
Capitulo 16, ar y eb
57
Inmunodeficiencia Primaria Capitulo 16
-
Upload
juliana-maggioni -
Category
Documents
-
view
305 -
download
3
Transcript of Capitulo 16, ar y eb

Inmunodeficiencia Primaria
Capitulo 16

INTEGRANTES:
Reveca Pinto Benítez Laura Barrios Gisell Velazquez Magnolia Benítez Paéz

Inmunodeficiencia Primaria
Se debe a los defectos intrínsecos de las cé lulas del sistema inmunitario

Las inmunodeficiencias se deben:
Las inmunodeficiencias específicas Las inmunodeficiencias inespecíficas

Las infecciones que contraen los pacientes inmunodeprimidos se puede dividir en dos categorias:
Con defectos de las inmunoglobulinas , las proteínas del complemento o los fagocitos.
Con defectos de la inmunidad celular ( cé lulas T).

Anomalías de las cé lulas B
Tienen como consecuencia la aparició n de infecciones piogénicas recurrentes tales como:
Neumonías; Otitis media; Sinusitis

La agammaglobulinemia Ligada al cromosoma x
Tienen un defecto en las primeras fases de maduració n de las cé lulas B.
Los varones afectados carecen total o casi totalmente de células B en la sangre y en los tejidos linfoides

Su estado de salud se puede mantener estable mediante la administració n por vía intravenosa de grandes dosis de gammaglobulinas.

Déficit de las subclases IgA e IgG.
Pacientes con déficit de IgA: desarrollan enfermedades por inmunocomplejos, carecen también de IgG2 e IgG4.
Estos déficit de determinadas clases y subclases de inmunoglobulinas son debidos a defectos en la diferenciació n final de cé lulas B

Síndrome de Inmunodeficiencia con aumento de IgM
No se produce el cambio entre isotipos de inmunoglobulinas.
Se caracteriza por el déficit de IgG e IgA en presencia de grandes cantidades de IgM policlonal.

En el 70% de los casos, la HIgM:
Se hereda con carácter recesivo ligado al cromosoma X, debido a mutaciones en el ligando de CD40

Los Individuos con Inmunodeficiencia Variable Común.
Existen defectos en la transmisió n de señ ales desde las cé lulas T a a las cé lulas B.
Presentan una agammmaglobulinemia adquirida durante la segunda o tercera décadas de su vida o incluso aún más tarde.
Afecta por igual a ambos sexos.

Pacientes con ALX e IDVC son muy susceptibles a:
Infecciones por organismos piogénicos
Producidas por el protozoo intestinal Giardia lamblia, que provoca una intensa diarrea.

Hipogammagloblinemia transitoria de la infacia
Se retrasa el momento en que comienza la síntesis de IgG.
Los lactantes normales comienzan a sintetizar sus propias IgG a los 3 mese de edad.
Algunos niñ os la síntesis normal de IgG se puede retrasar hasta los 36 meses, desde entonces los niñ os son altamente susceptibles a las infecciones piogénicas

Anomalías de las cé lulas T
Los defectos de la funció n de las cé lulas T tienen como consecuencia una mayor susceptibilidad frente a las infecciones oportunistas.
Las anomalías de las cé lulas T dan lugar a una inmunodeficiencia combinada en la que resultan afectadas tanto la inmunidad humoral como celular.

Imunodeficiencias primarias de las cé lulas T
Inmunodeficiencia combinada grave (IDCG) Déficit de adenosina desaminasa Déficit de nucleó sido de purina fosforilasa Déficit de mó leculas MHC de clase II Síndrome de DiGerge Ataxia-telangiectasia hereditaria (AT) Síndrome de Wiskott-Aldrich (SWA)

Inmunodeficiencia combinada grave
Existe déficit de linfocitos y falta de desarrollo del timo.
Desarrollan infecciones recurrentes desde el momento de su nacimiento

Estos niñ os suelen desarrollar:
Diarreas prolongadas provocadas por: rotavirus o por infecciones bacterianas del tracto gastrointestinal
Neumonías: causadas por el protozoo Pneumocytis carinii.

La ubicua levadura Candida albicans crece cn facilidad en su boca
o en la piel

La IDCG es incompatible con la vida
Los niñ os afectados suelen morir en el curso de los 2 primeros añ os de vida a no ser que el defecto sea corregido mediante un trasplante de médula ó sea.
Cuando se produce un trasplante de este tipo, los niñ os se convierten en quimeras linfocitarias y pueden sobrevivir y llevar una vida normal.

