
7.pdfCentro Médico EuroEspes Instituto para Enfermedades del Sistema Nervioso Central y Medicina...
100
www.gen-t.es JULIO 2011 • Nº 7 • P.V.P 5,00€ MEDICINA GENÓMICA, FARMACOGENÓMICA Y BIOTECNOLOGÍA DE LA SALUD Estados Depresivos Estados Depresivos
Transcript of 7.pdfCentro Médico EuroEspes Instituto para Enfermedades del Sistema Nervioso Central y Medicina...

www.gen-t.es
JULIO 2011 • Nº 7 • P.V.P 5,00€ MEDICINA GENÓMICA, FARMACOGENÓMICA Y BIOTECNOLOGÍA DE LA SALUD
EstadosDepresivos
EstadosDepresivos

Centro Médico EuroEspesInstituto para Enfermedades del Sistema Nervioso Central y Medicina GenómicaSanta Marta de Babío15165 Bergondo, La Coruña, EspañaTeléfono: 981 780 505 • Móvil: 608 322 207Fax: 981 780 [email protected] • www.euroespes.com
Plan PROFEPlan para la identificación precoz, la prevención y tratamiento del fracaso escolar en niños, adolescentes y jóvenes.
Tarjeta FarmacogenéticaLa personalización de los tratamientos, dando el fármaco adecuado en la dosis óptima a cada persona, para mejorar su eficacia y evitar efectos adversos.
Plan de Prevención del Riesgo Cerebral para DirectivosPrograma pionero en la prevención del riesgo cerebral en altos ejecutivos basado en los avances de la medicina genómica.
Plan de Prevención de la DemenciaProtocolo para el diagnóstico integral y el tratamiento multifactorial en pacientes con demencia (Alzheimer, vascular, carencial, metabólica).
Plan de Prevención Genética del Síndrome Metabólico y los Accidentes CerebrovascularesPrimer protocolo de prevención genética y medicina personalizada para combatir el Síndrome Metabólico que afecta a más de un 20% de la población.
Gracias por confiarnos su salud estos 20 años
escolar en niños, adolescentes y jóvenes.
personalizada para combatir el Síndrome Metabólico que afecta
población.
Síndrome Metabólico que afecta a más de un 20% de la población.
Síndrome Metabólico que afecta
Gracias por confiarnos su salud estos 20 años
EuroEspesSistema Nervioso Central
y Medicina Genómica
1991-2011XX Aniversario
más de 12.000 pacientes ya conocen su genómica

uando las relaciones hu-manas están mediatizadas por la perversión política y los conflictos de interés, se envi-lece la convivencia, surge la desconfianza y se degradan los valores. El debilitamiento moral
de una sociedad la hace más vulnerable a los ataques de oportunistas, chantajistas y depre-dadores sociales. La falta de liderazgo hace el resto: aumentan los complejos, se acentúan las sensaciones de persecución paranoide, se po-lariza la atención en la autodefensa, se relaja el principio de autoridad, y se busca el enemigo fuera para justificar las desgracias internas. Todas estas circunstancias hacen que el con-glomerado de parcelas que constituyen nuestra sociedad se vaya resquebrajando gradualmen-te, con el consecuente incremento de la des-confianza, la inseguridad, y la quiebra de una voluntad de crecimiento.
La gaseosa de los despropósitos políticos, agi-tada por la crisis económica, está a presión. La histórica hipocresía de nuestros líderes, acostumbrados a ocultar la podredumbre de su deficiente gestión (y moral) debajo de la al-fombra, ya no resiste por más tiempo el silencio cómplice de los adláteres de la España azul y la España roja, los que siempre sacan tajada según el color de la camiseta que gobierne. Por fin se empieza a hablar del despropósito de las autonomías y del fracaso colectivo de la política autonómica. Todo el mundo lo sabía (aunque muchos prefirieron negar la evidencia), pero nadie se atrevía a manifestar la inviabilidad del actual sistema de salud. Ya saltó la liebre de la alarma sanitaria; ya se abrió la caja de Pando-ra, el tarro de las esencias de nuestros grandes fracasos históricos: un marco laboral mutilado, un sistema financiero momificado, una políti-ca autonómica ruinosa, un sistema sanitario deficiente y económicamente insostenible, un modelo educativo no competitivo, un sistema judicial viciado y dependiente del bipartidismo reinante, y un aparato pseudodemocrático sub-sidiado.
¿Qué más necesitamos para despertar de nuestra prolongada siesta de autocomplacen-cia? La irresponsabilidad de la casta dirigente es tal que no se conforman con fracasar ellos, sino que en la caída quieren ir acompañados por la sociedad que les otorgó su confianza. Cuanto más hundida está una sociedad, me-nos vale (piensan lo mismo los tiburones con las empresas; primero las hunden y luego las redimen con una compra de calderilla). No es momento para dejarse embaucar por cantos de sirena ni por promesas redentoristas de brujos de salón, de los que se rasgan las vestiduras culpando a los anteriores; porque son los mis-mos; sólo han cambiado la camiseta. Son los que han alimentado al monstruo autonómico, los que han inflado a la gran vaca del estado para ordeñarle una leche artificial, los que han
puesto en quiebra el sistema financiero, los que han creado un país de funcionarios, los que han mantenido un estado de beneficencia corrupta, los que han inventado las fundaciones públicas con objetivos sectarios, los que compran a la prensa, los que se rodean de asesores inútiles para que les sirvan de parachoques, los que han explotado abusivamente las Spin-Offs para ocultar su incapacidad empresarial y jugar a lo privado con dinero público, los que han utiliza-do las corporaciones financieras para favorecer a sus amigos, los que nunca han entendido el principio de equidad cuando están en el poder, los que han utilizado su posición de privilegio para crear el monstruo administrativo, burocrá-tico y esperpéntico que tenemos que alimentar los ciudadanos. Son los mismos que se entre-tienen con la paja del ojo ajeno y no se preocu-pan de su ceguera; los que hurgan en la basura del vecino y viven en la mugre; los que van a misa de doce los domingos y crucifican a su ad-versario a la hora de comer; con la misma boca llena con la que rumian sus virtudes, salpican de calumnia y difamación a todo aquel que no comparte sus ideas. Son los que predican de-mocracia y ejercen de caudillos; los que desde lo público se sirven de lo privado para sus in-tereses; los que otorgan concursos bajo manga y prometen perseguir las corruptelas del poder; los que usan distinta vara de medir dependien-do del inquilino, pero se cuelgan la etiqueta de inspectores de lo justo; los que han hundido las
cajas de ahorros y ahora luchan por su resu-rrección para no ver debilitada su influencia y su nómina; los que evalúan los proyectos por el nombre de quien los presenta y no por la cali-dad de la propuesta; los que abanderan la pu-reza académica y viven permanentemente en la ciénaga del mercantilismo oportunista; son los mismos…en todas partes.
Una de las cosas buenas que tiene la globaliza-ción y el universo de las telecomunicaciones es que ya nadie puede escapar al ojo escrutador del curioso, del mercado o del depredador que sobrevuela el campo en busca de una presa fácil. Hoy, todo país, con sus virtudes y sus de-fectos, es analizado puntualmente por la red de intereses que le rodea. Hoy no se pueden tomar decisiones aleatorias basadas en la idiosincra-sia o el capricho político del momento, cuando esas decisiones repercuten en las personas y en terceros. En esta gran aldea interactiva, las decisiones que tome cualquier autoridad, sea del campo político, judicial, académico, laboral, educativo o empresarial, tienen repercusiones y consecuencias. Todo entra en la gran panta-lla analítica del ciberespacio; y pocas cosas se
pueden ocultar en una sociedad avanzada. Con lo cual el precio del progreso empieza a afec-tar al bolsillo de la intimidad, la confabulación, las maniobras conspirativas, el nivel de cono-cimiento, y la propia talla moral. En este esca-parate nudista resulta muy difícil al personaje público esconder sus vergüenzas; y ya la propia máquina judicial a nivel de estado (Islandia, Fin-landia, Italia) o a nivel transnacional (La Haya) empieza a pedir cuentas a quienes han tenido responsabilidades civiles y han violado la con-fianza de las personas, o han abusado de ellas, o han sido tan inmorales que no han tenido la decencia de declararse incapaces.
La política se ha convertido en un distractor po-blacional; amplificada por el eco mediático es una fuente de conflictividad social y un elemen-to de inestabilidad y desconfianza. España está exportando una imagen de división, fragmen-tación, insuficiencia, negligencia, vulnerabilidad económica, inadaptación educativa, fragilidad judicial, impotencia empresarial, anacronía laboral, endogamia académica, sectarismo re-gional e incapacidad cooperativa ante la ad-versidad de los tiempos, cuyas repercusiones son inimaginables para la bisoñez de los que se creen los reyes del mambo en los salones del poder. El coste del descrédito es mucho más alto que el de la deuda y tarda más en sufra-garse.
Es normal que en este mundo de “pepinos”, los alemanes -los listos de la clase- cuando les surge un problema de salud pública le echen la culpa al tonto del pueblo (ese vecino del sur, ese desaliñado contaminante, ese indigente endeu-dado, ese cuya situación lamentable -liderada por inútiles, agentes del parasitismo europeo- es una supuesta fuente de infección exporta-ble). Y no pasa nada. Ya no queda ni el orgullo de protestar frente a la injusticia o la acusación falsa. No se sabe utilizar el aparato del estado, sus instituciones académicas y científicas, para verificar la naturaleza de las acusaciones y ha-cer que aflore la verdad y se nos devuelva la dignidad vilipendiada en la prensa internacional (amén de las pérdidas económicas). Cada día nos demuestra cómo la insuficiencia intelectual no da tregua a la ineptitud. Estamos rebasando el límite de lo tolerable, por el bajo perfil de los de dentro y la desconsideración intencionada de los de fuera. Algunos debieran empezar a pensar que la sensación de asfixia puede aca-bar sobresaltando a la sociedad durmiente y enfureciendo a los pacíficos. Aunque estemos rodeados de “pepinos”, no todos vivimos intoxi-cados.
Pepinos, Parásitos e Intoxicaciones
C
EDITORIALpor Ramón Cacabelos
Julio 2011 3
La irresponsabilidad de la casta dirigente es tal que no se conforman con fracasar ellos, sino que en la caída quieren ir
acompañados por la sociedad que les otorgó su confianza

Gen-T Nº-7 Julio 2011
Editor-JefeRamón CaCabelos
DirecciónJavieR sánChez
Administración áuRea PeReiRo
Secretaria de Redacción RoCío maRtínez
Diseño y Producción JavieR masoliveR
PatRiCia Ria Ria odRíguez
Edición Internacional adam mCKayKayK
Relaciones Públicas gladys bahamonde
Personal Auxiliar amanda bello
CaRmen FRaile
Edición y Producción euRoesPes Publishing
ediF. euRoesPes, P1santa maRta de babío s/n
beRgondo, 15165-CoRuña
CONSEJO EDITORIAL: Antón Álvarez Farmacología Clínica y Experimental Pablo Bourkaib Nutrición, Nutracéutica y Nutrigenómica Ramón Cacabelos Medicina Genómica Pablo CarnotaOftalmología Iván Carrera Neurociencias Básicas Juan Carlos Carril Genómica Humana y Genética Forense Dolores Corzo Bioquímica Médica y Tecnología Analítica Lucía Fernández-Novoa Genómica Médica José Augusto García-Agúndez Farmacogenómica Salvador Harguindey Cáncer José Iglesias Pediatría Francisco Javier Jiménez-Gil Neurología Valter Lombardi Biotecnología de la Salud Antonio Moreno Neuroimagen Rodolfo Rodríguez Neurocirugía Ramón Segura Cirugía Vascular José Miguel Sempere Inmunología Masatoshi Takeda Psiquiatría y Psicogeriatría Iván Tellado Diagnóstico Digital Juan Carlos Yáñez Cardiología.
COLABORADORES: Xavier Alcalá, Pablo Álvarez de Linera, Jack de la Torre, Jesús Figueroa, Günter Freeman, José Manuel Garaeta, Luís García Mañá, Ruth Llovo, Irene Lourido, Manuela Márquez, José María Martín, Ricardo Martínez, Kiko Novoa, Luís A. Outeiriño, Ricardo Palleiro, Víctor Pichel, Andreas Pfützner, José Antonio Quesada, Antón Reixa, Fernando Sánchez Dragó, Sergio L. Sánchez Suárez, Ana Isabel Vallejo, Carmen Vigo.
Gen-T no se responsabiliza de las opiniones y criterios emitidos por los autores, reservándose la propiedad de los trabajos publicados. Queda expresamente prohibida la reproducción parcial, literaria o iconográfica de cualquier contenido sin previa autorización del editor.
ISSN: 1888-7937 Depósito Legal: C 713-2007 Impreso en España
Estados Depresivos¿Quién no habrá dicho alguna vez: “Estoy deprimido”, “Está con depresión”? En este nuevo número de Gen-T, The EuroEspes Journal, presentamos un artículo que trata precisamente de la depresión en sus distintas formas, y de la relación que tiene la genética con este trastorno que posiblemente tenga poco que ver con esa sensación de desgana que notamos de vez en cuando y tan imponderadamente tildamos de “depresión”.
otro tema que tratamos en estas páginas es la epilepsia, un mal que ha afectado a muchos personajes famosos a lo largo de la historia, y cuyas causas, gracias a la genética, están saliendo a la luz.
ofrecemos también artículos sobre la prevención del cáncer colorrectal, sobre las enfermedades cardiovasculares y sobre los progresos en la genómica a lo largo de la última década.
Que todo sea de su agrado.
EN PORTADA
www.gen-t.es
JULIO 2011 • Nº 7 • P.V.P 5,00€ MEDICINA GENÓMICA, FARMACOGENÓMICA Y BIOTECNOLOGÍA DE LA SALUD
EstadosDepresivosDepresivos
EstadosDepresivos
Estados DepresivosEl abismo de la melancolía, el delirio de la manía, los ciclos del trastorno bipolar, la herencia y la esperanza de la farmacogenómica 42

Gen-T Nº-7 Julio 2011
Julio 2011 5
SUMARIO Opinión
03 Editorial
07 Pluma Invitada
Ciencia
09 Genética de la Epilepsia
19 Prevención de colitis experimental crónica inducida por dextrán sulfato sódico (DSS) en ratones tratados con FR-91
31 Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular: Factores de Riesgo Emergentes
42 Estados DepresivosEl abismo de la melancolía, el delirio de la manía, los ciclos del trastorno bipolar, la herencia y la esperanza de la farmacogenómica
75 La revolución genómica: del proyecto Genoma Humano a la megasecuenciación como herramienta diagnóstica
Sociedad
87 V Conferencia Anual EuroEspes
Noticias
92 Ciencias Médicas
94 Noticias EuroEspes
Res Sacra Consilium
96 Consejos a un presidente
Genética de la Epilepsia 09
La revolución genómica: del proyecto Genoma Humano a la megasecuenciación como herramienta diagnóstica 75
Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular: Factores de Riesgo Emergentes 31
19Prevención de colitis experimental crónica inducida por dextrán sulfato sódico (DSS) en ratones tratados con FR-91


os recursos marinos no siempre se en-cuentran en un estado óptimo para el consumo humano, debido fundamen-talmente a la presencia temporal de ciertas toxinas que resultan perjudicia-ciertas toxinas que resultan perjudicia-ciertas toxinas que resultan perjudiciales, en mayor o menor medida, para el
organismo humano. Cuando esto sucede la Adminis-tración establece prohibiciones para su extracción por el peligro que representan para la salud de los consumidores.
Se trata de un problema sanitario que afecta especial-mente a los bivalvos y que obliga al cierre temporal de los bancos marisqueros.
Cuando la prohibición se prolonga en el tiempo per-Cuando la prohibición se prolonga en el tiempo per-Cuando la prohibición se prolonga en el tiempo permite un mayor desarrollo de estos bivalvos que llegan a alcanzar optimas tallas y pesos, haciéndolos espe-cialmente atractivos para su comercialización por personas que se dedican al furtivismo ilegal.
Se puede decir, sin lugar a dudas, que una de las ame-nazas que penden sobre esta importante actividad extractiva de los productos del mar en las costas galle-gas, además de la sobreexplotación, es el furtivismo.
El furtivismo es así una actividad ilegal que no debe ser tolerada socialmente, como con frecuencia ocu-rre, ya que pone en peligro la correcta regeneración de los recursos marisqueros al no respetar la plani-ficación de las vedas establecidas por la Administra-ficación de las vedas establecidas por la Administra-ficación de las vedas establecidas por la Administración, ni tampoco los topes de capturas, los controles sanitarios, los tamaños mínimos ni los circuitos lega-sanitarios, los tamaños mínimos ni los circuitos lega-sanitarios, los tamaños mínimos ni los circuitos legales de comercialización.
Su perfil más nocivo es el llamado “furtivismo pro-fesional” protagonizado por personas que realizan esta actividad como medio de subsistencia o como complemento de sus ingresos.
Una variante del furtivismo profesional en la Rías Baixas sería aquel que alterna la actividad extractiva con otras actividades ilícitas, como puede ser la del narcotráfico, y que en periodos de inactividad de-lictiva se centra en la extracción de vieira o almeja, organizándose en grupos que se dotan de telecomu-nicaciones y de potentes embarcaciones deportivas, equipos de buceo y aprovecha las horas nocturnas para eludir la acción de los servicios oficiales de con-trol.
El furtivismo profesional, al estar familiarizado con las situaciones de riesgo en defensa de su medio de vida, no duda en hacer frente a los agentes de vigilan-cia con amenazas y agresiones constitutivas de diver-cia con amenazas y agresiones constitutivas de diver-cia con amenazas y agresiones constitutivas de diversos ilícitos penales.
Junto a este tipo de furtivismo conviven otros, de menor entidad pero igualmente perjudiciales, como son el “furtivismo doméstico”, realizado por perso-nas que residen en los municipios costeros, gene-ralmente personas mayores o marineros jubilados que recogen ejemplares para consumo propio, el “furtivismo vacacional”, el “furtivismo con carencias
de integración social y necesidades económicas” y el “furtivismo recreativo” realizado por personas que amparadas por su licencia de pesca capturan espe-cies para introducirlas en el mercado mediante a venta directa a restaurantes o particulares.
Finalmente tendríamos el llamado “furtivismo le-gal”. En esta tipología se incluirían los mariscadores legales que superan los topes máximos de capturas y recogen el producto en zonas prohibidas o fuera de los horarios y fechas autorizadas.
Pero además el furtivismo constituye un delito contra la salud pública cuando se introducen en los circui-tos de consumo capturas que no pasaron los precep-tivos controles sanitarios, poniendo así en peligro la salud de las personas, como ocurre con la extracción de vieiras tóxicas.
En el mes de julio de 2010, la Consellería del Mar procedió a la apertura de la ría de O Burgo-A Pasaxe al comprobar, tras los análisis periódicos pertinentes, que se redujera considerablemente la carga micro-biológica hasta niveles que permitían la comerciali-zación del marisco en fresco.
Esta zona de extracción permanecía clausurada desde el año 2007, fecha en la que se detectó la pre-sencia de la bacteria Escherichia coli (E. Coli) de gran riesgo para la salud pública como recientes aconteci-mientos se han encargado de demostrar.
Al estar en peligro la salud de los consumidores, el furtivismo constituye un delito contra la salud públi-ca que sería cometido por los productores, distribui-dores o comerciantes que ofrecieran en el mercado productos nocivos para la salud.
Para evitar este mal es necesario demandar el máximo esfuerzo por parte de la Administración para actuar sobre todo en el control de la demanda, que es la que va a regular en gran parte la posibilidad de poner en el mercado el marisco extraído irregularmente para ponerlo a disposición de los establecimientos de con-sumo sin las preceptivas garantías de seguridad.
El reciente documento elaborado por el SEPRONA, a instancias de la Fiscalía Superior de Galicia, sobre la incidencia del furtivismo en nuestra Comunidad Autónoma constituirá un buen referente a tener en cuenta a la hora de diseñar actuaciones dirigidas a la conservación, gestión y explotación duradera y soste-nible de los recursos marisqueros por parte de quien ostenta la capacidad de crear estructuras de control, inspección y sanción que permitan neutralizar eficaz-mente los circuitos de distribución de todo producto extraído o comercializado ilegalmente procedente del marisqueo furtivo.
El futuro de la pesca y el marisqueo en Galicia depen-derá así de nuestra capacidad de cambiar y desterrar socialmente hábitos ilícitos y de profesionalizar debi-damente las estructuras económicas de este impor-damente las estructuras económicas de este impor-damente las estructuras económicas de este importante sector productivo.
L
por D. Carlos Varela GarcíaFiscal Superior de Galicia
Julio 2011 7
FURTIVISMO Y SALUD PÚBLICA


9Julio 2011
a epilepsia es una enfermedad neurológica que afecta al sistema nervioso central (SNC), y es la más frecuente después de las cefaleas. Afecta a más de 50 millones de personas en el mundo con dos picos, uno en la infancia y otro en la vejez. Las cifras de prevalencia oscilan entre 1.5 y 57 casos por 1000 habitantes. En España la prevalencia de la epilepsia se sitúa en torno a 8/1000 habitantes (supondría aproximadamen-te 360000 casos en España). La incidencia anual de epilepsia en España es de 31 a 57/100000
(entre 12400 y 22000 casos nuevos cada año). La epilepsia es la tercera patología neurológica en frecuencia en personas mayores de 60 años, tras la enfermedad cerebrovascular y las demen-cias (Fig. 1).
KeywordsEpilepsia, genes, sistema nervioso central, farmacogenética.
L
Lucía Fernández-NovoaDepartamento de Genética Molecular
EuroEspes Biotecnología, Bergondo, Coruña
Genética de la Epilepsia
“Conviene que la gente sepa que nuestros placeres, gozos, risas y juegos no proceden de otro lugar sino del cerebro. Acerca de la ‘enfermedad sagrada’ no me parece más sagrada que las demás enfermedades, sino que tiene una causa natural. A mi parecer, aquellos que hicieron sagrada esta afección eran iguales que los actuales magos y purificadores, impostores y charlatanes que utilizan lo divino para ocultar su impotencia por no contar ninguna ayuda que ofrecer...”
Hipócrates en su libro “Sobre la enfermedad sagrada”

10
Genética de la Epilepsia
DefinicionesEs importante definir la terminología que engloba a la epilepsia y de este modo aclarar con-ceptos.
Crisis epiléptica. Manifestación clínica, ya sea moto-ra, sensitiva, sensorial, psíquica u otras, secundaria a una descarga anormal, sincronizada y excesiva de neuronas corticales; suele tratarse de episodios bruscos, breves, paroxísticos y autolimitados.
Epilepsia. Trastorno del sistema nervioso central caracterizado por la repetición de dos o más crisis epilépticas en ausencia de una causa inmediata aguda identificable que la provoque. Así, una úni-ca crisis o crisis epilépticas recurrentes secunda-ca crisis o crisis epilépticas recurrentes secunda-ca crisis o crisis epilépticas recurrentes secundarias a factores corregibles o evitables no permiten, sin más, el diagnóstico de epilepsia.
Síndrome epiléptico. Conjunto de signos y síntomas que definen un tipo determinado de epilepsia. Conjunto de entidades que agrupan a pacientes con características clínicas, electroencefalográfi-cas, etiológicas, fisiopatológicas y pronósticas co-munes.
Status epiléptico. Aquella crisis comicial cuya du-ración excede los 30 minutos o bien, varias crisis encadenadas sin recuperación del nivel de con-ciencia entre ellas. Puede ser convulsivo o no convulsivo (suelen presentarse como alteración del comportamiento o del nivel de conciencia), parcial o generalizado.
ClasificaciónEn 1970, Gastaut hizo la primera clasificación de la epilepsia, dividiéndola en dos grandes grupos, dependiendo de si el área cerebral afectada estaba limitada a un hemisferio cerebral (parcial), o a am-bos hemisferios (generalizada). Posteriormente, en 1981, la “International League Against Epilepsy” (ILAE) estableció una nueva clasificación (ILAE 1981), en la que se incluyen dos nuevos términos en las crisis parciales, según si se preservaba (crisis parciales simples), o no (parciales complejas), el nivel de conciencia. En 1989, nuevamente la ILAE realizó una modificación de la clasificación previa (ILAE 1989), donde se introdujeron los términos
“Síndrome epiléptico” y crisis focales (parciales), y “Síndrome epiléptico” y crisis focales (parciales), y se debe nombrar el lóbulo afectado. Otra novedad de esta clasificación, fue la estratificación etiológi-ca de los síndromes en sintomáticos, idiopáticos y criptogénicos (origen desconocido). El primero se refería a aquellos donde la causa de la epilepsia se conoce; el segundo a aquellos casos en los que no se encontraba razón alguna para la epilepsia; y el tercero, cuando a pesar de la negatividad de todas las pruebas, se sospechaba un origen sintomático. Posteriormente ha habido numerosos intentos de reclasificar los síndromes epilépticos (2001, 2006 y 2010), sin conseguir aportar ningún cambio sus-tancial a la clasificación de 1989, que sigue tenien-do vigencia hoy en día.
Clasificación internacional de las crisis epilépticas (modificaciones ILAE 2010)
Generalizadas: Se originan y se desarrollan en am-bos hemisferios. Se subdividen en:
u Mioclónicasu Tónicasu Atónicas
u Clónicasu Tónicasu Tonicoclónicasu Atónicasu Ausencias
u Típicasu Atípicasu Especiales (ausencias mioclónicas,
mioclonías de los párpados)
Focales: Se originan en un hemisferio pero pue-den afectar posteriormente a ambos hemisferios. Se clasifican en:
u Con alteración de la conciencia o discog-Con alteración de la conciencia o discog-Con alteración de la conciencia o discognitivas
u Sin alteración de la concienciau Auras (con sintomatología motora, auto-
nómica, sensitiva o psíquica)u Focales que evolucionan a generalizadas
Desconocidas:
u Espasmos
Aproximadamente un 30% de las personas afectadas de epilepsia presentan problemas relacionados con la medicación que pueden desencadenar en una epilepsia refractaria o farmacorresistente, que se produce cuando el tratamiento anticonvulsivante no controla las crisis o sus efectos secundarios son limitantes para un desarrollo normal de la persona

