueba de lisis del coáágulo poco gulo poco sensible.sensible.
En caso de sospecha diluir el plasma y En caso de sospecha diluir el plasma y trabajar con I externo.trabajar con I externo.

TratamientoTratamiento
CrioprecipitadosCrioprecipitados o concentrados de o concentrados de factor XIII. Vida media larga. La mfactor XIII. Vida media larga. La máás s larga de los factores de la coagulacilarga de los factores de la coagulacióón.n.
Vida media 7-10días.Vida media 7-10días.

DEFECTOS COMBINADOSDEFECTOS COMBINADOS
Se han observado una gran variedad de Se han observado una gran variedad de defectos combinados pero son defectos combinados pero son sumamentessumamentes raros.raros.
Deficiencia de V y de VIIIDeficiencia de V y de VIII Deficiencia de VII y de VIIIDeficiencia de VII y de VIII OtrosOtros

TRASTORNOS HEMORRAGICOS ADQUIRIDOS
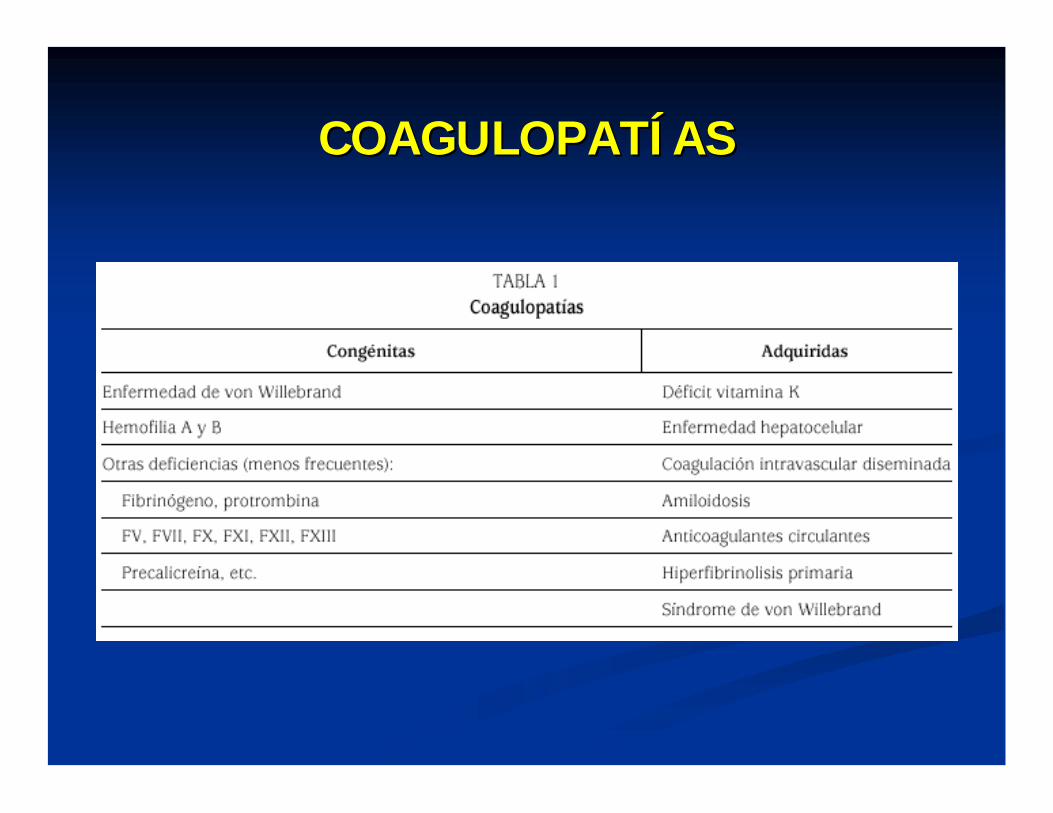
COAGULOPATCOAGULOPATÍÍASAS

DEFICIT DE VITAMINA KDEFICIT DE VITAMINA K
Afecta los factores: Afecta los factores: II, VII, IX y X. II, VII, IX y X. ProteProteíína C y Protena C y Proteíína Sna S