Déficit de las cé lulas TH
Se debe a un déficit de las moléculas MHC de clase II.
Enfermedad hereditaria, no va ligada al locus MHC situado en el brazo corto del cromosoma 6.
Los niñ os afectados padecen infecciones recurrente, especialmente del tracto gastrointestinal

Déficit de moléculas MHC clase II.
Se produce como consecuencia de defectos en las proteínas promotoras que se unen a la regió n 5` no codificado de los genes MHC clase II.

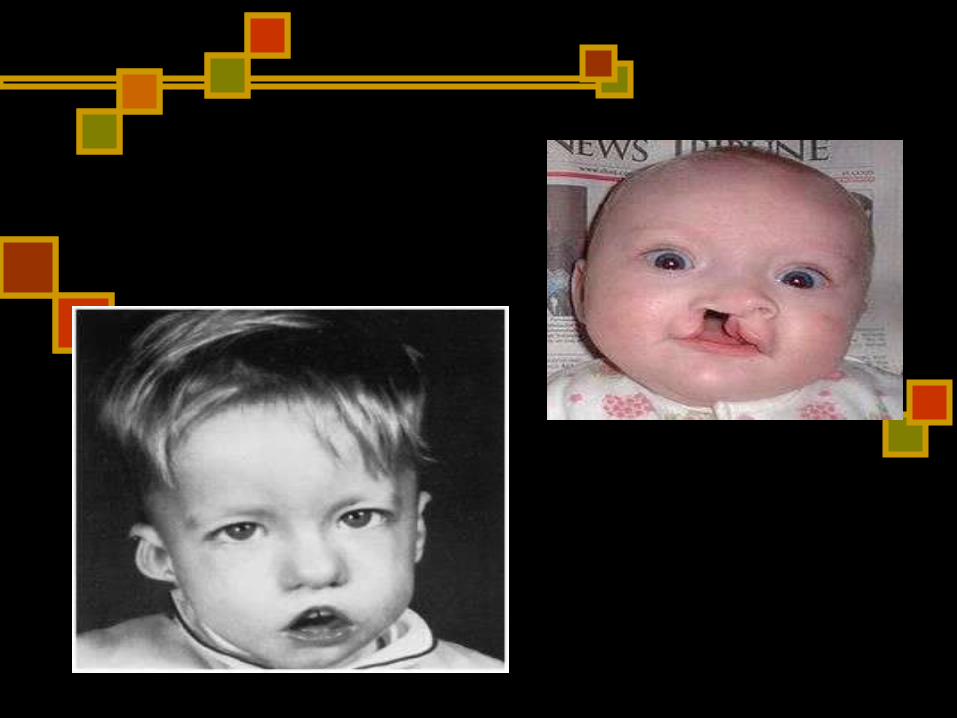
Síndrome de DiGeorge Se debe a un defecto en la
embriogénesis del timo. Los pacientes presentan rasgos faciales
característicos: Separació n excesiva de los ojos
(hipertelorismo) Posició n anormalmente baja de los
pabellones auriculares Acortamiento del surco subnasal del
labio superior

Síndrome Proliferativo ligado al cromosoma X
Aparece tras la infecció n con Virus de Epstein-Barr (VEB).
Los varones afectados parecen normales hasta que entran en contacto con el VEB, en cuyo caso desarrolla:
Mononucleosis infecciosa mortal Destrucció n completa de las cé lulas B, por lo que
aparece una agammmaglobulinemia Neoplasia linfática mortal Anemia aplásica.