Afecta a más de 50 millones de personas en el mundo con dos picos, uno en la infancia y otro
en la vejez
Julio 2011 11
ciencia
Clasificación internacional de las crisis epilépticas según etiología (modificacio-nes ILAE 2010)
Genéticas: De base genética definida.
Metabólico/Estructurales: Existe una etiología co-nocida causante de la crisis (ictus, traumatismos craneoencefálicos, infecciones).
Causa desconocida: Se sospecha de una causa sin-tomática pero no puede ser determinada.
Clasificación internacional de las epilep-sias y síndromes epilépticos (modificacio-nes ILAE 2010)
Síndromes Electroclínicos: Presentan caracterís-ticas electroclínicas comunes (edad de inicio, tipo de crisis, características eléctricas).
Neonatal: Epilepsia benigna neonatal familiar (BFNE)Principios de la encefalopatía mioclónica (EME)Síndrome de Ohtahara
Infancia: Migración de las crisis parciales de la infanciaEpilepsia mioclónica grave de la infancia (Síndrome de Dravet)Convulsiones benignas de la infanciaCrisis familiar benigna infantil
Infancia tardía: Convulsiones febriles plus (FS+) (puede comenzar en la infancia)Epilepsia benigna occipital (tipo Panayioto-poulos)Epilepsia mioclónica con atonía y crisis convulsivasEpilepsia benigna con puntas centrotem-porales (BECTS)Epilepsia nocturna del lóbulo frontal auto-sómica dominante (ADNFLE)Epilepsia occipital (tipo Gastaut)Epilepsia con ausencias mioclónicasSíndrome de Lennox-GastautEncefalopatía epiléptica con punta-onda y durante el sueño (CSWS) incluyendo: Sín-drome de Landau-Kleffner (LKS)Epilepsia de ausencia infantil (CAE)
Adolescencia:Epilepsia de ausencia juvenil (JAE)Epilepsia mioclónica juvenil (JME)Epilepsia mioclónica progresiva (PME)Epilepsia con crisis generalizadas tónico-clónicasEpilepsia parcial autosómica dominante con características auditivas (ADPEAF)
Constelaciones: No presentan características tan específicas como las anteriores, aunque si pre-sentan atributos distintivos.
Epilepsia del lóbulo temporal mesial con esclerosis del hipocampo (MTLE con HS)Síndrome de Rasmussen
Convulsiones Gelásticas con hamartoma hipotalámico
Epilepsias Estructurales/Metabólicas: Incluyen las epilepsias secundarias a lesiones específicas de carácter estructural o metabólico, pero que no presentan un patrón electroclínico específico.
Malformaciones del desarrollo cortical (he-mimegaencefalía, hetertopías, etc)Síndromes Neurocutáneos (el complejo de esclerosis tuberosa, de Sturge-Weber, etc)TumorInfecciónTraumaAngiomaInsultos perinatalesOtros
De causa desconocida:Convulsiones neonatales benignas (BNS)Convulsiones febriles (FS)
Genética de la EpilepsiaEstudios de agregación familiar indican que la epilepsia tiene un componente genético. El ries-go de padecer epilepsia es de 2 a 4 veces mayor en los familiares de primer grado de pacientes con epilepsia, con respecto a la población gene-ral. En estudios con gemelos se ha encontrado una mayor tasa de concordancia entre gemelos monozigóticos que en dizigóticos, tanto para la epilepsia generalizada como para la focal (aun-que la tasa de concordancia fue mayor en la ge-neralizada). Hoy en día la epilepsia se considera una enfermedad genéticamente compleja, en el que varios genes contribuyen cada uno con un pequeño efecto al desarrollo de la enfermedad,
Cartografía cerebral de epilepsia

Fig. 1. Epidemiología de la epilepsia en el mundo (Adaptado de Epilepsy Atlas © WHO
2005).
Número medio de personas con epilepsia por 1000 habitantes en
regiones OMS
Número medio de personas con epilepsia por 1000 habitantes en
cuanto a grupos de salarios
Número de personas con epilepsia en
regiones OMS
Los números son indicativos y se basan
en la información de las encuestas de los países
que han respondido
Fig. 1. Epidemiología de la epilepsia en el mundo
La epilepsia es un trastorno del sistema nervioso central caracterizado por la repetición de dos o más crisis epilépticas en ausencia de una causa inmediata aguda identificable que la provoque. Así, una única crisis, o crisis epilépticas recurrentes secundarias a factores corregibles o evitables, no permiten, sin más, el diagnóstico de epilepsia
(Adaptado de Epilepsy Atlas© WHO 2005)
12
Genética de la Epilepsia
de tal manera que de forma individual no ten-drían ese efecto. Estos genes a su vez están inte-ractuando con factores ambientales para produ-cir el fenotipo de enfermedad.
Aunque representan un porcentaje pequeño, existen formas monogénicas de epilepsia en las que se han identificado diferentes mutaciones en diferentes genes. Muchas de estas mutacio-nes se han localizado en genes que codifican para canales iónicos de sodio, potasio, calcio y cloro (Tabla 1). Los canales iónicos son impres-cindibles para la generación y transmisión de señales en el sistema nervioso central, partici-pan en varias funciones celulares como la comu-nicación neuronal, la contracción muscular, la conducción sensitiva y la secreción endocrina. Las mutaciones en los genes que codifican los canales iónicos se traducen en incrementos o disminuciones del flujo iónico, por lo que se ve-rán alteradas diversas funciones celulares.
Los canales de Na+ dependientes de voltaje son de los principales responsables de la rápi-da despolarización de la membrana neuronal presente en los procesos epilépticos. Las muta-ciones en las subunidades alfa (SCN1A) y en las subunidades beta (SCN1B) de estos canales de Na+ se asocian a determinadas formas de epi-lepsia (Tabla 1). Estos canales representan un importante sitio de unión para varios fármacos antiepilépticos: hidantoína, carbamazepina, ácido valproico y lamotrigina, entre otros. Las mutaciones en estos receptores son heterogé-neas y provocan la alteración de sus propieda-des pudiendo provocar ganancia o pérdida de función (Fig. 2 y 3).
La participación de los canales de Ca2+ depen-dientes de voltaje en las epilepsias proviene de la constatación de que las disminuciones acen-tuadas en la concentración extracelular de este ión pueden crear actividad epiléptica en ciertos tejidos cerebrales y estructuras del hipocampo. Los canales de Ca2+ dependientes de voltaje son altamente relevantes en los procesos funciona-les del sistema nervioso. Por ejemplo, la entra-da de Ca2+ presináptica provoca la liberación de neurotransmisores y la entrada postsináptica la despolarización sustentada de la neurona. Mu-taciones en genes codificantes para canales de Ca2+: CACNA1H y CACNA1H y CACNA1H CACNB4 se asocian con formas de epilepsia (Tabla 1).
El canal iónico de K+El canal iónico de K+El canal iónico de K participa en la repolariza-ción e hiperpolarización de las membranas celu-lares; se encuentra conformado por 4 subunida-des; la mutación de una ellas puede interferir en el control que este canal hace en la excitación celular. En las células excitables, la despolari-zación celular activa los canales de K+zación celular activa los canales de K+zación celular activa los canales de K y facilita la salida de K+la salida de K+la salida de K de la célula, lo que conduce a la repolarización del potencial de membrana. Además, los canales de K+Además, los canales de K+Además, los canales de K juegan un papel en el
Pídanos cita: +34 902 154 476 +34 981 780 505
Más información:
www.euroespes.com [email protected]
Centro Médico EuroEspes:Santa Marta de Babío s/n, 15165 Bergondo, La Coruña
Dar el fármaco adecuado, en la dosis óptima,para mejorar su e�cacia y evitar efectos adversos

ciencia
Pídanos cita: +34 902 154 476 +34 981 780 505
+34+34
Más información:
www.euroespes.com [email protected]
www.euroespes.com [email protected]
Centro Médico EuroEspes:Santa Marta de Babío s/n, 15165 Bergondo, La Coruña
Dar el fármaco adecuado, en la dosis óptima,para mejorar su e�cacia y evitar efectos adversos

Tabla 1. Tipos de epilepsias y genes asociados
Tipo de Epilepsia Modo de herencia Locus Gen Proteína
Epilepsia de ausencia infantil
ComplejaN/A
16p13.35q34-q3515q11.2-q125q348q24.31p34.2
CACNA1HGABRA1GABRB3GABRG2JRKSLC2A1
Calcium channel, voltage-dependent, t type, alpha-1h subunitGamma-aminobutyric acid (GABA) A receptor, alpha 1Gamma-aminobutyric acid (GABA) A receptor, beta 3Gamma-aminobutyric acid (GABA) A receptor, gamma 2Jerky homolog (mouse)Solute carrier family 2 (facilitated glucose transporter), member 1
Compleja
N/AAD
Epilepsia mioclónica juvenil
AD
Compleja
N/ACompleja
6p21.32q22-q233q27-q286p12.35q34-q358q24.38q24
BRD2 CACNB4CLCN2EFHC1GABRA1JRKKCNQ3
Bromodomain containing 2Calcium channel, voltage-dependent, beta 4 subunitChloride channel 2EF-hand domain (C-terminal) containing 1Gamma-aminobutyric acid (GABA) A receptor, alpha 1Jerky homolog (mouse)Potassium voltage-gated channel, KQT-like subfamily, member 3
Convulsiones febriles plus (FS +)
Compleja 1p|1p36.35q34 2q24.319q13.12q24.32q24
GABRDGABRG2 SCN1ASCN1BSCN2ASCN9A
Gamma-aminobutyric acid (GABA) A receptor, deltaGamma-aminobutyric acid (GABA) A receptor, gamma 2Sodium channel, voltage-gated, type I, alpha subunitSodium channel, voltage-gated, type I, betaSodium channel, voltage-gated, type II, alpha subunitSodium channel, voltage-gated, type IX, alpha subunit
AD
Síndrome de DravetAD
ARCompleja
5q342q24.319q13.18q24
GABRG2SCN1ASCN1BSCN9A
Gamma-aminobutyric acid (GABA) A receptor, gamma 2Sodium channel, voltage-gated, type I, alpha subunitSodium channel, voltage-gated, type I, betaSodium channel, voltage-gated, type IX, alpha subunit
Epilepsia benigna con puntas centro-temporales (BECTS)
AD Xq21.33-q23 SRPX2 Sushi-repeat-containing protein, X-linked 2
Síndrome de Lennox-Gastaut N/A 4q22.1-q23 MAPK10 Mitogen-activated protein kinase 10
Epilepsia benigna neonatal familiar (BFNE)
AD20q13.38q24
KCNQ2KCNQ3
Potassium voltage-gated channel, KQT-like subfamily, member 2Potassium voltage-gated channel, KQT-like subfamily, member 3
Epilepsia parcial autosómica domi-nante con características auditivas
(ADPEAF)AD 10q24 LGI1 Leucine-rich, glioma inactivated 1
Epilepsia de ausencia juvenil (JAE) Compleja3q27-q286p12.3
CLCN2EFHC1
Chloride channel 2EF-hand domain (C-terminal) containing 1
Epilepsia nocturna del lóbulo frontal autosómica dominante (ADNFLE)
AD8p2120q13.2-q13.31q21.3
CHRNA2CHRNA4CHRNB2
Cholinergic receptor, nicotinic, alpha 2 (neuronal)Cholinergic receptor, nicotinic, alpha 4Cholinergic receptor, nicotinic, beta 2 (neuronal)
Convulsiones benignas de la infancia AD 2q24.3 SCN2A Sodium channel, voltage-gated, type II, alpha subunit
Encefalopatía mioclónica
X recesivaN/AX dominanteADX dominante (en estudio)
AR
ADAR
AD/de novoCompleja
Xp21Xq11.1Xp2220q13.3Xq13.320p1219q13.3-q13.42q24.311p15.5
9q34.19q34.11
ARXARHGEF9CDKL5KCNQ2PCDH19PLCB1PNKPSCN2ASLC25A22
STXBP1SPTAN1
Aristaless related homeoboxCdc42 guanine nucleotide exchange factor (GEF) 9Cyclin-dependent kinase-like 5Potassium voltage-gated channel, KQT-like subfamily, member 2Protocadherin 19Phospholipase C, beta 1 (phosphoinositide-specific)Polynucleotide kinase 3'-phosphataseSodium channel, voltage-gated, type II, alpha subunitSolute carrier family 25 (mitochondrial carrier: glutamate), member 22Syntaxin binding protein 1Spectrin, alpha, non-erythrocytic 1 (alpha-fodrin)
AD (Autosómica dominante); AR (Autosómica recesiva)
14
Genética de la Epilepsia

Aunque representan un porcentaje pequeño, existen formas monogénicas de epilepsia en las que se han identificado diferentes
mutaciones en diferentes genes. Muchas de estas mutaciones se han localizado en genes que codifican para canales iónicos de sodio,
potasio, calcio y cloro
Julio 2011 15
ciencia
GABRB3, GABRG2 GABRD), que son receptores postsinápticos cuya estimulación produce la en-trada de cloro y por tanto la inhibición del im-pulso nervioso en la célula (Tabla 1).
Tratamiento de la EpilepsiaEl tratamiento de la epilepsia se basa en la utili-zación de medicamentos antiepilépticos (AED), cuyo mecanismo de acción consiste fundamen-talmente en la inhibición o facilitación de los ca-nales iónicos de Na+, Ca2+, K+, K+, K , Cl- y gabaérgicos. En la actualidad existen 3 generaciones de me-dicamentos antiepilépticos usados en la clínica; la primera incluye el fenobarbital, la fenilhidan-toína, las benzodiazepinas y la etosuximida. La segunda generación la componen la carbamaze-pina y el ácido valproico, y en la tercera están la vigabatrina, la gabapentina, el felbamato, la lamotrigina, el topiramato entre otros.
Fenobarbital: Su acción antiepiléptica más Fenobarbital: Su acción antiepiléptica más Fenobarbital:importante es el aumento de la actividad del receptor GABAA, prolongando la apertura del receptor del Cl- y, por tanto, la hiperpo- y, por tanto, la hiperpo- -larización. También disminuye la conduc-tancia de los canales de Na+, K+, K+, K y Ca2+.
mantenimiento del potencial de reposo celular, la frecuencia de disparo de las células automáti-cas, la liberación de neurotransmisores, la secre-ción de insulina, la excitabilidad celular, el trans-porte de electrolitos por las células epiteliales, la contracción del músculo liso y la regulación del volumen celular. También existen canales de K+K+K cuya activación es independiente de cambios del potencial de membrana que determinan el potencial de reposo y regulan la excitabilidad y el volumen extracelular. Mutaciones en dos ge-nes que codifican para canales de K+nes que codifican para canales de K+nes que codifican para canales de K , KCNQ2 y KCNQ2 y KCNQ2KCNQ3 se asocian a determinados tipos de epi-KCNQ3 se asocian a determinados tipos de epi-KCNQ3lepsias (Tabla 1).
El canal iónico de Cl- regula los potenciales de - regula los potenciales de -
membrana y sirve de señal celular. Permiten el paso pasivo de cloro por la capa bilipídica. Los canales se activan también por las concentra-ciones de magnesio y calcio y por el volumen celular. Mutaciones en el gen CLCN2, que co-difica para el receptor del canal de cloro, están implicadas en determinados tipos de epilepsia (Tabla 1).
En los genes codificantes para los canales ióni-cos relacionados con ligandos extracelulares también se han identificado mutaciones asocia-das a formas específicas de epilepsia (Tabla 1). Los genes CHRNA2, CHRNA4 y CHRNA4 y CHRNA4 CHRNB2 codifi-CHRNB2 codifi-CHRNB2can para receptores nicotínicos de acetilcolina, tienen un papel modulador y generan un poten-cial local que, cuando es de suficiente intensi-dad, desencadena la apertura de los canales de Na+ dependientes del voltaje.
Los receptores gabaérgicos median la acción inhibitoria del neurotransmisor ácido gamma-amino butírico (GABA). En la epilepsia se han identificado mutaciones en genes codificantes para receptores gabaérgicos tipo A (GABRA1,
Fig. 2. Mutaciones encontradas en el gen SCN1A asociadas con Convulsiones febriles plus (FS +)
(Adaptado de Metwally Y.)

16
Genética de la Epilepsia
Difenilhidantoína: Inhibe los canales de Na+, inhibe el flujo de calcio a través de las membranas neuronales mediante la modu-lación de los canales del calcio; por tanto disminuye la excitabilidad de la neurona.
Benzodiazepinas: Poseen una acción faciliBenzodiazepinas: Poseen una acción faciliBenzodiazepinas: -tadora de la transmisión gabaérgica me-diante el receptor GABAA.
Etosuximida: Inhibe los canales de CaEtosuximida: Inhibe los canales de CaEtosuximida: 2+ tipo T y se cree que también tiene acciones ga-baérgicas.
Carbamazepina: Inhibe los canales de Na+, por lo tanto la excitabilidad de la mem-brana neuronal. Posee otras propiedades como acción anticolinérgica, antidiurético de acción central, antiarrítmico, relajante muscular, antidepresivo, sedación y blo-queante neuromuscular.
Ácido valproico: Se cree que sus propiedades antiepilépticas son debidas a un aumento de la concentración del neurotransmisor GABA en las neuronas, debido a una dis-minución de su metabolismo o a una inhi-bición de la recaptación. También se cree que actúa inhibiendo los canales de Na+.
Vigabatrina: Pertenece al grupo de antie-pilépticos de tercera generación. Actúa aumentando la concentración del neuro-transmisor GABA, mediante la inhibición ireversible de su metabolismo o bloquean-do la recaptación de GABA.
Gabapentina: Es un análogo del neuroGabapentina: Es un análogo del neuroGabapentina: -transmisor GABA; se cree que se une a los canales de Ca2+ tipo N, además aumenta la concentración sináptica de GABA y dismi-nuye la liberación de neurotransmisores monoaminérgicos.
Felbamato: Su mecanismo de acción es des-conocido aunque estudios en animales e in vitro indican que puede actuar mediante la vitro indican que puede actuar mediante la vitro
Estudios de agregación familiar indican que la epilepsia tiene un componente genético. El riesgo de padecer epilepsia es de 2 a 4 veces mayor en los familiares de primer grado de pacientes con epilepsia, con respecto a la población general
Fig. 3. Mutaciones encontradas en el gen SCN1A asociadas con el Síndrome de Dravet
(Adaptado de Metwally Y.)

Julio 2011 17
ciencia
reducción de la neurotransmi-sión mediada por aminoácidos neuroexcitatorios, específica-mente de ácido glutámico, a través de un bloqueo selecti-vo de los receptores de tipo NMDA de este aminoácido.
Lamotrigina: Estudios in vitro indican que este medicamento inhibe los canales de Na+ esta-bilizando la membrana neuro-nal, hecho que todavía no se ha demostrado en humanos.
Topiramato: El mecanismo de acción del topiramato no se conoce con exactitud; se cree que puede actuar a través de di-ferentes mecanismos: bloqueo de canales de Na+; actuando sobre los receptores GABAA facilitando la acción de GABA; como antagonista de recepto-res AMPA y de receptores de kainato, inhibiendo la acción de neurotransmisores excita-dores; o inhibiendo la enzima anhidrasa carbónica.
Farmacogenética y Epilepsia
Aproximadamente un 30% de las personas afectadas de epilepsia presentan problemas relacionados con la medicación que pueden des-encadenar en una epilepsia refrac-taria o farmacorresistente, que se produce cuando el tratamiento an-ticonvulsivo no controla las crisis o sus efectos secundarios son limitan-tes para un desarrollo normal de la persona. Se considera refractario a terapia médica aquel paciente que haya utilizado al menos dos anti-convulsivos con indicación y dosis adecuadas en monoterapia o poli-terapia sin alcanzar un estado libre de crisis. La investigación farmaco-genómica ha permitido conocer muchos de los polimorfismos genéticos que pueden alterar la eficacia de los fármacos antiepilépticos, pro-duciendo una mala respuesta al tratamiento o la presentación de reacciones adversas, que pueden incluso ser muy severas, y que impiden el tratamiento eficaz de la enfermedad. Los genes implicados en la eficacia y seguridad de los antiepilépticos (Tabla 2) pertenecen a 4 categorías: a) Genes que codifican a pro-teínas transportadoras (ABCB1), cuya misión es transportar el medicamento a través de las
El tratamiento de la epilepsia se basa en la utilización de medicamentos antiepilépticos,
cuyo mecanismo de acción consiste fundamentalmente en la inhibición o facilitación
de los canales iónicos de Na+, Ca2+, K+, Cl- y gabaérgicos
Tabla 2. Farmacogenética de los antiepilépticos
Medicamento Gen/Polimorfismo Efecto
FenobarbitalABCB1CYP2C19
Transporte; Resistencia al fármacoMetabolismo
Difenilhidantoína
ABCB1/C3435TCYP2C9CYP2C19 HLA-B/HLA-B/HLA-B HLA-B*1502
Transporte; Resistencia al fármaco
Metabolismo
Reacción adversa
BenzodiazepinasABCB1CYP3A4NAT2
Transporte; Resistencia al fármaco
Metabolismo
EtosuximidaABCB1CYP3A4
Transporte; Resistencia al fármacoMetabolismo
Carbamazepina
ABCB1/C3435T, G2677T, C1236TABCC2/Val417IleABCC2/Val417IleABCC2CYP3A4 EPHX1/Tyr113His, His139ArgGSTM1/A304G, null (Del)GSTT1/Null (Del)HLA-B/HLA-B*1502HLA-B/HLA-B*1502HLA-BHSPA1L/C2437THSPA1L/C2437THSPA1LSCN1A/IVS5N+5G-ASCN1A/IVS5N+5G-ASCN1A (rs3812718)SCN2A
Transporte; Resistencia al fármaco
Metabolismo
Detoxificación
Reacción adversa
Inhibición canales de Na+
Ácido valproico
ABCB1/C3435TCYP2ACYP2B6CYP2C9CYP2C19CYP2E
Transporte; Resistencia al fármaco
Metabolismo
Vigabatrina ABAT Mecanismo de acción
Gabapentina
ABCB1/G2677TGABRR1GABRR2KCNH2SCN2ASLC22A4
Transporte; Resistencia al fármaco
Mecanismo de acción
Metabolismo
FelbamatoABCB1/C3435T, G2677T, C1236TCYP3A4
Transporte; Resistencia al fármacoMetabolismo
LamotriginaABCB1/rs3789243 and G2677TSCN2A
Transporte; Resistencia al fármacoInhibición canales de Na+
TopiramatoABCB1GRIK1/rs2832407SCN2A
Transporte; Resistencia al fármacoMecanismo de acciónInhibición canales de Na+

membranas celulares; b) Genes que codifican enzimas metabolizadoras, que se encargan de la eliminación del medicamento del organis-mo (CYP2C9, CYP2C9, CYP2C9 CYP2C19); c) Genes que codifiCYP2C19); c) Genes que codifiCYP2C19 -
can para proteínas que participan en el meca-nismo de acción del fármaco (SCNA1, SCNA2); y d) Genes que provocan reacciones alérgicas o de hipersensibilidad (HLA-B*1502).
El análisis genómico de determinadas regiones genéticas que influyen en la eficacia y seguri-dad del tratamiento antiepiléptico, representa un gran avance para el tratamiento de la enfer-un gran avance para el tratamiento de la enfer-un gran avance para el tratamiento de la enfermedad y para las personas afectadas. La utiliza-ción de estos análisis permite un acercamiento al conocimiento de la respuesta terapéutica del individuo a determinados medicamentos y la posibilidad de la aparición de efectos adver-la posibilidad de la aparición de efectos adver-la posibilidad de la aparición de efectos adversos al tratamiento.
18
Genética de la Epilepsia
¿Cómo actuar ante una crisis epiléptica?
xn Tranquilizar el entorno y conservar la calma en todo momentoxn Si se reconocen los signos y da tiempo, se puede ayudar al paciente a sentarse o ir a la cama, antes de que empiece la crisisxn No dejar solo al pacientexn Observar las características de la crisisxn Tender al paciente y girarlo hacia un lado. Así se evita la aspiración de vómitos o salivaxn Aflojar las ropas, proteger la cabeza, eliminar objetos duros o puntiagudos con los que pueda lastimarsexn No introducir los dedos ni objetos en la boca xn Controlar, pero permitir todo tipo de movimiento convulsivoxn No intentar la reanimación del enfermo, salvo en casos excepcionales que así lo aconsejenxn Se debe llamar al servicio de urgencias:
xy Si es la primera crisis que ha tenido en su vidaxy Si se ha hecho heridas durante la crisis o se ha golpeado la cabezaxy Si la crisis dura más de 5 minutosxy Si la persona está embarazada, es diabética o hipertensa
xn Observar las características de las crisis, para referirlo con detalle al médico
1. ILAE/IBE/WHO Global Campaign Against Epilepsy. Atlas: Epilepsy care in the world 2005. WHO Press, Switzerland, 2005.
2. García-Ramos García R, Gil Núñez AC, García Pastor A, Masjuan Vallejo J, Ramírez Moreno JM, Sánchez Sánchez C. Informe FEEN sobre la epilepsia en España. Fundación Española de Enfermedades Neurológicas. 2010.
3. Ottman R, Hirose S, Jain S et al. Genetic testing in the epilepsies-report of the ILAE Genetics Commission. Epilepsia 2010; 51:655-70.
4. Depondt C, Shorvon SD. Genetic association studies in epilepsy pharmacogenomics: lessons learnt and potential applications. Pharmacogenomics 2006; 7:731-45.
Referencias Bibliográficas:
Lucía Fernández-Novoa [email protected]
El análisis genómico de determinadas regiones genéticas que influyen en la eficacia y seguridad del tratamiento antiepiléptico, representa un gran avance para el tratamiento de la enfermedad y para las personas afectadas