VITAMINA KVITAMINA K
Se incorpora con la dieta.Se incorpora con la dieta. Es sintetizada por bacterias Es sintetizada por bacterias
intestinales. intestinales. Es liposoluble, por lo que necesita Es liposoluble, por lo que necesita
sales biliares para su absorcisales biliares para su absorcióónn
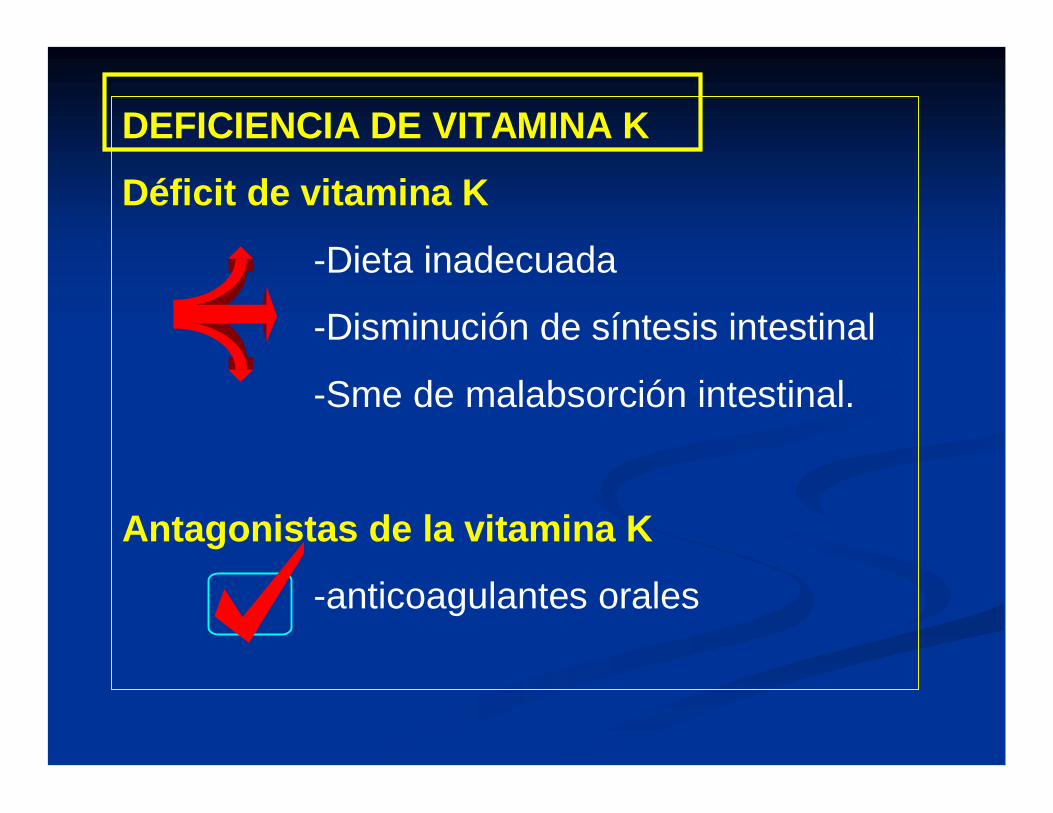
DEFICIENCIA DE VITAMINA K
Déficit de vitamina K
-Dieta inadecuada
-Disminución de síntesis intestinal
-Sme de malabsorción intestinal.
Antagonistas de la vitamina K
-anticoagulantes orales

DEFICIENCIA DE VITAMINA K
DISFUNCIÓN HEPÁTICA
-Inmadurez hepática
-Enfermedad hepática