Ataxia-telangiectasia
Exiten desperfectos cromosó micos que afectan a los genes de los TCR y de las inmunoglobulinas.
Los niñ os afectados comienzan a caminar de forma titubeante aproximadamente a los 18 meses de edad
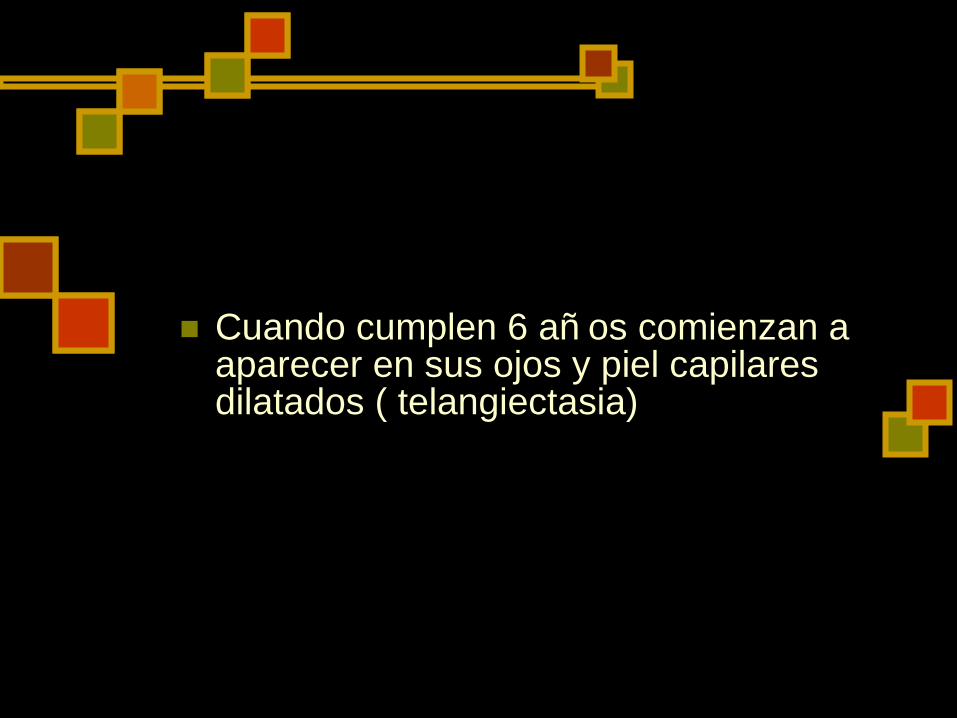
Cuando cumplen 6 añ os comienzan a aparecer en sus ojos y piel capilares dilatados ( telangiectasia)

Síndrome de Wiskott-Aldrich
existen anomalías de las cé lulas T y se detectan concentraciones anormales de inmunoglobulinas.

Los varones afectados:
Tienen plaquetas pequeñ as, su concentració n es más baja de lo normal (Trombocitopenia).
Desarrollan infecciones piogénicas y oportunistas.
Tienen cé lulas T con alteració n de su funcionamiento

Defectos Genéticos de las proteínas del complemento
La eliminació n de inmunocomplejos, La inflamació n; La fagocitosis Y la bacterió lisis.
Pueden verse afectadas por anomalías del complemento.

El Edema Angioneuró tico Hereditario (EAH)
El déficit más importante del sistema del complemento es el inhibidor C1.
Los pacientes con EAH presentan episodios recurrentes de Hinchazó n en determinadas regiones corporales

Exiten dos formas genéticamente determinadas de EAH
Tipo I, el gen inhibidor de CI es defectuoso y no llega a transcribir.
Tipo II, gen inhibidor de CI contiene mutaciones puntuales, lo que da lugar a la síntesis de moléculas defectuosas

Defectos genéticos de los fagocitos
Pueden provocar infecciones muy graves.
Existen dos defectos genéticos de los fagocitos que suelen dar lugar a infecciones graves y que muchas veces conducen a la muerte:
Granulomatosis Cró nica ( GC)
El defecto de adherencia leucocitaria (DAL)

Artritis Reumatoide

Artritis Reumatoide
Es una enfermedad cró nica que lleva a la inflamació n de las articulaciones y tejidos circundantes. También puede afectar otros ó rganos.