19Julio 2011
Abstractno de los principales tratamientos actualmente utilizados en humanos para combatir el cáncer es la quimioterapia. Un gran número de compues-tos con actividad antitumoral están presentes en la naturaleza, y muchos de sus derivados son producidos por microorganismos. Sin embar-go, debido fundamentalmente a la toxicidad de los fármacos y a la resistencia a muchos agentes quimioterápicos que se observa durante el trata-miento, la búsqueda de nuevos medicamentos aún representa uno de los objetivos principales
de la terapia antitumoral. En modelos animales, la administración oral de dextrán sulfato sódico (DSS) durante un período relativamente corto determina colitis, con características similares a los daños clínicos e histopatológicos que se ob-servan en la colitis ulcerosa (UC). Los factores patogenéticos responsables de la colitis inducida por el DSS, y del sucesivo desarrollo del cáncer de colon, aún no han sido identificados. Hemos investigado los efectos del compuesto FR-91, un lisado estandarizado de células microbianas
U
Iván Carrera*, Valter R.M. Lombardi**, Ignacio Etcheverría**, Enrique Martínez^, Rafael Chacón^, Ramón Cacabelos***
* Departamento de Neurociencias, EuroEspes Biotecnología, A Coruña, España
** Departamento de Biotecnología de la Salud, EuroEspes Biotecnología, A Coruña, España
^ GEAMED España, Collado Villalba, Madrid, España
*** Instituto para Enfermedades del Sistema Nervioso Central y Medicina Genómica, Centro de Investigación Biomédica EuroEspes, Bergondo, A Coruña, España
Prevención de colitis experimental crónica inducida
por dextrán sulfato sódico (DSS) en ratones tratados
con FR-91

20
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
que pertenecen al género Bacillus, que en ante-riores estudios ha demostrado una significativa actividad inmunomoduladora, en la prevención de la carcinogénesis colorrectal pre-maligna. La colitis ha sido inducida en ratones durante un período de cinco semanas mediante administra-período de cinco semanas mediante administra-período de cinco semanas mediante administración oral de una solución de DSS al 2%. Los cam-bios morfológicos en la mucosa del colon fueron evaluados mediante tinción con hematoxilina-eosina (H&E) y mediante métodos inmunohisto-químicos. Se ha demostrado, en células crípticas y adenocarcinomatosas del epitelio displásico in-testinal, la expresión de catenina-β, MLH-1, APC y p53, junto con un aumento en la expresión de IFN-γIFN-γIFN- . En este modelo, la mejor dosis-respuesta γ. En este modelo, la mejor dosis-respuesta γobservada ha sido la concentración del 20% del
FR-91, en la que no se han observado alteracio-nes histológicas o sólo modestas lesiones induci-das por el DSS. Estos resultados sugieren que el FR-91 posee unas importantes propiedades an-tiinflamatorias en el modelo de inducción con DSS, y que puede actuar como agente quimio-preventivo frente a procesos de carcinogénesis de cáncer de colon.
IntroducciónLa enfermedad inflamatoria intestinal (IBD) comprende un grupo de condiciones crónicas, que incluye la enfermedad de Crohn (CD) y la colitis ulcerosa (UC), que afectan al colon y al intestino delgado. Las dos enfermedades están caracterizadas por dolor agudo, vómito y diarrea a los que siguen períodos de remisión1. A día de
hoy no se ha identificado un agente etiológico individual, y por esta razón la patogénesis de la IBD es muy compleja e involucra al ambiente ex-terno, las bases genéticas, la microflora intestinal, y el sistema inmunológico2. Aunque nuevos y efi-caces tratamientos están disponibles, la mayoría de ellos son fármacos biológicos o inmunosupre-sores que a menudo se asocian con importantes efectos secundarios, en particular infecciones, y un aumento en el riesgo de manifestaciones tu-morales3, y en los costes médicos que requieren de continuos ajustes en los diferentes tratamien-tos. Consecuencia directa de esta situación es la necesidad de establecer lo antes posible pautas de predicción y de respuesta al tratamiento. Se ha realizado un gran número de intentos con el objetivo de identificar las características clínicas, los tratamientos combinados y los biomarcado-res genéticos y serológicos que permitan la pre-dicción de la respuesta frente a diferentes agen-tes biológicos. Pocos estudios se han publicado sobre cómo biomarcadores de la mucosa/tejido intestinal son capaces de predecir el comporta-intestinal son capaces de predecir el comporta-intestinal son capaces de predecir el comportamiento clínico de la enfermedad o su respuesta al tratamiento farmacológico4.
En los últimos años, se han desarrollado dife-rentes modelos animales de inflamación intes-tinal. Debido a su capacidad de interferir con la función de la barrera intestinal y estimular un proceso de inflamación local y sistémico, el dextrán sulfato sódico (DSS), un polisacárido parecido a la heparina5, se utiliza a menudo en un modelo de colitis en ratones que imita las ma-un modelo de colitis en ratones que imita las ma-un modelo de colitis en ratones que imita las manifestaciones clínicas e histológicas de la IBD con características de la UC. Okayasu y sus colabora-características de la UC. Okayasu y sus colabora-características de la UC. Okayasu y sus colaboradores6 han demostrado que la administración oral de 5% de DSS en el agua a ratones BALB/c es capaz de inducir colitis crónica después de diferentes ciclos con DSS. A este estudio le siguió otro de Cooper y colaboradores7 que utilizaron otro esquema experimental para la inducción de colitis crónica: (a) 7 días de administración de DSS seguidos de 7 días de agua (durante 1, 2 y
Fig. 1. Protocolo experimental para la inducción de colitis/cáncer de colon mediante DSS en ratones y para la administración del compuesto FR-91
El proceso de carcinogénesis ocurre en múltiples etapas en las que pueden observarse tres estadios: iniciación, promoción y progresión

Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
Julio 2011 21
ciencia
Colon proximal Colon medial Colon distal
Fig. 2. Secciones histológicas de tres diferentes porciones del colon de los ratones del estudio experimental para demostrar la presencia de lesiones evidenciadas mediante tinción con H&E. Las figuras A-C corresponden a secciones transversales de tres diferentes niveles colorrectales (proximal, medial y distal) de los ratones del grupo A tratados con FR-91 (10%). Puede notarse la ausencia de lesiones y la presencia de estructuras similares a las observadas en ratones normales. Las figuras D-F son secciones transversales pertenecientes a tres diferentes niveles colorrectales de los ratones del grupo B tratados con DSS (2%) durante cinco semanas. En cada sección estudiada, es posible observar múltiples lesiones histopatológicas, siendo par-ticularmente claro un grado severo de displasia (puntas de flecha en las figuras D y E) presente en el colon de los ratones de este grupo, junto a la presencia de criptas aberrantes (flecha en la figura D), pólipos adenomatosos en la figura F y ulceracio-nes incipientes en la figura F. Las figuras G-I son secciones transversales pertenecientes a tres diferentes niveles colorrectales de los ratones del grupo C tratados con DSS (2%) y FR-91 (5%) durante cinco semanas. A pesar de que las secciones del colon a los tres niveles presentan un bajo nivel de displasia y algunas ulceraciones puntuales de la mucosa, la estructura histológica parece en general funcional. Las figuras J-L son secciones transversales pertenecientes a tres diferentes niveles colorrecta-les de los ratones del grupo D tratados con DSS (2%) y FR-91 (10%) durante cinco semanas. Este grupo muestra una mejor organización histológica del colon con respecto a la observada en el grupo C, a pesar de que pueda observarse la presencia de algunas criptas displásicas principalmente a nivel de las secciones distales. Las figuras M-O son secciones transversales pertenecientes a tres diferentes niveles colorrectales de los ratones del grupo E tratados con DSS (2%) y FR-91 (20%) durante cinco semanas. Estas secciones muestran una organización epitelial normal, con células de las criptas bien diferenciadas y ausencia de lesiones con características atípicas. Barra de escala: 100 µm

La utilización de modelos experimentales animales representa una herramienta esencial para el desarrollo de nuevas terapias
22
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
de viruta bajo las condiciones estándar de labo-ratorio (dieta esterilizable, 35%±10% de hume-dad, 20-24ºC de temperatura, ventilación de 15 a 20 renovaciones de aire por hora, y ciclos de 12 horas de luz/oscuridad). Todos los ratones fueron mantenidos en cuarentena 3 semanas después de su llegada y sucesivamente distribui-dos de forma aleatoria con respecto a su peso en grupos experimental y grupos control. A todos los ratones se les permitió libre acceso al pellet comercial A04 (dieta de mantenimiento, SAFE, Francia) y a las botellas de agua o agua más tratamiento. Todos los procedimientos uti-lizados han seguido las normas establecidas por la Directiva del 24 de Noviembre de 1986 de la Unión Europea (86/609/EEC), por el Decreto Real Español 1201/2005 en materia de expe-rimentación animal, y la ley 32/2007 de 7 de noviembre, y fueron aprobados por el Comité Ético del Centro de Investigación de EuroEspes Biotecnología.
Diseño del estudio
El diseño del presente estudio se centró en primer lugar en la inducción de la displasia asociada a la colitis y/o en características del cáncer mediante la administración oral de DSS a los ratones y, sucesivamente, en su tratamien-to mediante diferentes concentraciones orales del compuesto FR-91 (un lisado estandarizado de células bacterianas pertenecientes al género Bacillus) (Fig. 1). A la edad de 7 semanas, los animales fueron separados en dos grupos con-trol (A y B, n=4 en cada grupo) y tres grupos experimentales (C-E, n=6 en cada grupo). En las primeras dos semanas, se administró agua con las diferentes concentraciones de FR-91 (5% en el grupo C, 10% en los grupos A y D, 20% en el grupo E) por vía oral a todos los animales de los grupos A, C-E. En la tercera semana, agua con 20 g/L (2%) de DSS (DSS; masa molecu-lar 5000; D4911, Sigma-Aldrich; MO, USA) fue administrada por vía oral a los animales de los grupos B y E. Para las sucesivas comparaciones entre grupos, los grupos control A y B recibie-ron DSS o tratamiento sólo con FR-91, respecti-vamente, como controles no tratados. Todos los animales fueron sacrificados al final de la expe-rimentación (octava semana), a la edad de 15 semanas.
Inducción de la colitis
La colitis experimental fue inducida en los rato-nes de los grupos B, C, D, y E mediante repetida administración de DSS al 2% (peso/volumen; 20 g/L) en el agua de bebida ad libitum duran-te cinco semanas. Esta dosis es suficiente, como se ha demostrado en estudios anteriores2, para inducir una moderada hasta severa colitis, sin ocasionar mortalidad en los ratones tratados. Ninguno de los ratones tratados murió antes del final del estudio experimental (día 50).
3 ciclos); (b) 7 días de administración oral de DSS seguidos de 14 días y 21 días de agua. Los resultados de este estudio demostraron que la colitis crónica inducida después de la adminis-tración de DSS durante 7 días podía representar un modelo muy útil para el estudio de los efec-tos farmacológicos en enfermedades inflamato-rias humanas y de los mecanismos moleculares de la inflamación, aportando también nume-rosas informaciones descriptivas de las lesiones histológicas, y demostrando que los principales cambios histológicos se observaban como pér-dida en la cripta focal, con sucesivos signos de inflamación aguda y crónica.
En el presente estudio se ha investigado el efec-to del extracto FR-91 en la reducción de la coli-tis crónica experimental inducida mediante DSS en ratones de la cepa Swiss. También se ha inves-tigado la posibilidad de que en la fase crónica se pudiera observar una regulación en la expre-sión de genes apoptóticos y una desregulación del balance T-colaboradores 1/T-colaboradores 2 (Th1/Th2) y cómo esta situación podría facili-tar una regeneración de la mucosa intestinal.
Materiales y MétodosAnimales
26 ratones hembra de la cepa Swiss, libres de enfermedad (7 semanas de edad, suministradas por el animalario de la Universidad de Santiago de Compostela, España) fueron mantenidas (2 o 3 por jaula) en jaulas de plástico con lecho
Fig. 3. Análisis de imágenes de las áreas colorrectales dañadas en las tres porciones del colon en cada grupo experimental. La cuantificación en píxeles del área inmunoreactiva afectada se ha realizado mediante un programa in-formático. En el grupo 5 se han observado diferencias significativas (p<0.05) respecto a los otros grupos tratados (3 y 4)
0
5000
10000
15000
20000
25000
30000
Gr Ap Gr Am Gr Ad GR Bp Gr Bm Gr Bd GR Cp GR Cm Gr Cd Gr Dp Gr Dm Gr Dd Gr Ep Gr Em Gr Ed
Pixeles de las áreas colorrectales dañadas
B-Catenin
BCL2
APC
MLH1
p53

Tabla 1. Puntuación de las lesiones colorrectales observadas en los cinco grupos experimentales de ratones. Los datos se expresan como media ± desviación estándar
n Segmento del colon afectado Ulceración Severidad Hiperplasia Área involucrada Puntuación total
Grupo A 4 NormalGrupo A 4 NormalGrupo A 4 Normal 0 00 0 0 0 0
Grupo B 4 Proximal, Medial, DistalGrupo B 4 Proximal, Medial, DistalGrupo B 4 Proximal, Medial, DistalGrupo B 4 Proximal, Medial, Distal 1 2.6±0.23 2.8±0.43 3.2±0.33 9.6±0.991 2.6±0.23 2.8±0.43 3.2±0.33 9.6±0.991 2.6±0.23 2.8±0.43 3.2±0.33 9.6±0.991 2.6±0.23 2.8±0.43 3.2±0.33 9.6±0.991 2.6±0.23 2.8±0.43 3.2±0.33 9.6±0.991 2.6±0.23 2.8±0.43 3.2±0.33 9.6±0.99
Grupo C 6 Medial, DistalGrupo C 6 Medial, DistalGrupo C 6 Medial, DistalGrupo C 6 Medial, Distal 0 1.5±0.18 2.7±0.31 2.4±0.17 6.6±0.660 1.5±0.18 2.7±0.31 2.4±0.17 6.6±0.660 1.5±0.18 2.7±0.31 2.4±0.17 6.6±0.660 1.5±0.18 2.7±0.31 2.4±0.17 6.6±0.660 1.5±0.18 2.7±0.31 2.4±0.17 6.6±0.660 1.5±0.18 2.7±0.31 2.4±0.17 6.6±0.66
Grupo D 6 Medial, DistalGrupo D 6 Medial, DistalGrupo D 6 Medial, DistalGrupo D 6 Medial, Distal 0 0.6±0.14 1.1±0.13 0.9±0.20 2.6±0.470 0.6±0.14 1.1±0.13 0.9±0.20 2.6±0.470 0.6±0.14 1.1±0.13 0.9±0.20 2.6±0.470 0.6±0.14 1.1±0.13 0.9±0.20 2.6±0.470 0.6±0.14 1.1±0.13 0.9±0.20 2.6±0.470 0.6±0.14 1.1±0.13 0.9±0.20 2.6±0.47
Grupo E 6 NormalGrupo E 6 NormalGrupo E 6 Normal 0 00 0 0 0 0
FR-91 posee unas importantes propiedades antiinflamatorias en el modelo de inducción con
Dextrán sulfato sódico en ratones
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
Julio 2011 23
ciencia
Preparación de los tejidos
Los ratones fueron anestesiados con éter, per-Los ratones fueron anestesiados con éter, per-Los ratones fueron anestesiados con éter, perfundidos con solución salina y fijados con una solución al 4% de paraformaldehido en tampón fosfato 0.1 M (pH 7.4). La totalidad de la re-gión colorrectal (desde la unión ileocecal hasta el canal anal) fue retirada, medida, examinada macroscópicamente, lavada con solución salina, e inmediatamente fijada utilizando la misma so-lución de paraformaldehido durante un tiempo de 48 horas. La región colorrectal fue dividida en tres segmentos de la misma longitud (proximal, mediano y distal), determinada bajo un estereo-microscopio de disección (Leica, M125). Los seg-microscopio de disección (Leica, M125). Los seg-microscopio de disección (Leica, M125). Los segmentos colorrectales fueron protegidos durante el proceso de congelación mediante inmersión en una solución al 30% de sucrosa en tampón fosfato 0.1 M, incrustados en el compuesto OCT (Tissue Tek, Torrance, CA), y congelados en isopentano enfriado con nitrógeno líquido. Series paralelas de secciones transversales de un espesor de 14-16 µm, que se obtuvieron utilizando un criostato (Starlet 2212, Bright, UK), fueron montadas en portas Superfrost Plus (Menzel Gläser®).
Tinción histológica e inmunohistoquímica
El examen rutinario histológico se realizó me-diante tinción de las secciones con hematox0ilina-eosina (H&E), en las que diferentes alteraciones morfológicas que se observan durante el desa-morfológicas que se observan durante el desa-morfológicas que se observan durante el desarrollo del cáncer colorrectal (como por ejemplo cripta absceso, displasia de la mucosa, adenoma y adenocarcinoma), fueron identificadas y diag-y adenocarcinoma), fueron identificadas y diag-y adenocarcinoma), fueron identificadas y diagnosticadas de acuerdo con estudios anteriores (Tanaka y colaboradores)8. Se utilizaron técnicas inmunohistoquímicas para detectar la expresión de marcadores histopatológicos como el APC (se utiliza para detectar los niveles endógenos de proteínas supresoras tumorales), catenina-β(asociada con complejos APC), la p53 (asociada con mecanismos de apoptosis relacionados con el ciclo celular), BCL-2 (regulador de la apop-tosis) y MLH1 (parte del complejo proteico de reparación de mal apareamiento del ADN). Para eliminar la peroxidasa endógena, las secciones primero fueron tratadas con H2O2, lavadas dos
veces en tampón fosfato a pH 7.4 (10 minutos/la-veces en tampón fosfato a pH 7.4 (10 minutos/la-veces en tampón fosfato a pH 7.4 (10 minutos/lavado), y, para bloquear las uniones inespecíficas, fueron sucesivamente tratadas durante una hora con una solución de tampón fosfato 0,1 M con 0.2% de Tween 20 y 15% de suero normal de ca-0.2% de Tween 20 y 15% de suero normal de ca-0.2% de Tween 20 y 15% de suero normal de cabra (Dako, Glostrup, Dinamarca). Todos los anti-cuerpos policlonales primarios de conejo como anti-catenina-β, anti-p53, anti-BCL-2, anti-MLH1 y anti-APC fueron purificados mediante cromato-grafía de afinidad, utilizando inmunógenos espe-cíficos de ratón (Bioworld Technology, MN, USA;
Catálogo Nº BS3603, BS3736; BS1511, BS2418, BS1017, respectivamente) a la dilución de 1:200 durante toda una noche. Después de dos lavados de 10 minutos cada uno con tampón fosfato, se añadió un segundo anticuerpo biotinilado (IgG de cabra anti-conejo; Dako, dilución 1:100) du-rante una hora. Se lavaron dos veces con tampón fosfato y se utilizó el kit Vectastain ABC (Vector Laboratories, Burlingame, CA) durante 1 hora, y se efectuaron dos lavados con tampón fosfato. Como control negativo, se utilizaron secciones en las que no se añadió uno de los anticuerpos (pri-mario, secundario o terciario) y en las que no se observó ninguna reacción positiva. En la última fase, la reacción inmunológica se reveló utilizan-do una solución de diaminobencidina al 0.005% (DAB; Sigma-Aldrich9 y 0.003% de H2O2). Todas las diluciones fueron realizadas utilizando tam-pón fosfato y 0.2% Tween 20, y las incubaciones se realizaron a temperatura ambiente en cáma-se realizaron a temperatura ambiente en cáma-se realizaron a temperatura ambiente en cámaras húmedas. Al final del proceso, las secciones fueron deshidratadas, montadas y cubiertas con cubreobjetos.
Caracterización y especificidad de los anticuerpos
De acuerdo con la información técnica ofreci-da por el fabricante (Bioworld Technology, MN, USA), los anticuerpos primarios utilizados, an-tisueros producidos en conejos, son dirigidos