Situaciones clSituaciones clíínicas que cursan nicas que cursan con dcon dééficit de VITAMINA Kficit de VITAMINA K
Enfermedad hemorrEnfermedad hemorráágica del RNgica del RN Ictericia obstructivaIctericia obstructiva SSííndrome de mala absorcindrome de mala absorcióónn AlimentaciAlimentacióón parenteral sin aporte de n parenteral sin aporte de
vitamina K vitamina K EsterilizaciEsterilizacióón intestinal por tratamiento n intestinal por tratamiento
con ATBcon ATB Tratamiento con anticoagulantes oralesTratamiento con anticoagulantes orales

Enfermedad hemorrEnfermedad hemorráágica del gica del RN(melenaRN(melena neonatorumneonatorum))
El RN tiene dEl RN tiene dééficit de ficit de vitvit. K debido a :. K debido a : Escasa reservaEscasa reserva Falta de flora intestinalFalta de flora intestinal Bajo contenido de vitamina K en la Bajo contenido de vitamina K en la
leche maternaleche materna Inmadurez hepInmadurez hepááticatica

El reciEl reciéén nacidon nacido
En el primer dEn el primer díía de vida tiene entre 25a de vida tiene entre 25--75% de factores 75% de factores vitvit. K dependientes. K dependientes
Entre el 2Entre el 2--3 d3 díía de vida bajan aa de vida bajan aúún mn máás s aumentando el riesgo de hemorragiaaumentando el riesgo de hemorragia
En el 7mo. dEn el 7mo. díía vuelven a sus valores a vuelven a sus valores inicialesiniciales
Al mes llegan a los valores del adultoAl mes llegan a los valores del adulto

En el reciEn el reciéén nacidon nacido
El dEl dééficit puede agravarse por:ficit puede agravarse por:
PrematurezPrematurez Enfermedad subyacenteEnfermedad subyacente Madre tratada con AC orales, Madre tratada con AC orales,
anticonvulsivantes, salicilatos.anticonvulsivantes, salicilatos.

INCIDENCIAINCIDENCIA
BAJA. PROFILAXIS.BAJA. PROFILAXIS.
MANIFESTACIONES CLMANIFESTACIONES CLÍÍNICAS NICAS COMIENZAN AL 2COMIENZAN AL 2--3 D3 DÍÍA DE VIDA A DE VIDA APARICIAPARICIÓÓN BRUSCA N BRUSCA HEMORRAGIAS GASTROINTESTINAL HEMORRAGIAS GASTROINTESTINAL Y EN EL CORDY EN EL CORDÓÓN UMBILICALN UMBILICAL

DIAGNDIAGNÓÓSTICO Y TRATAMIENTOSTICO Y TRATAMIENTO TP, APTT, ALTERADOS. FIBRINTP, APTT, ALTERADOS. FIBRINÓÓGENO GENO
Y PLAQUETAS NORMALES.Y PLAQUETAS NORMALES.
La profilaxis de las hemorragias del RN se realiza con la administración intramuscular de 1 mg de vit K1 en dosis única. Pacientes malnutridos o sometidos a terapéutica antibiótica de amplio espectro deberían recibir 5 mg de vit K1 2 veces por semana. En situaciones de urgencia puede ser necesario tratamiento sustitutivo con PFC o CCP (Prothromplex®)
RESPUESTA AL TRATAMIENTO BUENA, ENTRE LAS 4RESPUESTA AL TRATAMIENTO BUENA, ENTRE LAS 4--8 HS SE 8 HS SE OBSERVA ELEVACIOBSERVA ELEVACIÓÓN DE FACTORES.N DE FACTORES.

Situaciones clSituaciones clíínicas que cursan nicas que cursan con dcon dééficit de VITAMINA Kficit de VITAMINA K
Ictericia obstructiva: Ictericia obstructiva: es liposoluble, es liposoluble, para su absorcipara su absorcióón necesita sales n necesita sales biliares. Toda patologbiliares. Toda patologíía a intraintra o o extrahepextrahepáática que produzca tica que produzca disminucidisminucióón de la n de la secresisecresióónn de las de las mismas hacia la luz intestinal se mismas hacia la luz intestinal se acompaacompañña de disminucia de disminucióón en la n en la absorciabsorcióón.n.