Causas La causa de la artritis reumatoidea (AR) se
desconoce. Es una enfermedad autoinmunitaria, lo cual significa que el sistema inmunitario del cuerpo ataca por error al tejido sano.
La artritis reumatoidea se puede presentar a cualquier edad. Las mujeres resultan afectadas con mayor frecuencia que los hombres.
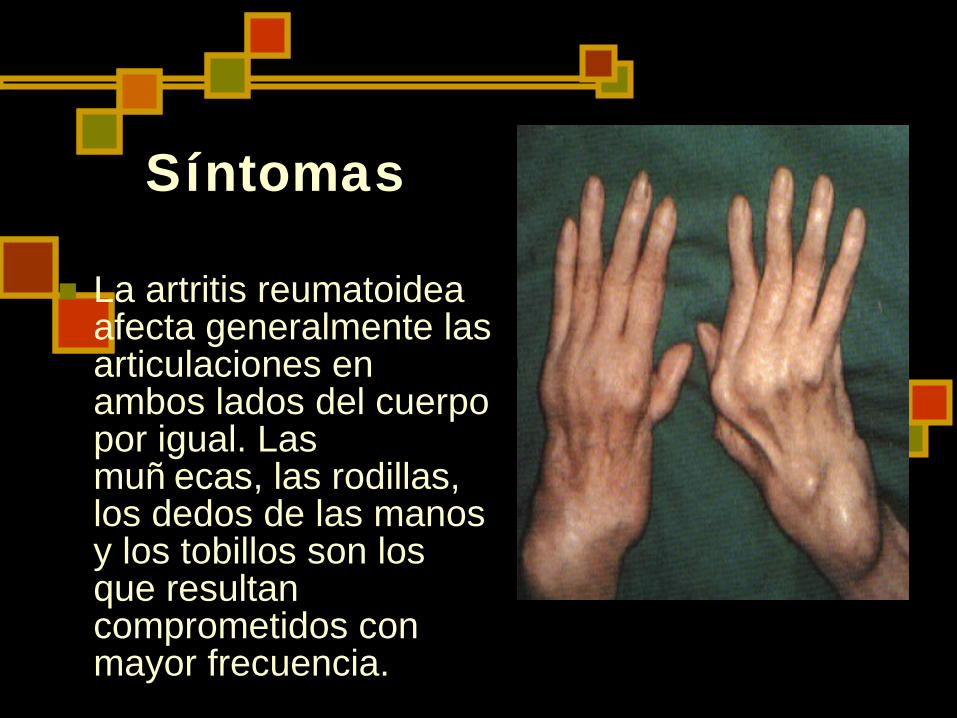
Síntomas
La artritis reumatoidea afecta generalmente las articulaciones en ambos lados del cuerpo por igual. Las muñ ecas, las rodillas, los dedos de las manos y los tobillos son los que resultan comprometidos con mayor frecuencia.

Dos pruebas de laboratorio que a menudo ayudan en el diagnó stico son:
Prueba del factor reumatoideo Prueba de anticuerpos antipéptidos
cíclicos citrulinados (anticuerpos anti-PCC)

Otros exámenes que se pueden hacer abarcan:
Conteo sanguíneo completo Proteína C reactiva Tasa de sedimentació n eritrocítica Ecografía o resonancia magnética (RM)
de las articulaciones Radiografías de las articulaciones Análisis del líquido sinovial

Tratamiento
La artritis reumatoidea generalmente requiere tratamiento de por vida que incluye medicamentos, fisioterapia, ejercicio, educació n y posiblemente cirugía. El tratamiento agresivo y oportuno para este tipo de artritis puede retardar la destrucció n de la articulació n.

Medicamentos
La artritis reumatoidea generalmente requiere tratamiento de por vida que incluye medicamentos, fisioterapia, ejercicio, educació n y posiblemente cirugía. El tratamiento agresivo y oportuno para este tipo de artritis puede retardar la destrucció n de la articulació n.

Medicamentos antinflamatorios:
abarcan ácido acetilsalicílico (aspirin) y antinflamatorios no esteroides (AINES), como ibuprofeno y naproxeno.

Medicamentos antipalúdicos:
este grupo de medicamentos abarca hidroxicloroquina (Plaquenil) y sulfasalazina (Azulfidine), por lo general, usados en combinació n con metotrexato. Pueden pasar semanas o meses para ver algún beneficio de estos medicamentos.

Corticosteroides:
estos medicamentos funcionan bien para reducir la hinchazó n e inflamació n articular. Sin embargo, debido a los efectos secundarios a largo plazo, los corticosteroides se deben tomar só lo por un corto tiempo y en dosis bajas cuando sea posible

CIRUGÍ A
Ocasionalmente, se necesita cirugía para corregir las articulaciones que sufrieron dañ o grave. La cirugía puede abarcar:
Extirpació n del revestimiento articular (sinovectomía).
Artroplastia total en casos extremos; puede incluir artroplastia total de rodilla, artroplastia de cadera, artroplastia del tobillo, artroplastia del hombro y otras.