Tabla 2. Niveles (pg/mL) de IL-1α, IL-2, IL-5, IL-6, IL-10, IFN-γ, TNF-α , GM-CSF, IL-4 y IL-17 en el suero de ratones. Los datos se expresan como media ± desviación estándar
IL-1α IL-2 IL-5 IL-6 IL-10 IFN-γ TNF-α GM-CSF IL-4 IL-17
FR-91 0 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 00 0 20±1.5 25±2.5 0 0 0 78±3.8 0 0
DSS 0 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.20 0 78±4.5 48±5.4 175±4.5 363±13.4 125±9,7 0 0 1±0.2
5% 0 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.90 55±3.3 78±3.9 53±7.6 96±10.1 0 0 0 0 24±2.9
10% 0 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.70 34±3.8 37±2.1 149±9 97±8.6 0 0 41±5.7 0 12±1.7
20% 0 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.50 0 38±3.9 47±7.2 55±5.8 15±2.1 0 75±4.4 0 17±2.5
En la formación y desarrollo de lesiones preneoplásicas y neoplásicas en el colon y recto, intervienen múltiples sustancias que se han introducido en el organismo
24
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
frente a epítopos de ratones y han sido purifi-cados mediante la técnica de cromatografía de afinidad utilizando epítopos inmunógenos espe-cíficos con un grado de pureza superior al 95% (mediante SDS-PAGE). Su especificidad ha sido comprobada mediante la técnica de Western blot; reconociendo sólo una banda proteica de aproxi-madamente ≈86-90 kD (catenina-β), ≈43-45 kD (p53), ≈26-28 kD (BCL-2), ≈84-86 kD (MLH1) y ≈270-280 kD (APC). Además, los anticuerpos uti-lizados tenían un amplio espectro de reactividad cruzada y se demostró su expresión en ratones, ratas y humanos. Las lesiones se asumieron como positivas para catenina-β /BCL-2/MLH1/APC si se podía detectar coloración citoplasmática/nu-clear, mientras que la p53 se consideró positiva al detectarse expresión nuclear. Para cuantificar el grado de daño del tejido, se analizaron al azar dos secciones microscópicas transversales por segmento y por animal. Un total de 6 secciones por animal fueron analizadas. El análisis cuantita-por animal fueron analizadas. El análisis cuantita-por animal fueron analizadas. El análisis cuantitativo del daño del tejido se realizó en cada sección utilizando un software de análisis de área/pixel (Pixcavator v2.4), para cuantificar el número de píxeles dentro del área dañada para cada sección colorrectal. Por lo tanto, el software fue utilizado para analizar el área ocupada por lesiones, com-parado con la tinción de fondo y expresado en
unidades porcentuales. El área del daño colorrec-tal en los ratones de los cinco grupos experimen-tales fue analizado y representado en gráficas (Fig. 3 y 4). Además, para cuantificar el nivel del daño en los ratones de cada grupo, dos diferentes observadores evaluaron de forma individual e in-dependiente las secciones histológicas en doble ciego y obtuvieron un elevado nivel de similitud demostrando la misma puntuación observada en estudios anteriores9. Por lo tanto, tres secciones
de cada segmento colorrectal de cada ratón fue-ron codificadas y puntuadas para las lesiones de acuerdo a las lesiones ulcerosas (mucosa sin re-vestimiento epitelial: 0 no presente y 1 presente), severidad de las lesiones (inflamación y/o exten-sión fibrósica), hiperplasia (espesor del revesti-miento epitelial) y al área afectada involucrada, puntuada de la siguiente manera: 0: normal; 1: débil; 2: moderada; y 3: severa. Los resultados fueron representados en las gráficas de la fig. 4.
Imágenes
Las secciones fueron fotografiadas utilizando un microscopio Olympus (BX50) equipado con una cámara digital (DP10). Las fotografías fueron convertidas en escala de grises y ajustadas por cla-convertidas en escala de grises y ajustadas por cla-convertidas en escala de grises y ajustadas por claridad y contraste mediante el software Corel Draw (Corel, Ottawa, Canada), y las figuras se compu-sieron utilizando el software Corel Photo Paint.
Determinación del perfil de citoquinas Th1/Th2
La detección cuantitativa de GM-CSF, IFN-γLa detección cuantitativa de GM-CSF, IFN-γLa detección cuantitativa de GM-CSF, IFN- , IL-γ, IL-γ1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-17 y TNF-α se α se αrealizó utilizando el FlowCytomix Mouse Th1/Th2 10plex (BMS820FF) (Bender MedSystems®) que permitió analizar múltiples analitos en una alícuota individual de 50 µL de suero de ratón. Unas microbolas fueron coloreadas con dife-rentes concentraciones de dos flurocromos para generar distintos sets de bolitas. Cada bolita se cubrió con un anticuerpo específico para cada analito. El analito fue capturado utilizando un segundo anticuerpo biotinilado y marcado con estreptoavidina-ficoeritrina (S-PE). Las muestras se analizaron con un citómetro de flujo (FAC-Scan, Becton Dickinson). Para el análisis de los resultados, se utilizó el FlowCytomix Pro Software (Bender MedSystems®).
Análisis de los resultados
Los datos de las secciones colorrectales estudiadas, obtenidos mediante el software (Pixcavator 2.4), fueron analizados mediante análisis de varianza (ANOVA) para detectar los mayores efectos entre grupos para cada parámetro. Se realizaron compa-grupos para cada parámetro. Se realizaron compa-grupos para cada parámetro. Se realizaron comparaciones a posteriori utilizando el test de Bonferroa posteriori utilizando el test de Bonferroa posteriori -ni/Dunn, con un nivel de significación de P<0.05.

Para los biólogos experimentales los focos de criptas aberrantes FCA pueden ofrecer la
oportunidad de identificar las alteraciones moleculares más tempranas que conducen al
cáncer colorectal (CCR)
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
Julio 2011 25
ciencia
Inmunohistoquímica de catenina-β, p53, BCL-2, MLH1 y APC.
Las técnicas inmunohistoquímicas utilizadas para identificar los marcadores celulares de las lesio-nes colorrectales demostraron la expresión de catenina-β (regulador celular de adhesión), BCL-2 (regulador de apoptosis), MLH1 (reparador de mal emparejamiento del ADN), APC y p53 (pro-teínas supresoras de tumor) en todas las lesiones de colon en los ratones. Mediante el marcaje con el anticuerpo de la porción colorectal, se ha de-tectado la presencia de expresión endógena de
catenina-β principalmente en el citoplasma de células displásicas y adenocarcinomatosas. Una in-tensa expresión de catenina-β fue observada en el colon proximal y medial (Fig. 5F) de los ratones Fig. 5F) de los ratones Fig. 5Fdel grupo B (DSS), mientras una moderada hasta intensa inmunoreactividad se observó en los seg-intensa inmunoreactividad se observó en los seg-intensa inmunoreactividad se observó en los segmentos colorrectales (Fig. 5K) de los ratones del Fig. 5K) de los ratones del Fig. 5Kgrupo C (DSS/5% FR-91). Las células β-catenina inmunoreactivas (ir) fueron localizadas en las cé-lulas displásicas de las criptas y en las células ade-nocarcinomatosas, en los estratos internos de las criptas (Fig. 5F, K). Las porciones colorrectales de Fig. 5F, K). Las porciones colorrectales de Fig. 5F, Klos ratones de los grupos A, D, y E demostraron una débil o nula presencia de reacción frente a la catenina-β en las células de las criptas (Fig. 5A, P, U), y estos valores se consideraron como valores U), y estos valores se consideraron como valores Ubasales de la expresión de la catenina-β. Se obser-. Se obser-. Se observaron células BCL-2-ir y p53-ir en las porciones colorrectales de los ratones pertenecientes a los grupos B (Fig. 5G, J) y C (Fig. 5G, J) y C (Fig. 5G, J Figuras 5L, O), en las que Figuras 5L, O), en las que Figuras 5L, Opudo observarse una intensa reacción en las cé-lulas adenocarcinomatosas y en las células de las criptas, particularmente evidente en los ratones
ResultadosEl tratamiento con FR-91 mejora la colitis agu-da y crónica inducida por DSS. Resultados pa-tológicos e inflamatorios.
En los grupos de ratones que recibieron repe-tidas dosis de DSS al 2% y baja o nula dosis de FR-91 (grupos B y C) se observó sangre en las heces durante la segunda mitad del experimen-to, mientras estos resultados no se observaron en los otros grupos. Macroscópicamente, se han identificado numerosas lesiones polipoi-deas hiperplásicas (epitelio con características morfológicas atípicas de un diámetro de 0,5 cm) en los ratones de los grupos B (4/4; 100%) y C (5/6; 83,3%), principalmente en las porcio-nes mediales y distales del segmento colorectal; sólo algunas se han observado en el grupo D (1/6; 16,6%). Notablemente, en ninguno de los ratones pertenecientes al grupo E (DSS/20% FR-91), así como en el grupo control (A; 10 FR-91), se observaron procesos ulcerosos en el seg-mento del colon analizado. Estas observaciones macroscópicas fueron confirmadas en el análi-sis histológico de la morfología intestinal en las que se pudo enfatizar la integridad de la mu-cosa, la inflamación de la mucosa y submucosa del colon, la presencia de epitelio displásico y la presencia de úlceras. El examen histológico de secciones transversales teñidas con H&E (Fig. 2) evidenció la presencia de úlceras con modera-das o severas alteraciones morfológicas (áreas multifocales de inflamación en la submucosa o úlceras que se extendían en grandes áreas), la presencia de moderadas hasta severas crip-tas hiperplásicas (el espesor del revestimiento epitelial era dos o tres veces más que lo nor-epitelial era dos o tres veces más que lo nor-epitelial era dos o tres veces más que lo normal, con marcada hipercromasia de las células y múltiples criptas con formaciones ramifica-das), displasia epitelial (alteración en la dife-renciación de las células epiteliales que pue-den progresar hacia un carcinoma invasivo) y la presencia de grandes áreas con pérdidas de criptas. Sin embargo, la severidad de estas lesiones colorrectales fue significativamente di-ferente en los ratones de los distintos grupos, siendo los ratones pertenecientes al grupo B (DSS) los que presentaron el mayor grado de alteración, mientras los ratones pertenecientes al grupo C (DSS/5% FR-91) evidenciaron alte-raciones moderadas hasta severas y los ratones del grupo D (DSS/10% FR-91) presentaron escasos signos de alteración. No se observaron lesiones características de colitis tanto en el grupo E (DSS/20% FR-91) como en el grupo control (A; 10% FR-91) (Fig. 2), de la misma forma que se ha descrito anteriormente con el examen macroscópico. Los resultados de las puntuaciones histológicas obtenidos mediante el examen de las lesiones colorrectales se pre-sentan en la tabla 1 y en las figuras 3 y 4.
criptas, particularmente evidente en los ratones
Fig. 4. Análisis de imágenes del valor medio de inmunoreactividad del área del colon dañada. En el grupo 5 se han observado diferencias significativas (p<0.05) respecto a los otros grupos tratados (3 y 4), y valores similares también al grupo control (grupo 1)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
Gr A Gr B Gr C Gr D Gr E
Áreas del colon inmunorreactivas
B-Catenin BCL2 APC MLH1 p53

26
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
B-CateninaColon medial
BCL-2Colon medial
MLH-1Colon distal
APCColon distal
p53Colon distal
Fig. 5. Inmunohistoquímica de los marcadores patológicos del colon en diferentes secciones trasversales del colon. En las figuras A-E se muestran detalles de las secciones transversales a nivel del colon medial y distal de los ratones del grupo A, que demuestran una escasa inmunoreactividad a catenina-β, p53 o ausencia frente a BCL-2, MLH-1 y APC. Se puede observar que estas secciones tam-bién muestran una organización epitelial normal con células de las criptas bien diferenciadas. Las figuras F-J se corresponden con detalles de las secciones transversales de los ratones del grupo B, mostrando una intensa inmunoreactividad con los marcadores uti-lizados. Es posible observar la presencia de numerosas células inmunoreactivas (flechas en las figuras F-I) y su masiva localización a nivel de las diferentes lesiones patológicas, así como la presencia de un nivel severo de displasia (puntas de flecha en la figura F), pólipos (punta de flecha en la figura J) y adenomas. Las figuras K-O muestran detalles de las secciones transversales de los ratones del grupo C, mostrando la presencia de escasa o moderada inmunoreactividad con los marcadores utilizados en los segmentos colorrectales. Es posible observar la presencia de algunas células inmunoreactivas a la catenina-β (puntas de flecha en la figura K), MLH-1 (flecha en la figura M), APC (flechas en la figura N) y p53 (flechas en la figura O). Las figuras P-T y U-Y se corresponden con las secciones transversales de los ratones del grupo D (P-T) y grupo E (U-Y) mostrando ausencia o inmunoreactividad basal (flechas en las figuras S y X) de los segmentos colorrectales estudiados con los marcadores celulares utilizados. Barra de escala: 100 µm
del grupo B. La inmunoreactividad a la BCL-2 y a la p53 estaba ausente en las secciones colo-rrectales de los ratones de los grupos A, D, y E estudiados (Fig. 5B, Q, E, T, Y). Una fuerte tinción Fig. 5B, Q, E, T, Y). Una fuerte tinción Fig. 5B, Q, E, T, Ya MLH1-ir se observó en células de los segmentos colorrectales en los ratones del grupo B (Fig. 5H) Fig. 5H) Fig. 5Hy del grupo C (Fig. 5M), demostrando una intenFig. 5M), demostrando una intenFig. 5M -sa reacción en las células adenocarcinomatosas y
en las células de las criptas del epitelio displásico, particularmente intensa en los ratones del grupo B. No se observaron células MLH1-ir en las sec-ciones colorrectales de los ratones de los grupos A, D y E (Fig. 5C, R, W). La inmunoreactividad obFig. 5C, R, W). La inmunoreactividad obFig. 5C, R, W -servada frente a la proteína APC fue muy intensa en todas la porciones colorrectales de los ratones de los grupos B (Fig. 5I) y C (Fig. 5I) y C (Fig. 5I Fig. 5N), a pesar Fig. 5N), a pesar Fig. 5N

Los intestinos grueso y delgado actúan como barreras importantes en contra de agentes
exógenos, en particular frente a substancias con capacidad mutagénica y carcinogénica
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
Julio 2011 27
ciencia
de que las células de la cripta de los ratones del grupo B demostraban una mayor positividad. Las porciones colorrectales de los ratones de los gru-pos A, D, y E demostraron sólo una débil reactivi-dad frente a la proteína APC en sus células de las criptas (Fig. 5D, S, X). La incidencia de lesiones Fig. 5D, S, X). La incidencia de lesiones Fig. 5D, S, Xdel colon demostrada con estos marcadores ce-lulares en el grupo E fue del 8.2% ±4%, mientras fue del 38.2% ±6%, 33.3% ±6% y 20.4% ±3% en los grupos B, C y D, respectivamente (Fig. 3 y 4). Una incidencia histológica similar fue observada en los ratones del grupo E cuando se comparó con el grupo control A (7.8% ±2%). Significativas diferencias fueron observadas entre los grupos experimentales A/E y B/C/D mediante análisis estadístico ANOVA de dos factores de los datos de los marcadores inmunohistológicos en la por-de los marcadores inmunohistológicos en la por-de los marcadores inmunohistológicos en la porción colorrectal (Fig. 4). El grupo E fue el grupo más resistente a las lesiones inducidas por DSS en la porción colorrectal, indicado por el valor más bajo de reactividad observada por cada biomarca-bajo de reactividad observada por cada biomarca-bajo de reactividad observada por cada biomarcador utilizado, valor similar al observado en el gru-po control A. Los grupos B, C, y D demostraron mayor susceptibilidad a las lesiones inducidas por DSS, demostrado por la presencia de valores más elevados de reactividad. Diferencias significativas entre grupos fueron observadas entre los grupos A/E y B/C/D en la susceptibilidad al DSS, corre-lacionándose con las diferencias entre los valores medio de consumo de FR-91.
Producción de citoquinas inflamatorias inducidas por DSS
Se han considerado los mecanismos potencia-Se han considerado los mecanismos potencia-Se han considerado los mecanismos potenciales que pueden estar relacionados con la colitis que se obtiene mediante tratamiento con DSS y el efecto del compuesto FR-91 en los grupos de ratones tratados. Como se evidencia en la Tabla 2, el tratamiento con DSS aumentó sólo la sín-tesis y liberación de la citoquina proinflamatoria IFN-γIFN-γIFN- . No se observaron cambios significativos en γ. No se observaron cambios significativos en γla síntesis y liberación de IL-1β, TNF-α, IL-6, IL-10 e IL-17 tanto en los grupos tratados como en los grupos control.
DiscusiónEl ambiente externo, el perfil genético, el siste-ma inmunológico y la flora intestinal son factores que contribuyen a formar una compleja red en la que todos están integrados y participan en la formación de reacciones de inflamación crónica que son características de la enfermedad inflama-que son características de la enfermedad inflama-que son características de la enfermedad inflamatoria intestinal (IBD). Entre todos estos factores, los más importantes son los genéticos y los que contribuyen a un correcto funcionamiento del sistema inmunológico, que actúan a nivel de la mucosa intestinal con el objetivo de prevenir sus alteraciones y/o rupturas y el consecuente desa-alteraciones y/o rupturas y el consecuente desa-alteraciones y/o rupturas y el consecuente desarrollo de procesos de inflamación crónica. Dife-rentes medicamentos son capaces de bloquear
moléculas clave que contribuyen a la aparición de procesos de inflamación crónica utilizando diferentes caminos. Sin embargo, debido a que los mecanismos patogenéticos de la IBD son muy complejos e involucran diferentes factores, es ra-complejos e involucran diferentes factores, es ra-complejos e involucran diferentes factores, es razonable pensar que los caminos que dan lugar a la inflamación puedan desempeñar diferentes papeles en diferentes pacientes. Estos factores de-berían ser considerados a la hora de elegir nue-vos procedimientos terapéuticos disponibles en la actualidad para ser utilizados en pacientes con IBD, de manera especial cuando se utilizan agen-tes biológicos o inmunosupresores. Estos trata-tes biológicos o inmunosupresores. Estos trata-tes biológicos o inmunosupresores. Estos tratamientos muy fuertes pueden dar lugar a efectos secundarios, en particular a infecciones y a un au-mento del riesgo de padecer procesos tumorales. Además, a veces no son útiles para el tratamiento de determinados subgrupos de pacientes. Como consecuencia directa de estas situaciones, nos en-frentamos a una creciente necesidad de nuevos compuestos que sean capaces de mejorar el efec-to terapéutico y, al mismo tiempo, de reducir la aparición de efectos secundarios.
Se han identificado más de 20.000 compuestos bioactivos sintetizados por microorganismos, y más de 10.000 de estos metabolitos secundarios son producidos por actinomicetos, representan-do alrededor de un 45% de todos los compues-tos con actividad biológica conocida10. Entre los actinomicetos, alrededor de 8.000 compuestos están sintetizados por la especie Streptomyces11Streptomyces11Streptomyces . La función principal de estos compuestos es su actividad antibiótica. Además, muchos de estos compuestos, como por ejemplo las antraciclinas

El compuesto FR-91 posee la capacidad de prevenir la formación de lesiones pretumorales en modelos murinos de colitis, inhibiendo el desarrollo de tumores colorrectales
28
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
(aclarubicina, daunomicina y la doxorubicina), péptidos (bleomicina y actinomicina D), enidi-nas (neocarzinostatina), antimetabolitos (pentos-tatina), carzinofilina, mitomicinas y otros12,13 han sido testados para la inhibición de la carcinogé-nesis inducida químicamente en modelos anima-les tanto in vitro como in vivo13.
En el presente estudio, hemos investigado el efecto del FR-91, un lisado estandarizado de células bacterianas pertenecientes al género Ba-cillus que en anteriores estudios, ha demostrado tener significativas propiedades inmunorregula-doras14 en la inflamación del colon, inducida tras la exposición a un tratamiento de 8 semanas con DSS en agua de beber, y en particular, en la capa-cidad de interferir en la aparición y/o progresión del cáncer de colon.
Los resultados obtenidos demuestran que un tra-tamiento de seis semanas con este compuesto da lugar a una ligera reducción en las lesiones colo-rrectales a baja dosis (5% FR-91) y una moderada o completa reducción a dosis más elevadas (10-20% FR-91). Además, los datos histopatológicos han demostrado que el FR-91 no presenta efectos patológicos sobre la organización morfológica del tejido del colon de los ratones, datos que han po-dido observarse cuando el FR-91 fue administrado durante todo el período experimental.
Los datos obtenidos sugieren que el FR-91 puede ser un importante agente quimiopreventivo fren-te a procesos tumorales de colon en ratones.
En nuestros experimentos inducimos un amplio espectro de lesiones colorrectales con el fin de evaluar los efectos anticancerígenos del FR-91 en ratones con colitis ulcerosa crónica. La ad-ministración repetida de DSS como inductor de lesiones en modelos murinos de colitis ha sido ampliamente descrita15-20, siendo esencial para la comprensión de las complejas interacciones en-tre ambiente, genética, y disfunción de la barre-ra epitelial en la IBD8,9,21. En el presente estudio, el uso de DSS en el agua durante ocho semanas ocasionó un daño epitelial y una importante res-puesta inflamatoria, consiguiéndose por lo tanto un eficaz modelo de daño agudo al colon para testar el tratamiento de seis semanas con FR-91. Nuestros resultados demuestran que la mejor do-sis-respuesta se obtuvo en el grupo E utilizando la concentración de FR-91 al 20%, grupo en el que no se observaron ni alteraciones histológicas ni otro tipo de lesiones.
La tinción histológica rutinaria9,22-24 y con mar-cadores pre-tumorales9,20,23,25, utilizada para eva-luar la severidad de las lesiones, ha confirmado el efecto protector del FR-91 frente a la reacción inflamatoria ocasionada por el tratamiento con DSS. También analizamos concentraciones más bajas de FR-91 en el agua en ratones tratados du-rante ocho semanas, obteniendo una aparición gradual de lesiones directamente proporcional a la disminución de la concentración del FR-91, y obtuvimos resultados histológicos similares a los ratones de los grupos no tratados. Estos datos sugieren, por lo tanto, una acción quimiopreven-tiva frente a los procesos de cancerogénesis, sin interferir con las estructuras epiteliales sanas del colon de los ratones.
Se han observado alteraciones en la producción de muchas citoquinas en pacientes con IBD26. A pesar de que el impacto real de estos descubri-mientos en la patogénesis de la IBD se manten-ga aún controvertido, no está claro si se trata de factores primarios o secundarios involucrados en la regulación del sistema inmunológico de la mu-cosa intestinal. De todas formas estas citoquinas pueden ser consideradas como marcadores muy interesantes a la hora de diferenciar entre pacien-tes. En fases activas de IBD, un desequilibrio entre células reguladoras y células efectoras, que mayo-ritariamente involucran células efectoras T (Th1 y Th2) y células reguladoras (Tregs, Th3). La enfer-medad de Crohn (CD) está asociada con un per-fil de citoquinas Th1, que incluye IFN-γ, TNF-α, y IL-12, mientras la UC está asociada a un tipo de respuesta modificada Th2 que incluye IL-15 e IL-1027. Estas observaciones han sido recientemente complementadas con el descubrimiento del eje IL-23/IL-17 como parte de la respuesta inmuno-lógica T efectora, y parece estar involucrado en la IBD. Los niveles de expresión de IL-23 e IL-17 se encuentran elevados en pacientes con IBD28.
A pesar de que la identificación de la acción me-tabólica específica del FR-91 en la prevención y tratamiento de la colitis necesita ulteriores in-vestigaciones, hemos demostrado que el com-puesto FR-91 posee la capacidad de prevenir la formación de lesiones pretumorales en modelos murinos de colitis, inhibiendo el desarrollo de tumores colorrectales. El FR-91 parece ser un compuesto que requiere la debida atención en ulteriores estudios de prevención de procesos tu-morales del colon.
AbreviaturasCL, criptas de Lieberkühn; d, porción distal del colon; GI, glándulas intestinales; Gr A-E, grupos A-E; LP, lámina propia; m, porción intermedial del colon; MM, muscularis mucosa; p, porción proximal del colon; TM, túnica muscularis ex-terna. TS, túnica serosa.
“AntiGan® es un nuevo complejo lipoproteico extraido de la especieC. conger con propiedades inmunopotenciadoras y reguladoras delcrecimiento celular”

Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
Julio 2011 29
ciencia
“AntiGan® es un nuevo complejo lipoproteico extraido de la especieC. conger con propiedades inmunopotenciadoras y reguladoras delcrecimiento celular”