Situaciones clSituaciones clíínicas que cursan nicas que cursan con dcon dééficit de VITAMINA Kficit de VITAMINA K
SSííndrome de mala absorcindrome de mala absorcióón n Diversas patologDiversas patologíías como as como SprueSprue, , Fibrosis quFibrosis quíística del stica del pancreaspancreas se se asocia a dasocia a dééficit por falta de aporte.ficit por falta de aporte.
Pacientes con cirugPacientes con cirugíía abdominal a abdominal sometidos a alimentacisometidos a alimentacióón parenteral n parenteral sin aporte de vitamina K msin aporte de vitamina K máás s esterilizaciesterilizacióón intestinal por tratamiento n intestinal por tratamiento con ATB.con ATB.

Situaciones clSituaciones clíínicas que cursan nicas que cursan con dcon dééficit de VITAMINA Kficit de VITAMINA K
Anticoagulantes oralesAnticoagulantes oralesConstituye uConstituye una de las causas mna de las causas máás s comunes de sangrado comunes de sangrado A)PorA)Por sobredosificacisobredosificacióónnB)TratamientoB)Tratamiento concomitante con concomitante con medicamentos que potencian la accimedicamentos que potencian la accióón n del anticoagulantedel anticoagulanteC)PorC)Por envenenamientoenvenenamiento

HepatopatHepatopatííasas El hEl híígado juega un papel central en la gado juega un papel central en la
hemostasia: hemostasia: a)sa)sííntesisntesis de muchos factores de muchos factores b)sb)sííntesisntesis de inhibidores. de inhibidores. c)sc)sííntesisntesis de componentes del sistema de componentes del sistema fibrinolfibrinolíítico. tico. d)depurad)depura factores activados, enzimas factores activados, enzimas fibrinolfibrinolííticas, etc.ticas, etc.

HepatopatHepatopatííasas
Los pacientes con Los pacientes con hepatopathepatopatííasaspueden presentar : pueden presentar :
CID CID FIBRINOLISIS PRIMARIA FIBRINOLISIS PRIMARIA PLAQUETOPENIA PLAQUETOPENIA

Deficiencia en sintesis de factores•Factores vitamina K dependientes : II, VII, X, IX.
Proteína C y S
•Factor V, XI, XII, XIII
•Deficit de fibrinógeno y disfibrinogenemia(incremento de ácido sálico del fibrinógeno queimpide polimereización de fibrina.
•Disminución de inhibidores naturales de la
coagulación: AT, Proteína C y S

•Sistema fibrinolítico: deficit de α2 antiplasmina
• Disminución del aclaramiento de factoresactivados circulantes
•CID
•Otros: Pérdida o consumo de factores en ellíquido ascítico.
Deficiencia en sintesis de factores

LaboratorioLaboratorio TP y APTT alargados.TP y APTT alargados.
TT alargado TT alargado
Reserva hepReserva hepáática: dosaje de V (se tica: dosaje de V (se prefiere al VII aunque prefiere al VII aunque ééste es de menor ste es de menor vida media)vida media)
..:El V permite diferenciar entre ictericia obstructiva (disminución de los vitamina K dependientes) y transtornodel parénquima hepático
..:El V permite diferenciar entre ictericia obstructiva (disminución de los vitamina K dependientes) y transtornodel parénquima hepático

TratamientoTratamiento
Vitamina K, a veces el parVitamina K, a veces el paréénquima no nquima no responde, si el dresponde, si el dééficit incluye a los no ficit incluye a los no vitamina K las pruebas no corrigenvitamina K las pruebas no corrigen
Plasma fresco congeladoPlasma fresco congelado Concentrados de factoresConcentrados de factores AntifibrinolAntifibrinolííticosticos


