El síndrome de Behcet

Es una enfermedad reumática cró nica que causa una inflamació n de los vasos sanguíneos (vasculitis) de causa desconocida, que puede afectar a casi cualquier parte del organismo (distribució n generalizada o sistémica) y está catalogada como una enfermedad rara.

caracterizado por tres síntomas:
Estomatitis aftosa, úlceras genitales y uveitis, inflamació n de los capilares que
riegan el ojo localizados en la úvea.
Es más frecuente en hombres que en mujeres.
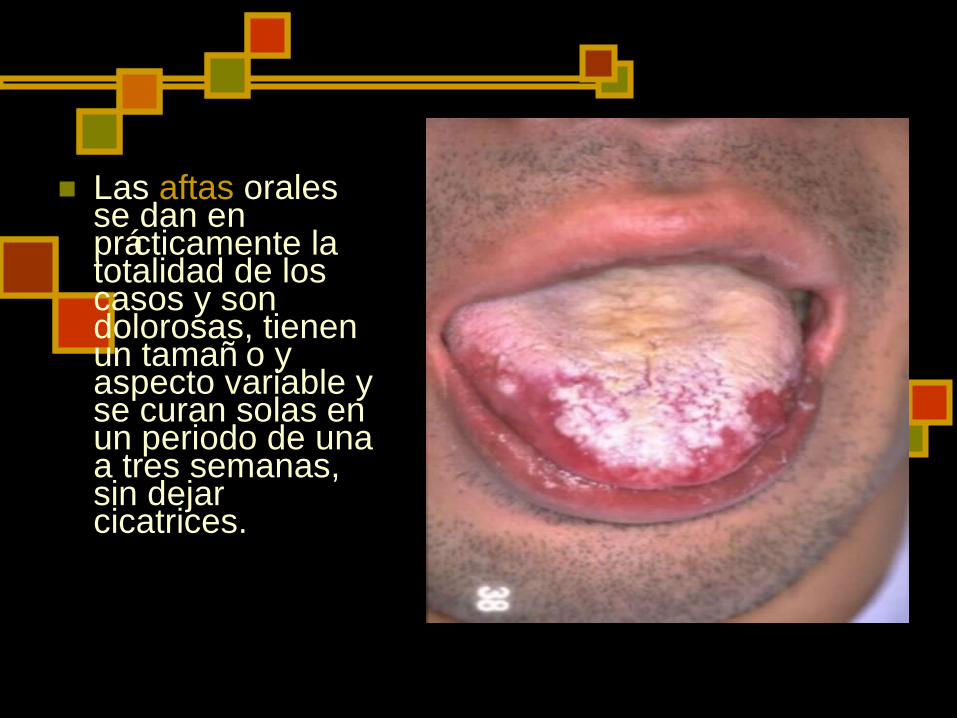
Las aftas orales se dan en prácticamente la totalidad de los casos y son dolorosas, tienen un tamañ o y aspecto variable y se curan solas en un periodo de una a tres semanas, sin dejar cicatrices.
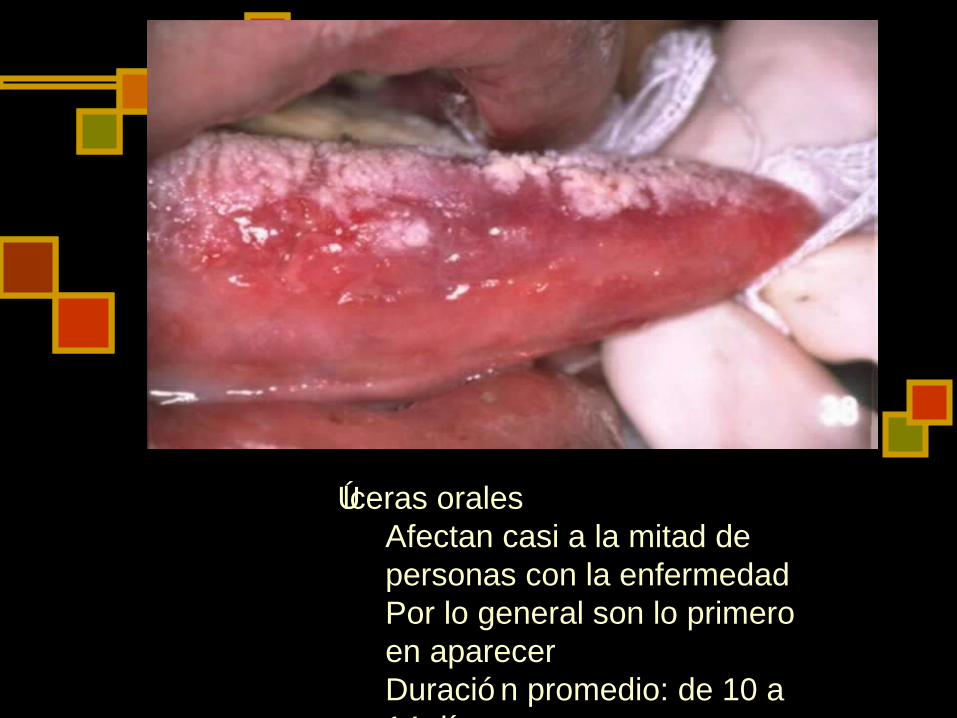
Úlceras orales Afectan casi a la mitad de personas con la enfermedad Por lo general son lo primero en aparecer Duració n promedio: de 10 a 14 días Pueden ser dolorosas Pueden causar cicatrizació n