3030
Prevención de colitis experimental crónica inducida por DSS en ratones tratados con FR-91
Conclusiones
Los protocolos experimentales de inducción de cáncer de colon abren una serie de posibilidades para el biólogo experimental. Por un lado, permiten avanzar en el
estudio de los mecanismos de formación de focos de criptas aberrantes (FCA), lesiones pre-neoplásicas que se cree corresponden a las primeras manifestaciones
de procesos carcinogénicos posteriores. El protocolo de inducción de inflamación crónica mediante administración de DSS, utilizado en el presente estudio, permite
además la posibilidad de examinar el efecto de agentes preventivos putativos que inhiban la formación de estas displasias colónicas y avanzar así dentro de nuevas
intervenciones terapéuticas. El FCA es especialmente válido como biomarcador de efecto para ensayar productos naturales a los que se puede atribuir propiedades
antitumorales. Por otra parte, en el presente estudio, se ha propuesto un modelo para evaluar la progresión de tumores inducidos por DSS y explorar posibilidades
de modulación del proceso, así como de prevención en la formación de neoplasias en colon, mediante el uso del FR-91. Los resultados presentados demuestran
diferencias significativas en los grupos tratados con FR-91 y apoyan la realización de ulteriores estudios, centrados en la prevención y/o tratamiento, para profundizar
en los mecanismos moleculares que subyacen al desarrollo del cáncer de colon.
1. 1. Keohane J, O’Mahony C, O’Mahony L et al. Irritable bowel syndrome-type symptoms Keohane J, O’Mahony C, O’Mahony L et al. Irritable bowel syndrome-type symptoms in patients with inflammatory bowel disease: a real association or reflection of occult in patients with inflammatory bowel disease: a real association or reflection of occult inflammation? Am J Gastroenterol 2010; 105:1788-94.inflammation? Am J Gastroenterol 2010; 105:1788-94.
2. 2. Scaldaferri F, Fiocchi C. Inflammatory bowel disease: progress and current concepts of Scaldaferri F, Fiocchi C. Inflammatory bowel disease: progress and current concepts of etiopathogenesis. J Dig Dis 2007; 8:171-8.etiopathogenesis. J Dig Dis 2007; 8:171-8.
3. 3. Harpaz N, Polydorides AD. Colorectal dysplasia in chronic inflammatory bowel disease: Harpaz N, Polydorides AD. Colorectal dysplasia in chronic inflammatory bowel disease: pathology, clinical implications, and pathogenesis. Arch Pathol Lab Med 2010; pathology, clinical implications, and pathogenesis. Arch Pathol Lab Med 2010; 134:876-95.134:876-95.
4. 4. Dotan I. Disease behavior in adult patients: are there predictors for stricture or fistula Dotan I. Disease behavior in adult patients: are there predictors for stricture or fistula formation? Dig Dis 2009; 27:206-11.formation? Dig Dis 2009; 27:206-11.
5. 5. Elson CO. Experimental models of inflammatory bowel disease. Gastroenterology 1995; Elson CO. Experimental models of inflammatory bowel disease. Gastroenterology 1995; 109:1344-67.109:1344-67.
6. 6. Okayasu I, Hatakeyama S, Yamada M et al. A novel method in the induction of reliable Okayasu I, Hatakeyama S, Yamada M et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990; experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990; 98:694-702.98:694-702.
7. 7. Cooper HS, Murthy SN, Shah RS et al. Clinicopathologic study of dextran sulfate sodium Cooper HS, Murthy SN, Shah RS et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest 1993; 69:238-49.experimental murine colitis. Lab Invest 1993; 69:238-49.
8. 8. Tanaka T. Colorectal carcinogenesis: Review of human and experimental animal studies. Tanaka T. Colorectal carcinogenesis: Review of human and experimental animal studies. J Carcinog 2009; 8:5.J Carcinog 2009; 8:5.
9. 9. Birkenmeier EH, Torrey A, Sundberg JP. Chromosomal location of modifier genes Birkenmeier EH, Torrey A, Sundberg JP. Chromosomal location of modifier genes determining sensitivity of mice to dextran sulphate sodium. In: Tytgat G, Bartelsman J, determining sensitivity of mice to dextran sulphate sodium. In: Tytgat G, Bartelsman J, van Deventer S (Eds). Inflammatory Bowel Disease. Kluwer Academic Publishers, Boston, van Deventer S (Eds). Inflammatory Bowel Disease. Kluwer Academic Publishers, Boston, 1995:401-7.1995:401-7.
10. Berdy J. Bioactive microbial metabolites. J Antibiot 2005; 58:1-26.10. Berdy J. Bioactive microbial metabolites. J Antibiot 2005; 58:1-26.
11. Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. 11. Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod 2007; 70:461-77.J Nat Prod 2007; 70:461-77.
12. Olano C, Méndez C, Salas JA. Antitumor compounds from actinomycetes: from 12. Olano C, Méndez C, Salas JA. Antitumor compounds from actinomycetes: from gene clusters to new derivatives by combinatorial biosynthesis. Nat Prod Rep 2009; gene clusters to new derivatives by combinatorial biosynthesis. Nat Prod Rep 2009; 26:628-60.26:628-60.
13. Demain AL, Sánchez S. Microbial drug discovery: 80 years of progress. J Antibiot 2009; 13. Demain AL, Sánchez S. Microbial drug discovery: 80 years of progress. J Antibiot 2009; 62:5-16.62:5-16.
14. Lombardi VR, Martínez E, Chacón R, et al. Effects of FR-91 on immune cells from healthy 14. Lombardi VR, Martínez E, Chacón R, et al. Effects of FR-91 on immune cells from healthy individuals and from patients with non-Hodgkin lymphoma. J Biomed Biotechnol 2009. individuals and from patients with non-Hodgkin lymphoma. J Biomed Biotechnol 2009. doi:10.1155/2009/187015.doi:10.1155/2009/187015.
15. Cho JY, Chang HJ, Lee SK et al. Amelioration of dextran sulfate sodium-induced colitis 15. Cho JY, Chang HJ, Lee SK et al. Amelioration of dextran sulfate sodium-induced colitis in mice by oral administration of beta-caryophyllene, a sesquiterpene. Life Sci 2007; in mice by oral administration of beta-caryophyllene, a sesquiterpene. Life Sci 2007; 80:932-9.80:932-9.
16. Araki Y, Mukaisyo K, Sugihara H et al. Increased apoptosis and decreased proliferation 16. Araki Y, Mukaisyo K, Sugihara H et al. Increased apoptosis and decreased proliferation of colonic epithelium in dextran sulfate sodium-induced colitis in mice. Oncol Rep 2010; of colonic epithelium in dextran sulfate sodium-induced colitis in mice. Oncol Rep 2010; 24:869-74.24:869-74.
17. Huang TC, Tsai SS, Liu LF et al. Effect of Arctium lappa L. in the dextran sulfate sodium 17. Huang TC, Tsai SS, Liu LF et al. Effect of Arctium lappa L. in the dextran sulfate sodium colitis mouse model. World J Gastroenterol 2010; 16:4193-9.colitis mouse model. World J Gastroenterol 2010; 16:4193-9.
18. Choi SY, Hur SJ, An CS et al. Anti-inflammatory effects of Inonotus obliquus in colitis induced 18. Choi SY, Hur SJ, An CS et al. Anti-inflammatory effects of Inonotus obliquus in colitis induced by dextran sodium sulfate. J Biomed Biotechnol 2010. doi:10.1155/2010/943516.by dextran sodium sulfate. J Biomed Biotechnol 2010. doi:10.1155/2010/943516.
19. Whittem CG, Williams AD, Williams CS. Murine colitis modeling using Dextran Sulfate 19. Whittem CG, Williams AD, Williams CS. Murine colitis modeling using Dextran Sulfate Sodium (DSS). J Vis Exp 2010. doi:10.3791/1652.Sodium (DSS). J Vis Exp 2010. doi:10.3791/1652.
20. Tanaka T, Kohno H, Suzuki R et al. Dextran sodium sulfate strongly promotes colorectal 20. Tanaka T, Kohno H, Suzuki R et al. Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc(Min/+) mice: inflammatory stimuli by dextran sodium sulfate carcinogenesis in Apc(Min/+) mice: inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int J Cancer 2006; 118:25-34.results in development of multiple colonic neoplasms. Int J Cancer 2006; 118:25-34.
21. Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of 21. Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol 2010; 8:564-77.IBD: lessons from mouse infection models. Nat Rev Microbiol 2010; 8:564-77.
22. Darsigny M, Babeu JP, Dupuis AA et al. Loss of hepatocyte-nuclear-factor-4alpha affects 22. Darsigny M, Babeu JP, Dupuis AA et al. Loss of hepatocyte-nuclear-factor-4alpha affects colonic ion transport and causes chronic inflammation resembling inflammatory bowel colonic ion transport and causes chronic inflammation resembling inflammatory bowel disease in mice. PLoS One 2009. doi:10.1371/journal.pone.0007609.disease in mice. PLoS One 2009. doi:10.1371/journal.pone.0007609.
23. Wang JG, Wang DF, Lv BJ et al. A novel mouse model for colitis-associated colon 23. Wang JG, Wang DF, Lv BJ et al. A novel mouse model for colitis-associated colon carcinogenesis induced by 1,2-dimethylhydrazine and dextran sulfate sodium. World J carcinogenesis induced by 1,2-dimethylhydrazine and dextran sulfate sodium. World J Gastroenterol 2004; 10:2958-62.Gastroenterol 2004; 10:2958-62.
24. Mähler M, Bristol IJ, Leiter EH et al. Differential susceptibility of inbred mouse strains to 24. Mähler M, Bristol IJ, Leiter EH et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol 1998; 274:44-51.dextran sulfate sodium-induced colitis. Am J Physiol 1998; 274:44-51.
25. Cruickshank SM, English NR, Felsburg PJ et al. Characterization of colonic dendritic cells 25. Cruickshank SM, English NR, Felsburg PJ et al. Characterization of colonic dendritic cells in normal and colitic mice. World J Gastroenterol 2005; 11:6338-47.in normal and colitic mice. World J Gastroenterol 2005; 11:6338-47.
26. Yamamoto T, Umegae S, Kitagawa T et al. Systemic and local cytokine production in 26. Yamamoto T, Umegae S, Kitagawa T et al. Systemic and local cytokine production in quiescent ulcerative colitis and its relationship to future relapse: a prospective pilot study. quiescent ulcerative colitis and its relationship to future relapse: a prospective pilot study. Inflamm Bowel Dis 2005; 11:589-96.Inflamm Bowel Dis 2005; 11:589-96.
27. Danese S, Fiocchi C. Etiopathogenesis of inflammatory bowel diseases. World J 27. Danese S, Fiocchi C. Etiopathogenesis of inflammatory bowel diseases. World J Gastroenterol 2006; 12:4807-12.Gastroenterol 2006; 12:4807-12.
28. Nielsen OH, Kirman I, Rüdiger N et al. Upregulation of interleukin-12 and-17 in active 28. Nielsen OH, Kirman I, Rüdiger N et al. Upregulation of interleukin-12 and-17 in active inflammatory bowel disease. Scand J Gastroenterol 2003; 38:180-5.inflammatory bowel disease. Scand J Gastroenterol 2003; 38:180-5.
Referencias Bibliográficas:
Valter R.M. Lombardi [email protected]

31Julio 2011
as enfermedades cardiovasculares (ECV) cons-tituyen una de las principales causas de morta-lidad y morbilidad en los países desarrollados y se ha demostrado la fuerte implicación del riesgo vascular en patologías cerebrovasculares, demencias, enfermedad de Alzheimer, etc. Di-versos informes y recomendaciones de exper-tos para la prevención de estas enfermedades nos presentan datos realmente escalofriantes, haciendo hincapié en la estrecha relación que presentan la mayoría de los casos de ECV con
hábitos de vida y factores bioquímicos y fisioló-gicos modificables. El primer paso para la inter-vención en el riesgo vascular es su conocimiento y para ello las escalas de estratificación de uso habitual a veces no son suficientemente preci-sas para tomar decisiones terapéuticas. Los fac-tores de riesgo vascular (FRV) “clásicos” dejan un vacío en el diagnóstico de la enfermedad vascular, precedida en ocasiones por una ate-rosclerosis subyacente progresiva. Estos hechos determinan el gran interés en el desarrollo
L
Lola CorzoDepartamento de Bioquímica Médica y Tecnología Analítica
Centro Médico EuroEspes, Bergondo, Coruña
Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular:
Factores de Riesgo Emergentes

Tabla 1.1. Factores de riesgo vascular (FRV) “Clásicos”
No Modificables• Edad Avanzada*
• ECV establecida*
• Factores genéticos
• Historia familiar de enfermedad coronaria precoz*
• Sexo masculino*
Modificables• DM (Diabetes Mellitus) **
• Dieta aterogénica
• Sobrepeso
• Inactividad física
• Tabaquismo*
• Hipertensión arterial*
• Hiperglucemia
• Dislipemia
• Estado trombogénico y hemostático***
* Factores de riesgo incluidos en el algoritmo ATP III-NCEP para estratificación de riesgo cardiovascular.
** Se ha demostrado que la modificación de la presión arterial y los lípidos en personas con diabetes ha redu-cido el riesgo cardiovascular. Ensayos clínicos indican que un mejor control de la glucosa reduce el riesgo cardiovascular, pero no de manera estadísticamente significativa.
*** El riesgo ha sido incluido según observaciones de efectos positivos de tratamientos anticoagulantes y antiagregantes plaquetarios en la reducción del riesgo de ECV.
Formación de placa ateromatosa
Tabla 1.2. Factores de riesgo vascular “Emergentes” (FRVE)
Modificables• Proteína C reactiva (ultrasensible)
• Homocisteína
• Lipoproteína (a)
• Fibrinógeno
32
Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular
de nuevos marcadores de predicción de riesgo para el tratamiento precoz y preventivo de estas enfermedades. De todos los propuestos por la comunidad científica, la medición de determi-nados marcadores emergentes, inflamatorios y trombogénicos, constituye una herramienta útil para el ajuste del riesgo vascular calculado me-diante factores de riesgo estándar, sobre todo en situaciones de riesgo límite o dudoso. Al tratarse de factores de riesgo modificables, nos permiten actuar sobre el riesgo vascular global de una per-sona, reduciendo las tasas de mortalidad y mor-bilidad para ECV, particularmente en sujetos de alto riesgo, según se ha demostrado en ensayos clínicos.
Introducción
Las ECV constituyen una de las principales causas de mortalidad y morbilidad en los paí-ses desarrollados y se ha demostrado la fuerte implicación del riesgo vascular en patologías cerebrovasculares, demencias, enfermedad de Alzheimer, etc.1 El informe SEA (Sociedad Espa-ñola de Arteriosclerosis) 20032 sobre enferme-dades cardiovasculares y sus factores de riesgo en España nos presenta datos importantes para entender esta problemática. A modo de resu-men, nos encontramos con que en la actualidad en España se producen más de 132.000 muertes y más de 5 millones de estancias hospitalarias al año por enfermedades cardiovasculares y en los próximos años se prevé un aumento en el núme-ro de hospitalizaciones por estas enfermedades. Los valores de colesterol sanguíneo en la mitad
de la población española son mayores de 200 mg/dL (elevados), la mayoría sin tratamiento hipolipemiante. La tasa de hipertensión arterial (HTA) es aproximadamente del 40%, alcanzan-do el 68% en las personas de más de 60 años. La prevalencia de tabaquismo es del 36%, de las más altas de Europa en jóvenes. El 63% de la población española mayor de 16 años consume alcohol de forma habitual. A todo ello hay que añadir que la dieta rica en grasas saturadas, la obesidad, la diabetes y el sedentarismo son fac-tores de riesgo en aumento en nuestra sociedad. A nivel europeo, la Sociedad Europea de Car-diología (ESC) publicó en el 2007 la Guía Euro-pea de Prevención Cardiovascular en la Práctica Clínica3 basándose en unos fundamentos simila-res que se detallan brevemente a continuación: res que se detallan brevemente a continuación:

La mayoría de los casos de ECV están estrechamente relacionados con hábitos de vida y factores bioquímicos y fisiológicos modificables
Figura 1. Perfil bioquímico de riesgo vascular propuesto
200 240
130 160
150
63 114
35
120 176
2.7 4.5
3.0
12.0 30.0
300
3.0
120
0.90
4.5
270
190
170
130
50
120
4.6
13
8
415
60
50
3.6
5.4
1.1
Julio 2011 33
ciencia
(i) La ECV es la mayor causa de muerte prema-tura en Europa y en todo el mundo occidental, así como una importante causa de discapacidad, que contribuye de forma sustancial al imparable aumento de los costes de asistencia sanitaria. (ii) La modificación del riesgo cardiovascular ha mostrado su capacidad de reducir la morta-lidad y la morbilidad por ECV, particularmen-te en sujetos de alto riesgo. (iii) La muerte por ECV ocurre con frecuencia de manera súbita y antes de acceder a los servicios sanitarios, por lo que muchas intervenciones terapéuticas son inaplicables o paliativas. (iv) La mayoría de los casos de ECV están estrechamente relacionados con hábitos de vida y factores bioquímicos y fi-siológicos modificables. (v) La aterosclerosis sub-yacente evoluciona insidiosa y progresivamente a lo largo de muchos años y suele estar avanzada cuando aparecen los síntomas clínicos. Estos da-tos se pueden extrapolar a nuestra población, y el Comité Español Interdisciplinario para la Pre-vención Cardiovascular (CEIPC) ha publicado una adaptación de esta guía para la población española (2008)4.
Estratificación del riesgo vascular
La primera medida a llevar a cabo para la pre-vención de la enfermedad vascular en atención primaria es la estratificación del riesgo vascular. Para ello se han desarrollado una serie de esca-las de riesgo, basadas en estudios poblacionales siendo la tabla SCORE (2003)5 la recomendada por la CEIPC para la población española. Su validación en 12 países europeos y la inclusión del riesgo no coronario le ofrecen ventajas fren-te a la tabla Framingham (Anderson, 1991)6
estudiada en la población americana, de alto riesgo, y limitada a riesgo coronario. La difícil extrapolación entre diferentes poblaciones, y la omisión de factores de riesgo como anteceden-tes familiares de enfermedad ECV prematura, sedentarismo-obesidad, casos de aterosclero-sis subclínica y otros factores implicados en la enfermedad vascular limitan la predicción de riesgo de estas escalas. La utilización de factores de riesgo vascular (FRV) mayores o “clásicos” (Tabla 1.1) como edad avanzada, diabetes me-llitus, tabaquismo, historia familiar de enferme-dad coronaria precoz, HTA y dislipidemia en la predicción del riesgo vascular deja un 20% de casos de enfermedad vascular sin explicación plausible. Es, por tanto, necesario el estudio de otros FRV adicionales para mejorar el manejo y detección del riesgo vascular calculado ha-bitualmente por escalas de riesgo. Varios FRV han sido propuestos para apoyar los criterios clásicos en la detección de aterosclerosis sub-clínica. En nuestro perfil (Fig.1), además de in-dicar el riesgo SCORE, hemos seleccionado los
considerados de mayor utilidad por la comuni-dad científica. Estos parámetros denominados marcadores emergentes de riesgo vascular incluyen marcadores emergentes de riesgo vascular incluyen marcadores emergentesparámetros lipídicos no incluidos en escalas clásicas: lipoproteína (a), apolipoproteína A-1, apolipoproteína B, biomarcadores inflamato-rios como la proteína C reactiva ultrasensible y fibrinógeno; y marcadores nutricionales asocia-dos con aterotrombosis prematura tal como la
homocisteína, además de los clásicos colesterol HDL (cHDL) y LDL (cLDL) con medición direc-ta. Con claridad se puede observar que la gran mayoría de estos nuevos factores de riesgo para enfermedad vascular son modificables (son modificables (son modificables Tabla 1.2), es decir, que se puede actuar sobre ellos para reducir el riesgo vascular de una persona.

La modificación del riesgo cardiovascular ha mostrado su capacidad de reducir la mortalidad y la morbilidad por ECV, particularmente en sujetos de alto riesgo
Tabla 2. Principales factores de riesgo cardiovascular2
Factores de riesgo causales (mayores independientes)
Tabaco
Presión arterial elevada
Colesterol sérico total y colesterol-LDL elevados
Colesterol-HDL bajo
Diabetes mellitus
Edad avanzada
Factores de riesgo predisponentes
Obesidad*
Obesidad abdominal
Inactividad física*
Historia familiar de enfermedad coronaria prematura
Características étnicas
Factores psicosociales
Factores de riesgo condicionales
Triglicéridos séricos elevados
Pequeñas partículas LDL
Homocisteína sérica elevada
Lipoproteína a sérica elevada
Factores protrombóticos (ej. fibrinógeno)
Marcadores de la inflamación (proteína C-reactiva)
*Estos factores son también denominados factores de riesgo mayores por la Asociación Americana del Corazón.
LDL: Lipoproteínas de baja densidad; HDL: Lipoproteínas de alta densidad.
34
Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular
En general, la pérdida de peso, la dieta, el ejercicio, y la suspensión del tabaco llevan a la disminución de su concen-tración sérica y a la reducción del riesgo cardiovascular.
Fibrinógeno: es una glucoproteína sinte-Fibrinógeno: es una glucoproteína sinte-Fibrinógeno:tizada por el hígado que participa acti-vamente en la coagulación sanguínea. Existen cuatro mecanismos a través de los cuales el aumento de fibrinógeno puede promover la enfermedad arterial: la aterogénesis, la agregación plaqueta-ria, la formación de trombos de fibrina y el aumento de la viscosidad plasmá-tica. Niveles plasmáticos >300 mg/dL podrían utilizarse para definir un ries-go arteriosclerótico aumentado. Como ocurre con otros factores de riesgo, una sola determinación puede no reflejar de forma precisa los niveles basales de un paciente. No hay tratamiento específico disponible para disminuir sus niveles. Existen fármacos que reducen los nive-les plasmáticos de fibrinógeno, como la heparina, pero también tienen otras inci-dencias cardiovasculares. En fumadores, suspender el hábito de fumar disminuye las concentraciones.
En la actualidad, es posible realizar una estratifi-cación de riesgo vascular más precisa para iden-tificar a las personas de alto riesgo, en quienes debemos extremar los aspectos educativos y las medidas de tratamiento y control, tanto en la fase de prevención primaria (cuando no se han producido eventos cardio- ni cerebrovascula-res), como en la secundaria (después de eventos previos). Los factores de riesgo cardiovascular también se pueden clasificar en causales, predis-ponentes y condicionales, según su implicación en la enfermedad vascular (Tabla 2).
Para conocer en más detalle cada uno de los marcadores emergentes propuestos y su utilidad en el conocimiento del riesgo vascular (Tabla 3) presentamos una pequeña monografía de cada uno de ellos:
Proteína C reactiva (PCR-us) ultrasensible: es una proteína de fase aguda sintetiza-da por el hígado en situaciones de infla-mación aguda. Diversas investigaciones prospectivas demostraron una asocia-ción directa entre los niveles de PCR-us y el riesgo de enfermedad vascular (in-farto de miocardio, accidente cerebro-vascular (ACV) y enfermedad vascular periférica), independiente de otros factores de riesgo coronario. La presen-cia de niveles elevados de esta proteína (> 3 mg/L) incrementa el valor predic-tivo del perfil lipídico (colesterol total y HDL) para determinar el riesgo de un primer infarto de miocardio o ACV en individuos aparentemente de bajo ries-go. La PCR-us es una proteína reactante de fase aguda, por lo que una única de-terminación de PCR-us puede no reflejar el nivel basal de PCR-us de un paciente. Valores >3.0 mg/L indican riesgo vascu-lar alto aunque valores persistentes muy elevados de PCR-us (>10 mg/L) pueden enmascarar patologías no vasculares y habría que descartarlas7. No ha sido eva-luada una terapia específica para redu-cir los niveles de PCR-us; sin embargo muchas intervenciones conocidas para reducir el riesgo vascular han sido aso-ciadas con la disminución de PCR-us. Se ha demostrado que las estatinas dismi-nuyen los niveles de PCR en un 15-25% 6 meses después de iniciada la terapia.
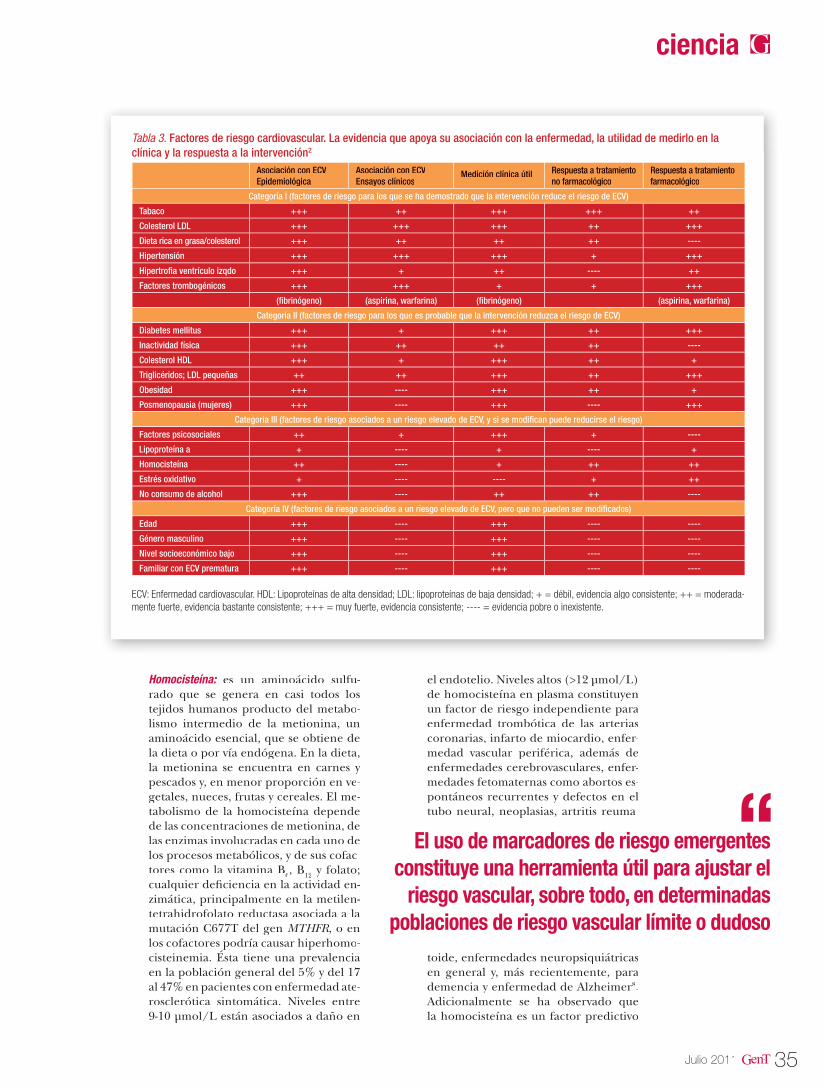
El uso de marcadores de riesgo emergentes constituye una herramienta útil para ajustar el
riesgo vascular, sobre todo, en determinadas poblaciones de riesgo vascular límite o dudoso
Tabla 3. Factores de riesgo cardiovascular. La evidencia que apoya su asociación con la enfermedad, la utilidad de medirlo en la clínica y la respuesta a la intervención2
Asociación con ECV Epidemiológica
Asociación con ECV Ensayos clínicos
Medición clínica útil Respuesta a tratamiento no farmacológico
Respuesta a tratamiento farmacológico
Categoría I (factores de riesgo para los que se ha demostrado que la intervención reduce el riesgo de ECV)
Tabaco +++ ++ +++ +++ ++
Colesterol LDL +++ +++ +++ ++ +++
Dieta rica en grasa/colesterol +++ ++ ++ ++ ----
Hipertensión +++ +++ +++ + +++
Hipertrofia ventrículo izqdo +++ + ++ ---- ++
Factores trombogénicos +++ +++ + + +++
(fibrinógeno) (aspirina, warfarina) (fibrinógeno) (aspirina, warfarina)
Categoría II (factores de riesgo para los que es probable que la intervención reduzca el riesgo de ECV)
Diabetes mellitus +++ + +++ ++ +++
Inactividad física +++ ++ ++ ++ ----
Colesterol HDL +++ + +++ ++ +
Triglicéridos; LDL pequeñas ++ ++ +++ ++ +++
Obesidad +++ ---- +++ ++ +
Posmenopausia (mujeres) +++ ---- +++ ---- +++
Categoría III (factores de riesgo asociados a un riesgo elevado de ECV, y si se modifican puede reducirse el riesgo)
Factores psicosociales ++ + +++ + ----
Lipoproteína a + ---- + ---- +
Homocisteína ++ ---- + ++ ++
Estrés oxidativo + ---- ---- + ++
No consumo de alcohol +++ ---- ++ ++ ----
Categoría IV (factores de riesgo asociados a un riesgo elevado de ECV, pero que no pueden ser modificados)
Edad +++ ---- +++ ---- ----
Género masculino +++ ---- +++ ---- ----
Nivel socioeconómico bajo +++ ---- +++ ---- ----
Familiar con ECV prematura +++ ---- +++ ---- ----
ECV: Enfermedad cardiovascular. HDL: Lipoproteínas de alta densidad; LDL: lipoproteínas de baja densidad; + = débil, evidencia algo consistente; ++ = moderada-mente fuerte, evidencia bastante consistente; +++ = muy fuerte, evidencia consistente; ---- = evidencia pobre o inexistente.
Julio 2011 35
ciencia
Homocisteína: es un aminoácido sulfu-Homocisteína: es un aminoácido sulfu-Homocisteína:rado que se genera en casi todos los tejidos humanos producto del metabo-lismo intermedio de la metionina, un aminoácido esencial, que se obtiene de la dieta o por vía endógena. En la dieta, la metionina se encuentra en carnes y pescados y, en menor proporción en ve-getales, nueces, frutas y cereales. El me-tabolismo de la homocisteína depende de las concentraciones de metionina, de las enzimas involucradas en cada uno de los procesos metabólicos, y de sus cofac-tores como la vitamina B6, B12 y folato; cualquier deficiencia en la actividad en-zimática, principalmente en la metilen-tetrahidrofolato reductasa asociada a la mutación C677T del gen MTHFR, o en MTHFR, o en MTHFRlos cofactores podría causar hiperhomo-cisteinemia. Ésta tiene una prevalencia en la población general del 5% y del 17 al 47% en pacientes con enfermedad ate-rosclerótica sintomática. Niveles entre 9-10 µmol/L están asociados a daño en
el endotelio. Niveles altos (>12 µmol/L) de homocisteína en plasma constituyen un factor de riesgo independiente para enfermedad trombótica de las arterias coronarias, infarto de miocardio, enfer-coronarias, infarto de miocardio, enfer-coronarias, infarto de miocardio, enfermedad vascular periférica, además de enfermedades cerebrovasculares, enfer-enfermedades cerebrovasculares, enfer-enfermedades cerebrovasculares, enfermedades fetomaternas como abortos es-pontáneos recurrentes y defectos en el tubo neural, neoplasias, artritis reuma-
toide, enfermedades neuropsiquiátricas en general y, más recientemente, para demencia y enfermedad de Alzheimer8. Adicionalmente se ha observado que la homocisteína es un factor predictivo

Cuanto más alto es el riesgo vascular, mayor es el beneficio terapéutico
Procesamiento en laboratorio
36
Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular
muy importante de mortalidad en pa-cientes con cardiopatía coronaria com-probada. Valores de referencia:
9.0-10.0 µmol/L Posible daño endotelio<10.0 µmol/L Deseable<12.0 µmol/L Normal
12.0-30.0 µmol/L Moderada30.0-100.0 µmol/L Intermedia
>100.0 µmol/L Severa
El tratamiento es indicado en pacientes con niveles mayores de 10 µmol/L9. Se deben descartar enfermedades conco-mitantes antes del inicio terapéutico. Para ello se recomienda medir niveles sanguíneos de vitamina B12, ácido fólico, función renal básica, y función tiroidea. Una vez descartadas otras causas, el tra-tamiento de la hiperhomocistinemia se basa generalmente en suplemento vitamí-nico con ácido fólico (cereales, verduras verdes, frutas cítricas, frijoles e hígado), vitamina B6 (cereales integrales, hígado, frutos secos y levadura de cerveza) y vita-mina B12 (carne, huevo, pescado, lácteos, ciertas algas y derivados de la soja enri-quecidos). Normalmente se alcanzan disminuciones del 25% de los niveles de homocisteína en individuos normales y hasta del 50% con hiperhomocisteine-mia utilizando dosis habituales.
Lipoproteína(a) [Lp(a)]: es una proteína plasmática cuya estructura semeja a las lipoproteínas de baja densidad (LDL). Debido a su homología con el plasminó-geno, la Lp(a) compite por los mismos sitios de unión en la molécula y en las proteínas de membrana celular interfi-riendo en la fibrinolisis y acentuando el riesgo trombótico. La Lp(a) es un nue-vo factor de riesgo independiente para la enfermedad de la arteria coronaria (EAC), especialmente en los hombres de raza blanca hipercolesterolémicos, y actúa como marcador predictivo de la severidad angiográfica en varones jóve-nes con inicio de esta patología. En los adultos ancianos, un nivel elevado de lipoproteína(a) es un factor de riesgo independiente de accidente cerebrovas-cular o accidente isquémico transitorio y de muerte por enfermedad vascular10. El efecto aterotrombogénico de la Lp(a) depende de su concentración plasmática, bastante estable e independiente de la
dieta y el ejercicio físico. El polimorfismo genético de esta lipoproteína constituye también un importante riesgo para la arteriosclerosis. Valores plasmáticos ma-yores de 30 mg/dL representan un incre-mento de dos veces el riesgo relativo para la enfermedad aterosclerótica. La mag-nitud del riesgo atribuible para infarto miocárdico y cerebral fue similar al apor-tado por la presencia de hipercolestero-lemia (9.3% y 10.3%, respectivamente)11. Valores >30 mg/dL se consideran de ries-go vascular elevado. El conocimiento de su concentración plasmática permite se-leccionar una subpoblación de pacientes dislipidémicos con mayor riesgo vascular que pudieran beneficiarse con un trata-miento hipocolesterolemizante y un se-guimiento particularmente intensivo12. Para reducir los niveles de lipoproteína (a) se han utilizado terapias de reem-plazo hormonal a base de estrógenos en mujeres post-menopáusicas y ácido nico-tínico en hombres y mujeres en las que la terapia con estrógenos está contrain-dicada, consiguiendo reducciones del 25% y 35%, respectivamente. La adición de neomicina a estos tratamientos puede lograr una reducción de casi un 50%.
Apolipoproteína A-1 (apo A-1): es la princi-Apolipoproteína A-1 (apo A-1): es la princi-Apolipoproteína A-1 (apo A-1):pal proteína encontrada en las lipopro-teínas de alta densidad (HDL), aunque la apo A-1 está también presente en los quilomicrones. El principal papel de las apolipoproteínas A-1 es la activación de la lecitin-colesterol-acil-tranferasa

Formacion placa aterosclerótica
LDL: Low density lipoprotein (lipoproteínas de baja densidad)LDLox: LDL oxidado
FT: Factor tisularWSMC: Vascular smooth muscle cells (células de músculo liso vascular)
Julio 2011 37
ciencia
importante información para el cálculo de riesgo vascular. Un informe reciente de la American College of Cardiology Foundation14 recomienda añadir apo B a la valoración del riesgo vascular en per-sonas con alto riesgo cardiometabólico.
Apolipoproteína E (apo E): es una proteína Apolipoproteína E (apo E): es una proteína Apolipoproteína E (apo E):de 299 aminoácidos sintetizada prin-cipalmente en el hígado y en menor medida en el cerebro, bazo, glándulas suprarrenales, ovarios, riñones, células musculares y en macrófagos. La apoli-poproteína E tiene muchas funciones
(LCAT) y la extracción del colesterol li-bre de los tejidos extra-hepáticos. La apo A-1 puede ser descrita como una proteí-na no aterogénica. Existe una correla-ción inversa entre los niveles de apo A-1 y el riesgo cardiovascular; incluso varios grupos científicos le han otorgado mayor valor pronóstico que al colesterol HDL (cHDL)13.
Apolipoproteína B (apo B): es la proteína Apolipoproteína B (apo B): es la proteína Apolipoproteína B (apo B):mayoritaria encontrada en las lipopro-teínas LDL pero puede ser encontrada en todas las formas de partículas po-tencialmente aterogénicas (LDL, VLDL (lipoproteínas de muy baja densidad) e IDL (lipoproteínas de densidad me-dia)). En humanos se encuentran dos formas de apo B; la más común es la B-100, o B grande, la cual constituye las apo B encontradas en las lipoproteínas sintetizadas por el hígado. La apo B es la principal proteína transportadora de colesterol en la sangre y es el ligando por el cual se internaliza el colesterol en las células a través del LDL-receptor. Por lo tanto, la concentración de apo B indica el número total de partículas peligrosas y es muy útil para la evaluación global del riesgo vascular. Existe una correlación di-recta entre los niveles de apo B y el riesgo cardiovascular. La incorporación de apo B y del índice apoB/apoA1 nos aporta
Placa ateromatosa

El mejor conocimiento de los factores de riesgo de la enfermedad vascular y de la capacidad de actuación sobre ellos nos ayudará a su mejor prevención y es hoy uno de los principales retos de la comunidad científica
Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular
incluyendo el transporte de triglicéri-dos al tejido hepático (como parte de VLDL), y la distribución de colesterol entre las células (como parte de HDL). Apo E es un ligando para el receptor LDL. Se trata de una proteína polimór-LDL. Se trata de una proteína polimór-LDL. Se trata de una proteína polimórfica que se presenta en tres isoformas: Apo E2, E3, E4. Apo E3 es la más común
con una prevalencia en la población caucásica del 77%. Apo E4 es la segun-da isoforma más común (15%) y E2 es la más rara (8%). El polimorfismo es la más rara (8%). El polimorfismo APOE influye tanto en procesos fisioló influye tanto en procesos fisiolóAPOE influye tanto en procesos fisiolóAPOE -gicos como patológicos, en los niveles gicos como patológicos, en los niveles
séricos de colesterol total, cLDL y séricos de colesterol total, cLDL y en la concentración de apo B. en la concentración de apo B.
La apolipoproteína E ha sido La apolipoproteína E ha sido asociada con la formación asociada con la formación de la lesión aterosclerótica de la lesión aterosclerótica
inhibiendo la agregación inhibiendo la agregación plaquetaria15,16. Niveles . Niveles disminuidos de apo E se disminuidos de apo E se relacionan con el polimorrelacionan con el polimor-relacionan con el polimor-relacionan con el polimor
fismo E4 y con niveles altos y con niveles altos E4 y con niveles altos E4de colesterol total y triglide colesterol total y trigli-céridos, facilitando una céridos, facilitando una aterosclerosis prematura. aterosclerosis prematura.
El polimorfismo E4E4 ha sido E4 ha sido E4también implicado en divertambién implicado en diver-también implicado en diver-también implicado en diver
sas enfermedades incluyendo sas enfermedades incluyendo enfermedades cardiovasculaenfermedades cardiovascula-
res, enfermedades neurodegeres, enfermedades neurodege-nerativas como la enfermedad de nerativas como la enfermedad de
Alzheimer y otras. Apo E2 está Alzheimer y otras. Apo E2 está asociada con la disbetalipoasociada con la disbetalipo-
proteinemia familiar proteinemia familiar (hiperlipoproteine(hiperlipoproteine-
mia tipo III) y no mia tipo III) y no se une al receptor se une al receptor lipídico. Los ranlipídico. Los ran-gos de referencia gos de referencia son usados para son usados para las isoformas las isoformas más comunes. más comunes. En valores fueEn valores fue-ra de rango es ra de rango es recomendable recomendable determinar el determinar el
genotipo. Estugenotipo. Estu-dios recientes han dios recientes han
demostrado que la atorvastatina causa una disminución significativa en los ni-veles plasmáticos de apoE en pacientes con hiperlipidemia combinada17.
Colesterol HDL directo: la lipoproteína HDL está compuesta por partículas he-terogéneas que incluyen colesterol y va-rían según tamaño, contenido lipídico y proteico. La lipoproteína HDL actúa eliminando el colesterol de las células periféricas hacia el hígado, donde el colesterol es convertido en ácidos bi-liares y excretado al intestino. Existe una relación inversa entre los niveles de cHDL en suero y la prevalencia/in-cidencia de enfermedades cardiovascu-lares. La medida directa del cHDL nos ofrece una mejorada exactitud y repro-ducibilidad comparada con otros méto-dos utilizados. Según la NCEP-ATPIII18, se consideran valores bajos y por tanto de riesgo los inferiores a 40 mg/dL en hombres y 45 mg/dL en mujeres. Nive-les mayores a 60 mg/dL se consideran un factor positivo. Una dieta rica en aceite de oliva, nueces y aguacate pue-de ser muy beneficiosa para aumentar los niveles de cHDL, sin aumentar los de cLDL. La pérdida de peso y el ejerci-cio físico también son recomendables. El consumo moderado de alcohol (2 copas de vino/día) puede aumentar en un 5 a 10% el cHDL y además, dis-minuye la tendencia a formar coágulos en el interior de los vasos sanguíneos. Dentro de las alternativas farmacológi-cas disponibles, los fibratos y la niacina pueden aumentar los niveles de cHDL hasta en un 30%, las estatinas entre 5% y 10% y las resinas secuestrantes de sa-les biliares entre 3% y 5%.
Colesterol LDL directo: la lipoproteína Colesterol LDL directo: la lipoproteína Colesterol LDL directo:LDL es sintetizada en el hígado por la acción de varias enzimas lipolíticas sobre las VLDL ricas en triglicéridos. Los receptores específicos de LDL facilitan la eliminación del LDL del plasma por las células del parénquima hepático. Se ha demostrado que la ma-yoría del colesterol depositado en las placas de ateroma procede de LDL. Por esta razón el cLDL se considera el marcador individual predictivo más importante para la aterosclerosis y el marcador diana en las terapias hipoli-pemiantes para la prevención o reduc-ción de la aterosclerosis. El método directo de determinación nos ofrece grandes ventajas frente al habitual-mente utilizado sistema de estimación

Los factores de riesgo vascular “clásicos” dejan un hueco en el diagnóstico de la enfermedad
vascular, precedida en ocasiones por una aterosclerosis subyacente progresiva
Julio 2011 39
ciencia
muscular y otros. Su principal función es suministrar energía a la célula. La hi-pertrigliceridemia (>150 mg/dL) está considerada como un factor de riesgo independiente3,18. Frecuentemente va asociada a otros factores de riesgo vas-cular. Una alimentación equilibrada, sin exceso de calorías, es fundamen-tal en la regulación de los niveles de triglicéridos. Reducir los hidratos de carbono refinados (azúcar, gaseosas comunes, golosinas y miel) y aumentar las grasas poliinsaturadas como omega 3 (alto contenido en el pescado azul), disminuyendo grasas animales y grasa trans es aconsejable para evitar o redu-cir la hipertrigliceridemia. El aumento de ejercicio físico y la disminución del consumo de alcohol, si éste es abun-dante, serían otras medidas terapéuti-cas. Cuando estas recomendaciones no son suficientes para regular la concen-tración de triglicéridos hay que recu-rrir a terapias farmacológicas a base de ácido nicotínico, fibratos. Varios estu-dios clínicos demostraron disminución significativa del riesgo de enfermedad coronaria al reducir los niveles de tri-glicéridos con medicamentos.
Ratios o Índices de riesgo, basados en los mar- basados en los mar- basados en los marcadores del metabolismo lipídico, son de gran utilidad en el manejo del riesgo vascular por potenciar, en muchos casos, la información que dan los marcadores individualmente. Los más utilizados en la práctica clínica son:
Índice Colesterol Total/Colesterol HDL: Valores Índice Colesterol Total/Colesterol HDL: Valores Índice Colesterol Total/Colesterol HDL:normales: Hombres: 3.5-5.0. Mujeres: 3.4-4.5.
Índice apoB/apoA1: Los resultados de cuatro Índice apoB/apoA1: Los resultados de cuatro Índice apoB/apoA1:estudios epidemiológicos prospectivos, in-cluido el AMORIS (Suecia)20, han demostra-do que la apo B es superior al colesterol total o al cLDL para predecir el riesgo de enfer-medad cardiovascular, y que la relación de apoB/apoA1 es superior a la relación coles-terol total/HDL, o a la de LDL/cHDL como índice general de riesgo y como índice de riesgo de eventos cardiovasculares para los pacientes en tratamiento con estatinas. Se consideran valores de riesgo aterogénico ín-dices mayores de 0.9.
por fórmulas matemáticas. Según la NCEP-ATPIII18 y las European Guideli-nes3,4 se definen niveles con riesgo vas-cular alto los superiores a 130 mg/dL, en población sin otro factor de riesgo. Valores de referencia:
<100 mg/dL Óptimo100-129 mg/dL Próximo al óptimo130-159 mg/dL Límite Alto160-189 mg/dL Alto
≥190 mg/dL Muy Alto
Un cambio en el estilo de vida sería la primera opción para reducir los niveles de cLDL. Se aconseja una dieta baja en grasas saturadas y colesterol, ingesta de derivados vegetales como esteroles y estanoles (principalmente encontrados en frutas, verduras, nueces, semillas, cereales, legumbres y aceites vegetales, como la soja), ingesta de alimentos ricos en fibras solubles, reducción de peso y aumento de la actividad física. Si esta estrategia no fuera suficiente, habría que recurrir a tratamiento farmacológi-co con estatinas, que consiguen una re-ducción del 18%-55% en los niveles de cLDL, u otras opciones farmacéuticas recomendadas por su médico. Cuanto más alto es el riesgo, mayor es el bene-ficio. Una reducción del 10% del coles-terol total en sangre es seguido de una reducción del 25% en la incidencia de EAC después de 5 años y una reducción de 1 mmol/L (40 mg/dL) de cLDL se acompaña de una disminución del 20% de eventos coronarios19.
Triglicéridos: son ésteres de glicerol y Triglicéridos: son ésteres de glicerol y Triglicéridos:ácidos grasos que provienen de la dieta o son sintetizados, principalmente, en el hígado. Los triglicéridos se transpor-el hígado. Los triglicéridos se transpor-el hígado. Los triglicéridos se transportan por el plasma en las lipoproteínas y son utilizados por el tejido adiposo,
Riesgo cerebrovascular

HepatoSar® es el primer nutracéuttiicococompmpueueststoo por un 57% de una esttructtuurra lilipopoprprototeieicca natural de origen mmarinno((SS. pilcharrdus) y un extracto veggettal dee Cynaarara sccolymus L. estandarizaadoo al 55%%,quuee apportta propiedades muy bbeneeficioosas s soobrbrbbrbbrrb ee la ffunción hepatobiliary lalaaaa ddddddigii esstión de las grasas.
Favorece la función Hepatobiliar y la Digestión de las Grasas
40
Nuevas Estrategias para el Diagnóstico Bioquímico del Riesgo Vascular
Lola [email protected]
Conclusiones
La prevención de la enfermedad vascular se ha convertido en uno de los objetivos principales en la clínica diaria. La utilización de los factores de riesgo clásicos
y las escalas para la estratificación del riesgo vascular de uso habitual deja un vacío en el diagnóstico de la enfermedad vascular. Con el fin de realizar una
detección del riesgo vascular más precisa se han estudiado nuevos marcadores bioquímicos, íntimamente ligados al riesgo aterosclerótico y vascular, a los que
se les ha denominado “emergentes”. A pesar de que existe una gran controversia en los estudios publicados, el uso de los marcadores de riesgo emergentes
descritos constituye, según recomendaciones internacionales18, una herramienta útil para el ajuste del riesgo vascular y su monitorización, sobre todo en
situaciones de riesgo límite o de dudosa necesidad de una terapia hipolipemiante. El riesgo vascular debe ser evaluado inicialmente mediante los llamados
factores de riesgo mayores y es decisión del médico modificar este riesgo según la información adicional obtenida de los factores emergentes y la historia del
paciente. La inclusión de la aterosclerosis subclínica como factor de riesgo en la estratificación del riesgo vascular, es aconsejable en personas menores de
40 años o mayores de 65 años, dónde la capacidad predictiva de las escalas de uso habitual disminuye. El mayor conocimiento de los factores de riesgo de la
enfermedad vascular y de la capacidad de actuación sobre ellos es, sin duda, la mejor arma para su prevención y para combatir la alta morbilidad y mortalidad
derivada de estas enfermedades. Nuevos estudios epidemiológicos a gran escala son necesarios para valorar la inclusión de éstos u otros factores noveles en
las fórmulas de cálculo habituales del riesgo vascular.
1. de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disor-der: the role of constitutive nitric oxide. Brain Res Rev 2000; 34:119-36.
2. Sociedad Española de Arteriosclerosis (SEA). Las enfermedades cardiovasculares y sus factores de riesgo en España: Hechos y cifras. Informe SEA 2003.
3. The European Society of Cardiology. European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Fourth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Pre-vention in Clinical Practice (Constituted by representatives of nine societies and by invited experts). Eur Heart J 2007; 28:2375-414.
4. Comité Español Interdisciplinario para la Prevención Cardiovascular (CEIPC). Guía Euro-pea de Prevención Cardiovascular en la Práctica Clínica. Adaptación Española del CEIPC 2008. Ministerio de Sanidad y Consumo.
5. Conroy RM, Pyörälä K, Fitzgerald AP et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J 2003; 24:987-1003.
6. Anderson KM, Wilson PWF, Odell PM, Kannel WB. Un update coronary risk profile. A statement for health professionals. Circulation 1991; 83:356-62.
7. Pearson TA, Mensah GA, Alexander RW et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the centers for disease control and prevention and the American Heart Association. Circulation 2003; 107:499-511.
8. van de Kommer TN, Dik MG, Comijs HC et al. Homocysteine and inflammation: Predictors of cognitive decline in older persons? Neurobiol Aging 2010; 31:1700-9.
9. Lentz SR, Haynes WG. Homocysteine: is it a clinically important cardiovascular risk fac-tor? Cleveland Clinic Journal of Medicine 2004; 71:729-34.
10. Ariyo AA, Thach C, Tracy R. Lp(a) Lipoprotein, vascular Disease, and mortality in the elderly. N Engl J Med 2003; 349:2108-15.
11. Bostom AG, Cupples LA, Jenner JL et al. Elevated plasma lipoprotein(a) and coronary heart disease in men aged 55 years and younger. A Prospective Study. JAMA 1996; 276:544-8.
12. Kamstrup PR, Benn M, Tybjærg-Hansen A et al. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population. The Copenhagen city heart study. Circula-tion 2008; 117:176-84.
13. Florvall G, Basu S, Larsson A. Apolipoprotein A1 is a stronger prognostic marker than are HDL and LDL cholesterol for cardiovascular disease and mortality in elderly men. J Gerontol A Biol Sci Med Sci 2006; 61:1262-6.
14. Brunzell JD, Davidson M, Furberg CD et al. Lipoprotein management in patients with cardio-metabolic risk: Consensus conference report from the American Diabetes Association and the American College of Cardiology Foundation. J Am Coll Cardiol 2008; 51:1512-24.
15. Riddell DR, Owen JS. Inhibition of ADP-induced platelet aggregation by apoE is not media-ted by membrane cholesterol depletion. Thromb Res 1996; 81:597-606.
16. Raffai RL, Loeb SM, Weisgraber KH. Apolipoprotein E promotes the regression of atheros-clerosis independently of lowering plasma cholesterol levels. Arterioscler Thromb Vasc Biol 2005; 25:436-41.
17. Cohn JS, Tremblay M, Batal R et al. Effect of atorvastatin on plasma apoE metabolism in patients with combined hyperlipidemia. J Lipid Res 2002; 43:1464-71.
18. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults. JAMA 2001; 285:2486-97.
19. Baigent C, Keech A, Kearney PM et al. Efficacy and safety of cholesterol lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267-78.
20. Walldius G, Aastveit AH, Jungner I. Stroke mortality and the apoB/apoA-I ratio: results of the AMORIS prospective study. J Intern Med 2006; 259:259-66.
Referencias Bibliográficas:

HepatoSar® es el primer nutracéuttiicococompmpueueststoo por un 57% de una esttructtuurra lilipopoprprototeieicca natural de origen mmarinno((SS. pilcharrdus) y un extracto veggettal dee Cynaarara sccolymus L. estandarizaadoo al 55%%,quuee apportta propiedades muy bbeneeficioosas s soobrbrbbrbbrrb ee la ffunción hepatobiliary lalaaaa ddddddigii esstión de las grasas.
Favorece la función Hepatobiliar y la Digestión de las Grasas
Lola [email protected]

42
Introduccióna depresión y sus diferentes manifestaciones, en forma de quiebra del equilibrio emocional, repre-sentan trastornos psiquiátricos de alta prevalencia en la población Occidental. Decía Hagop S. Akis-kal que los trastornos del humor y las emociones son las enfermedades que más han afectado a la especie humana durante los últimos 2500 años. Hace más de una década, la Organización Mun-dial de la Salud (OMS) llegó a catalogar a la de-presión como el cuarto problema más importante de salud en el mundo. Los trastornos depresivos afectan a un 20% de las mujeres y a un 12% de
los hombres y son la causa del 50-70% de los sui-cidios. Desde 1960, Gerald Klerman preconizó un incremento de la depresión en fases tempranas de la vida, comprometiendo a niños y adolescentes. Junto a su enorme poder discapacitante, los episo-dios depresivos recurrentes, su cronificación y un alto número de fracasos terapéuticos, convierten a la depresión en un síndrome que merece un re-planteamiento global en cuanto a sus bases etiopa-planteamiento global en cuanto a sus bases etiopa-planteamiento global en cuanto a sus bases etiopatogénicas, sus factores de riesgo, su componente genético y su abordaje terapéutico en las socieda-genético y su abordaje terapéutico en las socieda-genético y su abordaje terapéutico en las sociedades desarrolladas.
L
Ramón Cacabelos, Rocío Martínez-Bouza, Iván Tellado, Margarita Alcaraz, Laura Nebril, Ángela Casas, Yolanda González, Lucía López, Verónica Couceiro, Carmen Fraile, Susana Rodríguez, Lola Corzo, Juan Carlos Carril, Valter Lombardi, Iván Carrera, Lucía Fernández-Novoa
Centro de Investigación Biomédica EuroEspes, Instituto para Enfermedades del Sistema Nervioso Central y Medicina Genómica, 15165-Bergondo, Coruña;
Cátedra EuroEspes de Biotecnología y Genómica, Universidad Camilo José Cela, Madrid.
DepresivosEstados

43Julio 2011
Depresivos
Desde un punto de vista conceptual y nosológico, existen diversas formas de presentación de los esta-existen diversas formas de presentación de los esta-existen diversas formas de presentación de los estados depresivos, cuya denominación ha ido evolucio-nando a lo largo de los siglos. Sintéticamente, podría decirse que las cuatro manifestaciones más conven-cionales de depresión son (i) la distimia o depresión reactiva, activada generalmente por eventos exter-reactiva, activada generalmente por eventos exter-reactiva, activada generalmente por eventos externos, (ii) la ciclotimia, estado emocional fluctuante entre la depresión y la euforia, reconocida por Emil Kraepelin y Ernst Kretschmer como un factor de predisposición depresiva, (iii) la depresión mayor o depresión endógena, asociada a diversos factores
etiopatogénicos y alto componente genético, y (iv) el trastorno bipolar, antiguamente conocido como psicosis maníaco-depresiva, en sus dos formas (a) trastorno bipolar tipo I, con estados fluctuantes de depresión y manía, y (b) trastorno bipolar tipo II, una condición más tenue en la que alternan estados depresivos con fases hipomaníacas, ambos con claro componente heredofamiliar. La experiencia clínica ha demostrado que la comorbilidad psiquiátrica en los estados depresivos es frecuente, especialmente cuando la depresión se coexpresa con la ansiedad y los trastornos del sueño; y de igual manera se
El abismo de la melancolía, el delirio de la manía, los
ciclos del trastorno bipolar, la herencia y la esperanza
de la farmacogenómica

(1) Neocortex; 2) Gyrus cinguli; (3) Striae longitudinales; (4) Corpus callosum; (5) Fornix; (6) Stria terminalis; (7) Nucleus anterior thalami; (8) Stria medullaris thalami; (9) Thalamus; (10)
Nucleus interstitialis striae terminalis; (11) Lamina medullaris interna; (12) Nucleus habenulae lateralis; (13) Nucleus habenulae medialis; (14) Tractus mamillothalamicus; (15) Lamina
medullaris externa; (16) Corpus geniculatum mediale+laterale; (17) Nucleus septi medialis; (18) Nucleus paraventricularis, pars parvocellularis; (19) Fasciculus telencephalicus medialis;
(20) Bandeletta diagonalis; (21) Bulbus olfactorius; (22) Nucleus oftactorius anterior; (23) Substantia perforata anterior; (24) Nucleus gyri diagonalis; (25) Ansa peduncularis+fibrae
amygdalofugales ventrales; (26) Nucleus centralis amydalae; (27) Nucleus basalis amygdalae; (28) Gyrus dentatus; (29) Cornu Ammonis; (30) Subiculum; (31) Gyrus parahippocampalis;
(32) Tractus habenulointerpeduncularis; (33) Fasciculus longitudinalis dorsalis; (34) Colliculus superior; (35) Colliculus inferior; (36) Griseum centrale mesencephali; (37) Nucleus raphes dorsalis; (38) Nucleus interpeduncularis; (39) Cortex cerebelli; (40) Locus coeruleus, rostral
extension (A6cg); (41) Locus coeruleus (A6); (42) Area subcoerulea (A6sc); (43) Nuclei lemnisci lateralis; (44) Locus coeruleus, caudal extension (A4); (45) Brachium conjunctivum;
(46) Nuclei centrales cerebelli; (47) Nuclei pontis; (48) Formatio reticularis metencephali; (49) Nucleus sensorius principalis nervi trigemini; (50) Nucleus cochlearis ventralis; (51)
Nucleus cochlearis dorsalis; (52) Formatio reticularis myelencephali; (53) Nucleus solitarius; (54) Nucleus dorsalis nervi vagi; (55) Nucleus spinalis nervi trigemini; (56) Cornu posterius
(Laminae IV, V, VI); (57) Cornu anterius(Adaptado de Rudolf Nieuwenhuys. Chemoarchitecture of the brain. Springer-Verlag, Berlín,
1985)
Fig. 1. Sistema noradrenérgico cerebral. El complejo del locus cerúleo
44
Estados Depresivos
ha podido comprobar que determinados factores ha podido comprobar que determinados factores metabólicos, cerebrovasculares y cardiovasculares se pueden eregir en factores de riesgo para desarrollar una depresión psicótica tardía (melancolía involu-una depresión psicótica tardía (melancolía involu-una depresión psicótica tardía (melancolía involutiva). Especial atención merece la depresión en la mujer, particularmente la depresión post-parto, la influencia de los cambios hormonales sobre la esta-influencia de los cambios hormonales sobre la esta-influencia de los cambios hormonales sobre la estabilidad emocional, la depresión premenopáusica y las depresiones estacionales.
Se han postulado diversos modelos teóricos para ex-Se han postulado diversos modelos teóricos para ex-Se han postulado diversos modelos teóricos para explicar las causas y patogenia de la depresión, hasta que se llegó al convencimiento de que las alteracio-nes emocionales tenían un sustrato neuroquímico caracterizado por disfunciones bioquímicas en los mecanismos de neurotransmisión mediados por
aminas biógenas, especialmente noradrenalina y serotonina, en los circuitos córtico-hipocámpicos y el sistema límbico, que regulan las emociones. Es-el sistema límbico, que regulan las emociones. Es-el sistema límbico, que regulan las emociones. Estudios convencionales de genética (genética pobla-tudios convencionales de genética (genética pobla-tudios convencionales de genética (genética poblacional, estudios gemelares, estudios de adopción) y la observación clínica clásica preconizaron que la depresión podría tener un fundamento genético. Con el desarrollo de diversas técnicas de biología y genética molecular, y la realización de múltiples estudios de ligamiento y asociación, se pudo com-estudios de ligamiento y asociación, se pudo com-estudios de ligamiento y asociación, se pudo comprobar que -igual que en la mayoría de las enfer-probar que -igual que en la mayoría de las enfer-probar que -igual que en la mayoría de las enfermedades complejas del sistema nervioso central- la depresión se ajustaba a un perfil de enfermedad po-ligénica y multifactorial en el que múltiples defectos en el genoma humano, en interacción con factores ambientales y fenómenos epigenéticos, podrían dar cuerpo de doctrina a un concepto etiopatogénico coherente. El desarrollo de fármacos para regular la neurotransmisión serotonérgica y noradrenérgica representó un claro avance en el abordaje terapéu-representó un claro avance en el abordaje terapéu-representó un claro avance en el abordaje terapéutico de la depresión, hasta llegar a convertirse en el trastorno psiquiátrico mayor con mejor respuesta al tratamiento farmacológico, junto con el tratamiento del insomnio y la ansiedad con benzodiacepinas. Sin embargo, el tratamiento crónico con antidepresivos demostró un limitado nivel de eficacia en el tiempo, con ciclajes a la hipomanía, con incremento de la ideación suicida, y con las reacciones adversas pro-pias del perfil farmacológico de cada categoría de antidepresivos. Con el advenimiento de la farmaco-genética en los últimos 20 años se ha podido com-genética en los últimos 20 años se ha podido com-genética en los últimos 20 años se ha podido comprobar que la inmensa mayoría de los antidepresivos se metabolizan a través de las enzimas del citocromo P-450, y que defectos genéticos en los genes de la familia CYP, que codifican a las enzimas hepáticas responsables de las reacciones metabólicas de fase I, daban lugar a perfiles fenotípicos de respuesta far-I, daban lugar a perfiles fenotípicos de respuesta far-I, daban lugar a perfiles fenotípicos de respuesta farmacológica desigual. Muchos casos de depresión re-sistente y un amplio número de reacciones adversas serias tienen que ver con el perfil farmacogenético de los pacientes, en los que los fármacos convencio-nales o no ejercen el efecto terapéutico deseado o multiplican su toxicidad, poniendo en peligro la vida del enfermo. Hoy día, la personalización del tratamiento antidepresivo, mediante el conocimien-to de las características genómicas de cada paciente, permite optimizar los recursos terapéuticos disponi-permite optimizar los recursos terapéuticos disponi-permite optimizar los recursos terapéuticos disponibles, mejorando el nivel de eficacia de los fármacos, reduciendo efectos secundarios y racionalizando el coste que supone un tratamiento crónico con anti-coste que supone un tratamiento crónico con anti-coste que supone un tratamiento crónico con antidepresivos.
Lecciones de la historiaDesde los tiempos de la hegemonía de los imperios griego y romano se conocen los términos de “melan-colía” y “manía”. Hipócrates (460-357 a.C.) describía la melancolía (bilis negra) como un estado de aver-la melancolía (bilis negra) como un estado de aver-la melancolía (bilis negra) como un estado de aversión alimentaria, inquietud, labilidad emocional, nerviosismo, alteración del sueño e irritabilidad. Los griegos, que importaron el término de los egipcios,

Los trastornos depresivos afectan a un 20% de las mujeres y a un 12% de los hombres y son la
causa del 50-70% de los suicidios
Julio 2011 45
ciencia
fueron los primeros en desarrollar una hipótesis bio-lógica de la melancolía, asociándola a la influencia del planeta Saturno. Aristóteles (384-322 a.C.) atri-del planeta Saturno. Aristóteles (384-322 a.C.) atri-del planeta Saturno. Aristóteles (384-322 a.C.) atribuía cualidades creativas al temperamento melan-cólico, y lo diferenciaba del colérico y el flemático, que consideraba temperamentos menos deseables. Galeno (131-201 d.C.) amplió el concepto griego de la melancolía, y Aureliano, citando a Sorano de Éfeso, estableció el vínculo del suicidio y los estados delusionales o ideas delirantes a los estados depresi-delusionales o ideas delirantes a los estados depresi-delusionales o ideas delirantes a los estados depresivos. Estos mismos autores manejaron el concepto de manía como un estado de excitabilidad o psicosis ex-manía como un estado de excitabilidad o psicosis ex-manía como un estado de excitabilidad o psicosis excitada, y Areteo de Capadocia, en el siglo II, estable-ció la relación entre melancolía y manía. Las ideas de griegos y romanos pasaron el umbral de la edad media gracias a las traducciones y reinterpretaciones de los árabes, especialmente Ishq Ibn Imran y Avice-na, que introdujeron la primera sospecha del com-na, que introdujeron la primera sospecha del com-na, que introdujeron la primera sospecha del componente genético de la melancolía o el daño prena-ponente genético de la melancolía o el daño prena-ponente genético de la melancolía o el daño prenatal debido a un defecto en el esperma paterno. En la era moderna, la obra de Robert Burton, Anatomy of Melancholy, publicada en 1621, dominó la esfera Occidental, unido al protagonismo que adquirieron otros alienistas británicos y, sobretodo, franceses. Según Germán Berrios, las nociones actuales sobre la depresión se consolidaron en la segunda mitad del siglo XIX. En 1890, Esquirol introdujo el con-cepto de Lypemania para enfatizar la naturaleza depresiva de la melancolía. Y todas las ideas del pa-depresiva de la melancolía. Y todas las ideas del pa-depresiva de la melancolía. Y todas las ideas del pasado convergieron en el concepto de enfermedad maníaco-depresiva acuñado por Kraepelin en 1921. El Manual de Régis de 1885 describía la depresión como un estado opuesto a la excitación o una pará-como un estado opuesto a la excitación o una pará-como un estado opuesto a la excitación o una parálisis mental. La autoridad de William Gull en 1894, Savage en 1898, Jastrow y Meyer en 1901, consagra-Savage en 1898, Jastrow y Meyer en 1901, consagra-Savage en 1898, Jastrow y Meyer en 1901, consagraron el término de depresión en la literatura médica, desplazando definitivamente al vocablo melancolía, depuesto a un nivel más poético o literario, pero des-depuesto a un nivel más poético o literario, pero des-depuesto a un nivel más poético o literario, pero desterrado de la terminología médica. Hubo intentos de resurrección con variantes como melancolía ató-nita, agitada, estuporosa, hipocondríaca, puerperal, climatérica, senil y otras, que no cuajaron. Cotard en 1882 prefería hablar de “delusión nihilista”, que tampoco cautivó a sus contemporáneos, hasta que la autoridad de Kraepelin impuso el término de “esta-autoridad de Kraepelin impuso el término de “esta-autoridad de Kraepelin impuso el término de “estados depresivos” en la octava edición de su Textbook en 1921, reconocido como la biblia de la psiquiatría moderna precontemporánea. Para la definición de trastorno bipolar o la enfermedad maniaco-depre-siva también hubo un largo debate histórico, que quizá empezó con Jules Falret (1794-1870) en la Sal-quizá empezó con Jules Falret (1794-1870) en la Sal-quizá empezó con Jules Falret (1794-1870) en la Salpêtrière, cuando describió la forme circulaire de maladie mentale; y siguió Baillarger (1809-1890) en 1854 con mentale; y siguió Baillarger (1809-1890) en 1854 con mentalesu folie à double forme, Billod en 1856 (folie à double forme, Billod en 1856 (folie à double forme à double phase), à double phase), à double phaseDelaye y Legrand du Saulle (folie alterneDelaye y Legrand du Saulle (folie alterneDelaye y Legrand du Saulle ( ), Kahlbaum folie alterne), Kahlbaum folie alterne(1828-1899) y Ritti en 1883. Krapelin (1856-1926) acabó con la polémica estableciendo el concepto de enfermedad maniaco-depresiva en su obra magna de 1921, y reservó el término de melancolía involu-de 1921, y reservó el término de melancolía involu-de 1921, y reservó el término de melancolía involutiva para los casos de depresión senil, introduciendo el término de demencia presenil basado en los estu-el término de demencia presenil basado en los estu-el término de demencia presenil basado en los estudios de su discípulo Alois Alzheimer. Kraepelin dejó
abierta la puerta nosológica a la depresión psicogé-nica, de carácter exógeno, para diferenciarla clara-nica, de carácter exógeno, para diferenciarla clara-nica, de carácter exógeno, para diferenciarla claramente de los trastornos depresivos endógenos, de carácter preponderantemente hereditario. Durante el siglo XIX, los autores ingleses más representativos en el campo de la depresión fueron Prichard, con su definición de manía en 1835; Bucknill y Tuke, que hicieron una clasificación primitiva de las enfer-que hicieron una clasificación primitiva de las enfer-que hicieron una clasificación primitiva de las enfermedades mentales en 1858; Maudsley, en 1895, el
abierta la puerta nosológica a la depresión psicogé
(1) Thalamus, periventricular region; (2) Nucleus interstitialis striae terminalis; (3) Nucleus septi lateralis; (4) Nucleus paraventricularis, pars magnocellularis; (5) Nucleus
paraventricularis, pars parvocellularis; (6) Area lateralis hypothalami; (7) Fasciculus telencephalicus medialis; (8) Fasciculus longitudinalis dorsalis; (9) Nucleus gyri diagonalis;
(10) Nucleus anterior hypothalami; (11) Nucleus dorsomedialis; (12) Area caudalis hypothalami; (13) Nucleus praeopticus medialis; (14) Nucleus supraopticus; (15) Nucleus
infundibularis; (16) Corpus amygdaloideum; (17) Eminentia mediana; (18) Formatio reticularis mesencephali; (19) Griseum centrale mesencephali; (20) Nucleus centralis
superior; (21) Locus coeruleus; (22) Cell group A7; (23) Formatio reticularis metencephali; (24) Nuclei parabrachiales; (25) Nucleus motorius nervi trigemini; (26) Nuclei pontis; (27)
Nucleus raphes magnus; (28) Cell group A5; (29) Nucleus nervi facialis; (30) Formatio reticularis myelencephali; (31) Cell group A1; (32) Cell group A2; (33) Nucleus dorsalis nervi
vagi; (34) Nucleus solitarius; (35) Substantia grisea centralis; (36) Substantia gelatinosa; (37) Nucleus intermediolateralis
(Adaptado de Rudolf Nieuwenhuys. Chemoarchitecture of the brain. Springer-Verlag, Berlín, 1985)
Fig. 2. Sistema noradrenérgico cerebral. Grupos neuronales troncoencefálicos con proyección a regiones talámica e hipotalámica

Fig. 3. Sistema serotonérgico cerebral
(1) Neocortex; (2) Gyrus cinguli; (3) Striae longitudinales +cingulum; (4) Nucleus caudatus; (5) Copus callosum; (6) Putamen; (7) Fornix; (8) Stria terminalis; (9) Thalamus; (10) Stria medullaris; (11) Nucleus habenulae medialis; (12) Nucleus septi medialis+ lateralis; (13) Nucleus dorsomedialis; (14) Area lateralis hypothalami; (15) Area tegmentalis ventralis;
(16) Nucleus accumbens; (17) Nucleus praeopticus medialis+lateralis; (18) Nucleus ventromedialis; (19) Fasciculus telencephalicus medialis; (20) Bulbus olfactorius; (21) Nucleus
olfactorius anterior; (22) Nucleus gyri diagonalis; (23) Nucleus suprachiasmaticus; (24) Ansa peduncularis+fibrae amygdalofugales ventrales; (25) Nucleus anterior hypothalami;
(26) Nucleus infundibularis; (27) Corpus mamillare; (28) Corpus amygdaloideum; (29) Gyrus parahippocampalis; (30) Gyrus dentatus; (31) Gyrus Ammonis; (32) Subiculum; (33)
Substantia nigra; (34) Griseum centrale mesencephali; (35) Nucleus raphes dorsalis (B7); (36) Nucleus tegmentalis dorsalis; (37) Colliculus superior; (38) Colliculus inferior; (39) Fasciculus
longitudinalis dorsalis; (40) Nucleus interpeduncularis; (41) Nucleus centralis superior (B6+B8); (42) Plexus supraependymalis; (43) Locus coeruleus; (44) Nucleus raphes pontis (B5); (45) Nuclei parabrachiales; (46) Formatio reticularis metencephali; (47) Ventriculus
quartus; (48) Cortex cerebelli; (49) Nuclei centrales cerebelli; (50) Nucleus raphes magnus (B3); (51) Nucleus raphes obscurus (B2); (52) Formatio reticularis myenlencephali; (53)
Nucleus raphes pallidus (B1); (54) Nucleus solitarius; (55) Nucleus dorsalis nervi vagi; (56) Nucleus spinalis nervi trigemini; (57) Substantia gelatinosa; (58) Cornu anterius; (58) Nucleus
intermediolateralis(Adaptado de Rudolf Nieuwenhuys. Chemoarchitecture of the brain. Springer-Verlag, Berlín,
1985)
46
Estados Depresivos
College of Physicians of London”. Otras importantes College of Physicians of London”. Otras importantes College of Physicians of London”contribuciones al entendimiento de la depresión y la enfermedad maniaco-depresiva vinieron ya en el siglo XX de la mano de médicos relevantes, como Edward Mapother en 1926, Robert Dick Gillespie (1897-1945), McCurdy (1886-1947), Karl Leonhard en 1957, Jules Angst en 1966, Carlo Perris en 1966, George Winokur, Paula Clayton, y Theodore Reich en 1969, entre otros. Repercusión importante en la psiquiatría americana tuvo la figura de Adolf Meyer (1866-1950), desde su cátedra de la Universidad Johns Hopkins, que como neo-kraepeliniano san-tificó el término de depresión en la psiquiatría mundial, hasta que se redefinió la taxonomía de los trastornos depresivos, tal como hoy los entende-mos, en el DSM-IV (Diagnostic and Statistical Manual of Mental Disorders) de la APA (of Mental Disorders) de la APA (of Mental Disorders) American Psychiatric Association), y el ICD-10 (International Statistical Clas-International Statistical Clas-International Statistical Classification of Diseases and Related Health Problems) de la sification of Diseases and Related Health Problems) de la sification of Diseases and Related Health Problems)Organización Mundial de la Salud en sus últimas ediciones revisadas.
FenotipoEl fenotipo es la expresión clínica de la enferme-dad. Los trastornos del humor o los trastornos emocionales suelen clasificarse en 4 categorías: (i) trastornos depresivos (depresión unipolar), (ii) trastornos bipolares, (iii) trastornos depresivos aso-ciados a una situación o condición médica, y (iv) trastornos depresivos causados por el uso de fár-trastornos depresivos causados por el uso de fár-trastornos depresivos causados por el uso de fármacos u otras sustancias o tóxicos. Los trastornos depresivos (depresión mayor, distimia, trastorno depresivo inespecífico) se distinguen de los trastor-depresivo inespecífico) se distinguen de los trastor-depresivo inespecífico) se distinguen de los trastornos bipolares en que no existe historia de episodios maníacos o hipomaníacos; en cambio, los trastor-maníacos o hipomaníacos; en cambio, los trastor-maníacos o hipomaníacos; en cambio, los trastornos bipolares (tipo I, tipo II, ciclotimia, trastorno bipolar inespecífico), casi siempre presentan episo-dios previos de manía, hipomanía, depresión ma-dios previos de manía, hipomanía, depresión ma-dios previos de manía, hipomanía, depresión mayor o situaciones mixtas, donde depresión y manía se entremezclan. El trastorno depresivo mayor se caracteriza por uno o más episodios depresivos de varias semanas de duración. El trastorno distímico se prolonga en el tiempo por periodos de uno o más años, con humor deprimido, sin características clínicas de depresión mayor. La categoría del tras-torno depresivo inespecífico se reserva a casos que no cumplen criterios de depresión mayor, trastor-no cumplen criterios de depresión mayor, trastor-no cumplen criterios de depresión mayor, trastorno distímico, trastorno de ajuste con humor depri-mido o trastorno depresivo con ansiedad asociada. El trastorno bipolar tipo I se presenta con uno o más episodios de manía y depresión intercalados. El trastorno bipolar tipo II cursa con episodios de depresión mayor y al menos un episodio hipoma-depresión mayor y al menos un episodio hipoma-depresión mayor y al menos un episodio hipomaníaco. El trastorno ciclotímico se presenta con pe-riodos múltiples de hipomanía (que no cumplen criterios de manía) y episodios de depresión (que no cumplen criterios de depresión mayor). Se ha-no cumplen criterios de depresión mayor). Se ha-no cumplen criterios de depresión mayor). Se habla de trastorno bipolar inespecífico en aquellos casos en los que se alternan episodios de depresión y manía que no cumplen criterios de trastorno bi-
padre de la tradición psiquiátrica británica, a quien la historia dedicó el nombre de un famoso hospital; y George Savage y Percy Smith, que en 1906 dirigie-ron el grupo de médicos que estableció la “Nomen-clature of Diseases” del Joint Committee of the Royal
El tratamiento crónico con antidepresivos demostró un limitado nivel de eficacia en el tiempo, con ciclajes a la hipomanía, con incremento de la ideación suicida, y con las reacciones adversas propias del perfil farmacológico de cada categoría de antidepresivos