Las aftas genitales se observan en más de la mitad de los casos tratados, se localizan en el glande y escroto en el varó n y en la vulva, vagina y cervix en la mujer, siendo dolorosas y tardando en cicatrizar en el varó n, y mucho menos molestas en las mujeres.

Obs:
Los síntomas de la enfermedad de Behcet pueden variar de leves a muy severos. Los síntomas tienden a aparecer, sanar, y después volver a aparecer (referido como un brote) frecuentemente durante meses o añ os.
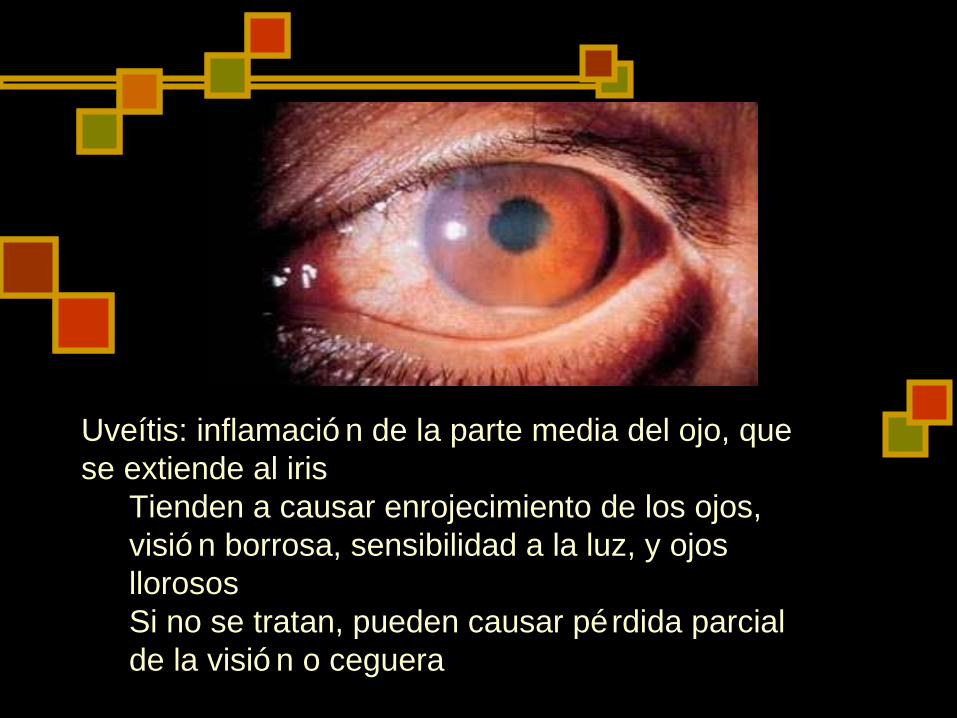
Uveítis: inflamació n de la parte media del ojo, que se extiende al iris
Tienden a causar enrojecimiento de los ojos, visió n borrosa, sensibilidad a la luz, y ojos llorosos Si no se tratan, pueden causar pérdida parcial de la visió n o ceguera

Los medicamentos están dirigidos a reducir la inflamació n o bien a intentar regular el sistema inmunitario. Algunos de los medicamentos usados son:
Corticoides locales, que pueden aplicarse directamente en las lesiones de la piel, boca u ojos para reducir la inflamació n y el dolor.
Antiinflamatorios no esteroideos y
analgésicos, como la aspirina, ibuprofeno y el paracetamol para aliviar la inflamació n articular y el dolor.

Corticoides orales, como la prednisona, para reducir la inflamació n.
Inmunosupresores, se utilizan para frenar la actividad inmunoló gica en las manifestaciones graves de la enfermedad y requieren una estricta vigilancia del tratamiento.

Gracias


