Julio 2011 47
ciencia
polar I o II. El trastorno depresivo asociado a una condición médica es aquel causado por una pato-logía concreta; y el trastorno depresivo causado por una sustancia específica viene determinado por el tipo de tóxico responsable.
Los episodios de humor se clasifican en depresivo mayor, maníaco, mixto e hipomaníaco. Los trastor-mayor, maníaco, mixto e hipomaníaco. Los trastor-mayor, maníaco, mixto e hipomaníaco. Los trastornos depresivos se subclasifican en depresivo mayor, distímico e inespecífico. Los trastornos bipolares puede subtipificarse en tipo I, tipo II, ciclotimia, y trastorno bipolar inespecífico. Los trastornos de-presivos también pueden clasificarse por la causa (médica o tóxica), por el tipo de episodio (leve, moderado, severo, con o sin características psicóti-cas, con remisión parcial o total, con características catatónicas, melancólicas, atípicas), por la estacio-nalidad (depresión estacional, depresión puerpe-ral), por la época de la vida (infantil, juvenil, senil), o por las características del ciclaje (de ciclaje rápido o ciclaje lento).
EpidemiologíaLa prevalencia de los estados depresivos varía en función del fenotipo clínico de las 4 formas prototí-función del fenotipo clínico de las 4 formas prototí-función del fenotipo clínico de las 4 formas prototípicas de depresión. La prevalencia del trastorno de-presivo mayor es del 10-25% en mujeres y del 5-12% en hombres. El trastorno distímico aparece en un 25-30% de las personas. La frecuencia del trastorno bipolar tipo I es del 0.4-1.6%; la del trastorno bipo-lar tipo II es inferior al 1%; y el trastorno ciclotími-co presenta una prevalencia del 0.4-1.0%. Existen claras diferencias en función de la edad, el sexo, el status marital, y la distribución geográfica. En estu-status marital, y la distribución geográfica. En estu-status marital, y la distribución geográfica. En estudios internacionales se pueden ver diferencias en la prevalencia de depresión llamativas, como es el caso de Beirut, donde la frecuencia del trastorno depresivo mayor es del 1.5-19%, o el caso de Nue-va Zelanda, con una prevalencia del 0.8-5.8%. En cuanto al género, las mujeres tienen mayor riesgo que los hombres. La raza no parece modificar la prevalencia de depresión en diferentes etnias. La depresión suele ser más frecuente en gente joven, aunque existe una depresión presenil, una depre-sión premenopáusica y una depresión senil. En ocasiones, a las depresiones de la senectud, con deterioro cognitivo, se las reconoce como pseudo-demencia depresiva, que en un 20% de los casos evolucionan hacia una demencia. El status marital es un elemento de riesgo, según el cual separados y divorciados tienden a presentar más casos de de-presión. Las experiencias traumáticas en la infancia también parecen condicionar, en cierto modo, la aparición de depresión en la juventud o en la ma-aparición de depresión en la juventud o en la ma-aparición de depresión en la juventud o en la madurez. El stress crónico es otro factor de riesgo im-portante. El medio urbano ofrece condiciones que favorecen la depresión, en contra del medio rural, donde la depresión es menos frecuente. De todos los factores epidemiológicos de riesgo, sin duda el más poderoso es la historia familiar de depresión.
Modelos EtiopatogénicosLos modelos con los que se ha pretendido explicar el síndrome depresivo a lo largo de los últimos 100 años pueden clasificarse en 5 grandes categorías: (i) modelos contemporáneos (1900-1990), que re-presentan una mezcla de teorías clínicas, heurísti-cas, instrumentales y biológicas primitivas; (ii) las hipótesis psicodinámicas derivadas del universo psicoanalítico, cuando la psiquiatría estaba domi-nada por los discípulos de Freud; (iii) los modelos neurobiológicos capaces de documentar la disfun-ción bioquímica existente en el cerebro de los pa-ción bioquímica existente en el cerebro de los pa-ción bioquímica existente en el cerebro de los pacientes depresivos; (iv) los modelos funcionales ba-cientes depresivos; (iv) los modelos funcionales ba-cientes depresivos; (iv) los modelos funcionales basados en la electrofisiología y la imagen funcional; y (v) la información genética que acredita el sustrato de vulnerabilidad genómica por el cual una perso-na es susceptible de padecer una depresión o un trastorno bipolar.
Fig. 4. Estructuras encefálicas potencialmente afectadas en los estados depresivos
Adaptado de Martinowich, Schloesser y Manji HK, 2009
Adaptado de Martinowich, Schloesser y Manji HK, 2009
Tálamo
Giro cinguladoCíngulo
Al hipocampo
Núcleos profundos del cerebeloNúcleos del rafe rostral
Complejo ponto-mesencefálico-tegmental
Sistema dopaminérgico nigroestrial
Sistema dopaminérgico mesolímbico y mesocorticalÁrea ventral tegmental
Sistema dopaminérgico tubero-infundibular
Sistema neuronal noradregérnico tegmento-lateralLocus coeruleus
Sustancia negra
Núcleos del rafe caudal
Corteza del cerebelo
Estriado
Corteza frontal
Cortezas olfatoria y entorrinal
Hipotálamo
AmígdalaHipocampo
Médula espinal
Núcleo accumbens(estriatum ventral)
Neocórtex
Adaptado de Martinowich, Schloesser y Manji HK, 2009
Fig. 5. Circuitos cerebrales que participan en la regulación de las emociones y que pueden alterarse en la depresión, la manía y el trastorno bipolar
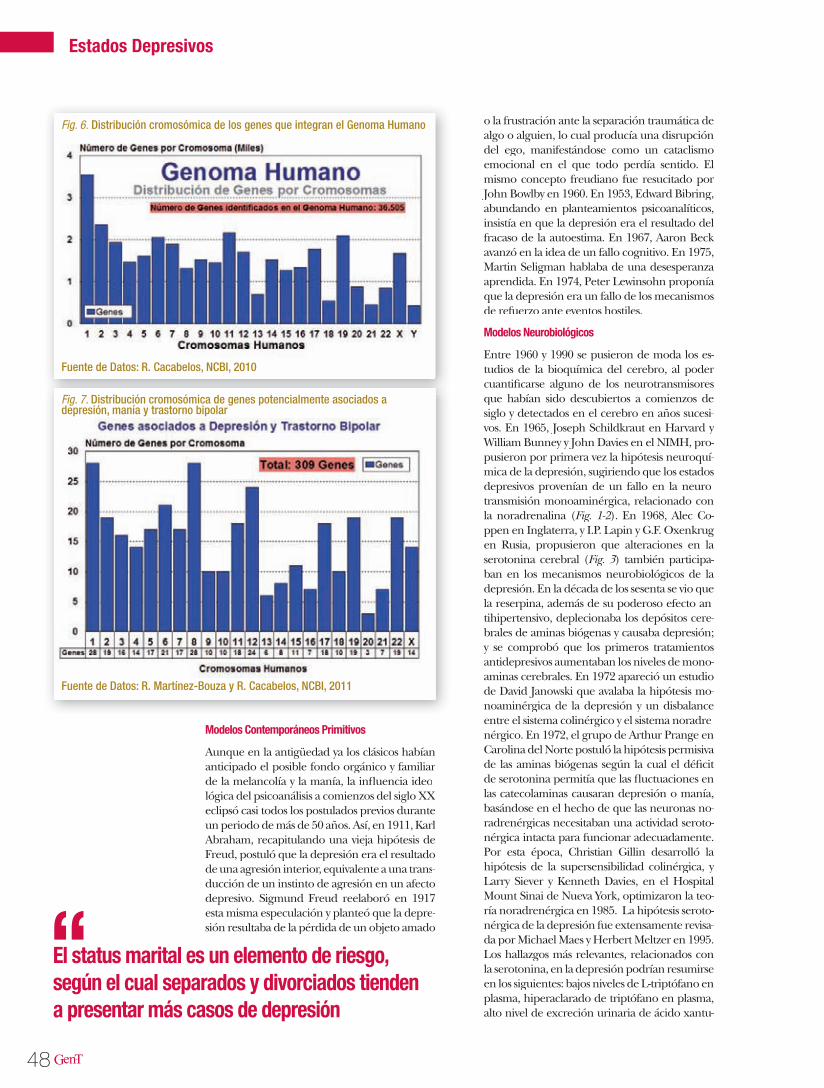
48
Estados Depresivos
Modelos Contemporáneos Primitivos
Aunque en la antigüedad ya los clásicos habían anticipado el posible fondo orgánico y familiar de la melancolía y la manía, la influencia ideo-lógica del psicoanálisis a comienzos del siglo XX eclipsó casi todos los postulados previos durante un periodo de más de 50 años. Así, en 1911, Karl Abraham, recapitulando una vieja hipótesis de Freud, postuló que la depresión era el resultado de una agresión interior, equivalente a una trans-de una agresión interior, equivalente a una trans-de una agresión interior, equivalente a una transducción de un instinto de agresión en un afecto depresivo. Sigmund Freud reelaboró en 1917 esta misma especulación y planteó que la depre-sión resultaba de la pérdida de un objeto amado
o la frustración ante la separación traumática de algo o alguien, lo cual producía una disrupción del ego, manifestándose como un cataclismo emocional en el que todo perdía sentido. El mismo concepto freudiano fue resucitado por John Bowlby en 1960. En 1953, Edward Bibring, abundando en planteamientos psicoanalíticos, insistía en que la depresión era el resultado del fracaso de la autoestima. En 1967, Aaron Beck avanzó en la idea de un fallo cognitivo. En 1975, Martin Seligman hablaba de una desesperanza aprendida. En 1974, Peter Lewinsohn proponía que la depresión era un fallo de los mecanismos de refuerzo ante eventos hostiles.
Modelos Neurobiológicos
Entre 1960 y 1990 se pusieron de moda los es-Entre 1960 y 1990 se pusieron de moda los es-Entre 1960 y 1990 se pusieron de moda los estudios de la bioquímica del cerebro, al poder cuantificarse alguno de los neurotransmisores que habían sido descubiertos a comienzos de siglo y detectados en el cerebro en años sucesi-siglo y detectados en el cerebro en años sucesi-siglo y detectados en el cerebro en años sucesivos. En 1965, Joseph Schildkraut en Harvard y William Bunney y John Davies en el NIMH, pro-pusieron por primera vez la hipótesis neuroquí-pusieron por primera vez la hipótesis neuroquí-pusieron por primera vez la hipótesis neuroquímica de la depresión, sugiriendo que los estados depresivos provenían de un fallo en la neuro-transmisión monoaminérgica, relacionado con la noradrenalina (Fig. 1-2). En 1968, Alec CoFig. 1-2). En 1968, Alec CoFig. 1-2 -ppen en Inglaterra, y I.P. Lapin y G.F. Oxenkrug en Rusia, propusieron que alteraciones en la serotonina cerebral (Fig. 3) también participaFig. 3) también participaFig. 3 -) también participa-) también participaban en los mecanismos neurobiológicos de la depresión. En la década de los sesenta se vio que la reserpina, además de su poderoso efecto an-la reserpina, además de su poderoso efecto an-la reserpina, además de su poderoso efecto antihipertensivo, deplecionaba los depósitos cere-brales de aminas biógenas y causaba depresión; y se comprobó que los primeros tratamientos antidepresivos aumentaban los niveles de mono-aminas cerebrales. En 1972 apareció un estudio de David Janowski que avalaba la hipótesis mo-noaminérgica de la depresión y un disbalance entre el sistema colinérgico y el sistema noradre-nérgico. En 1972, el grupo de Arthur Prange en Carolina del Norte postuló la hipótesis permisiva de las aminas biógenas según la cual el déficit de serotonina permitía que las fluctuaciones en las catecolaminas causaran depresión o manía, basándose en el hecho de que las neuronas no-radrenérgicas necesitaban una actividad seroto-nérgica intacta para funcionar adecuadamente. Por esta época, Christian Gillin desarrolló la hipótesis de la supersensibilidad colinérgica, y Larry Siever y Kenneth Davies, en el Hospital Mount Sinai de Nueva York, optimizaron la teo-ría noradrenérgica en 1985. La hipótesis seroto-nérgica de la depresión fue extensamente revisa-nérgica de la depresión fue extensamente revisa-nérgica de la depresión fue extensamente revisada por Michael Maes y Herbert Meltzer en 1995. Los hallazgos más relevantes, relacionados con la serotonina, en la depresión podrían resumirse en los siguientes: bajos niveles de L-triptófano en plasma, hiperaclarado de triptófano en plasma, alto nivel de excreción urinaria de ácido xantu-alto nivel de excreción urinaria de ácido xantu-alto nivel de excreción urinaria de ácido xantu
Fig. 6. Distribución cromosómica de los genes que integran el Genoma Humano
Fig. 7. Distribución cromosómica de genes potencialmente asociados a depresión, manía y trastorno bipolar
Fuente de Datos: R. Cacabelos, NCBI, 2010
Fuente de Datos: R. Martínez-Bouza y R. Cacabelos, NCBI, 2011
El status marital es un elemento de riesgo, según el cual separados y divorciados tienden a presentar más casos de depresión

Julio 2011 49
ciencia
rénico tras carga de triptófano, bajos niveles de triptófano plasmático en relación con antidepre-sivos serotonérgicos, inducción de síntomas de-presivos tras depleción experimental de triptófa-presivos tras depleción experimental de triptófa-presivos tras depleción experimental de triptófano, escasa captación de serotonina en plaquetas, bajo nivel de unión de imipramina y paroxetina a receptores plaquetarios, bajos niveles plas-a receptores plaquetarios, bajos niveles plas-a receptores plaquetarios, bajos niveles plasmáticos de serotonina, bajos niveles cerebrales de precursor (triptófano) y metabolito (ácido 5-hidroxi-indolacético, 5-HIAA) de serotonina (5-hidroxitriptamina), baja concentración de 5-HIAA en líquido cefalorraquídeo, hiperacti-5-HIAA en líquido cefalorraquídeo, hiperacti-5-HIAA en líquido cefalorraquídeo, hiperactividad de receptores 5-HT2 periféricos y centra-vidad de receptores 5-HT2 periféricos y centra-vidad de receptores 5-HT2 periféricos y centrales, baja actividad de receptores 5-HT1A, pobre respuesta de prolactina y cortisol a triptófano y antidepresivos. En los últimos 20 años pocos avances ha habido en torno a la hipótesis mono-aminérgica de la depresión, aunque la evidencia demuestra que el trastorno de las aminas bióge-nas en el cerebro no es necesario ni suficiente para que se manifieste una depresión, a pesar de que todos los antidepresivos se formulan sobre la base de una regulación de noradrenalina, do-pamina, serotonina, acetilcolina, GABA e hista-pamina, serotonina, acetilcolina, GABA e hista-pamina, serotonina, acetilcolina, GABA e histamina. El hecho de que estos neurotransmisores fuesen poderosos reguladores del sistema neu-fuesen poderosos reguladores del sistema neu-fuesen poderosos reguladores del sistema neuroendocrino, tras la caracterización de los fac-roendocrino, tras la caracterización de los fac-roendocrino, tras la caracterización de los factores hipotalámicos por Guillemin y Vale en la década de los ochenta, impulsó la investigación de la función neuroendocrina en la depresión. Prange y su grupo comprobaron alteraciones en el eje hipotálamo-hipofiso-tiroideo en la de-presión; Bernard Carroll, en Michigan, observó una disfunción hipotálamo-hipófiso-adrenal (sistema corticotropinérgico) en pacientes de-presivos y preconizó el test de la dexametasona como ayuda diagnóstica; y Charles Nemeroff,
en Emory, encontró alteraciones en los niveles de CRF (Corticotropin-releasing factor) en el lí-de CRF (Corticotropin-releasing factor) en el lí-de CRF (Corticotropin-releasing factor) en el líquido cefalorraquídeo de pacientes depresivos. Otras disfunciones endocrinas fueron detecta-Otras disfunciones endocrinas fueron detecta-Otras disfunciones endocrinas fueron detectadas en el sistema somatotropinérgico (GRF-GH)(test del GRF y respuesta de GH a clonidina) y en diversos neuropéptidos cerebrales, pero nunca se llegó a definir de forma concluyente si estas alteraciones neuroquímicas eran causa o consecuencia de la depresión. Florian Holsboer realizó diversos estudios neuroendocrinos y
Fig. 8. Distribución y frecuencia de genotipos APOE en trastornos del sistema nervioso central
Fig. 9. Resonancia Magnética Cerebral de una paciente de 39 años con Depresión Mayor Recurrente Familiar de 10 años de evolución
Fig. 10. Resonancia Magnética Cerebral de un paciente de 64 años con Trastorno Bipolar Familiar de más de 30 años de evolución en fase estacionaria semiasintomática durante los últimos 12 años, a tratamiento con dosis bajas de antidepresivos y carbonato de litio
Fuente de Datos: R. Cacabelos, Base de Datos CIBE, 2010
Fuente de Datos: R. Cacabelos, Base de Datos CIBE Fuente de Datos: R. Cacabelos, Base de Datos CIBE

50
Estados Depresivos
revisó la experiencia existente, pero ninguna conclusión fue lo suficientemente concluyente como para establecer que la disfunción neuroen-como para establecer que la disfunción neuroen-como para establecer que la disfunción neuroendocrina pudiera relacionarse con la depresión desde un punto de vista patogénico.
Otra constatación importante en el entorno bio-Otra constatación importante en el entorno bio-Otra constatación importante en el entorno biológico de la depresión es la alteración del sistema inmunológico, caracterizado por un status de inmunodepresión y disregulación de los factores mediadores de la inmunidad y la inflamación. Michael Irwin revisó la psiconeuroinmunología de la depresión y el papel del stress en la inmu-de la depresión y el papel del stress en la inmu-de la depresión y el papel del stress en la inmunidad celular y humoral. Existen predictores de alteraciones inmunes causadas por stress, tales como experiencias vitales tempranas y determi-como experiencias vitales tempranas y determi-como experiencias vitales tempranas y determinadas conductas sociales; se observaron intere-santes relaciones entre la inmunidad, la función endocrina y la depresión; pero nada ha sido con-endocrina y la depresión; pero nada ha sido con-endocrina y la depresión; pero nada ha sido concluyente hasta la fecha.
En los últimos años, la biología molecular está dando un nuevo impulso al conocimiento de los mecanismos etiopatogénicos responsables del fracaso emocional que caracteriza a la depresión y al trastorno bipolar, con nuevos factores neu-y al trastorno bipolar, con nuevos factores neu-y al trastorno bipolar, con nuevos factores neurobiológicos emergentes que, probablemente, en un horizonte de 10 años, nos permitan en-en un horizonte de 10 años, nos permitan en-en un horizonte de 10 años, nos permitan entender con cierta fiabilidad los disturbios neuro-tender con cierta fiabilidad los disturbios neuro-tender con cierta fiabilidad los disturbios neuroquímicos que alteran el equilibro emocional en los estados depresivos. Algunos de estos nuevos factores, que también participan en otros trastor-factores, que también participan en otros trastor-factores, que también participan en otros trastornos neuropsiquiátricos, incluyen los siguientes: melatonina, GABA, glutamato, interleukinas, receptores estrogénicos, chaperonas, sinaptofisi-receptores estrogénicos, chaperonas, sinaptofisi-receptores estrogénicos, chaperonas, sinaptofisinas, ceramida, BDNF, GSK3b, alteraciones en la plasticidad sináptica, defectos en la neurogénesis del hipocampo, y déficit de CREB (Fig. 4-5). Fig. 4-5). Fig. 4-5
Modelos Funcionales
Diversos modelos funcionales evidencian una profunda disrupción de los ritmos biológicos en los trastornos depresivos: alteración en rit-en los trastornos depresivos: alteración en rit-en los trastornos depresivos: alteración en ritmos circadianos, alteración en los biorritmos hormonales, alteración del ciclo sueño-vigilia. Alec Coppen y D.M. Shaw postularon en 1963 la existencia de un trastorno del balance elec-trolítico neuronal. La depleción de los niveles de sodio intraneuronales podría contribuir a alterar la excitabilidad neuronal causando tan-to depresión como manía. Joseph Mendels y Peter Whybrown capitalizaron en 1968 la idea del trastorno electrolítico intraneuronal. Ro-bert Post en 1990 y Frederick Goodwin y Kay Jamison, también en los años 90, enfatizaron en la hipótesis neurofisiológica de la depresión y el papel del stress como agente diatético para disparar mecanismos de disregulación bioquí-disparar mecanismos de disregulación bioquí-disparar mecanismos de disregulación bioquímica centroencefálica en la depresión. Poco se ha avanzado en estos modelos, aunque es evi-dente una severa alteración de la actividad bio-eléctrica cerebral en los pacientes depresivos y maníacos. David Kupfer propuso el test de latencia REM como ayuda diagnóstica, puesto que esta latencia se encuentra acortada en de-presivos; y Daniel Kripke sugirió que la terapia lumínica podría beneficiar a los pacientes con depresión. Los modelos funcionales tienen un vínculo claro con alteraciones en los centros hipotalámicos que regulan los ritmos hormo-nales y los biorritmos circadianos. Esta circuns-tancia también apunta a una disfunción entre hipotálamo, sistema límbico y glándula pineal, con participación de los mecanismos regula-con participación de los mecanismos regula-con participación de los mecanismos regulados por la melatonina. De hecho, la modula-dos por la melatonina. De hecho, la modula-dos por la melatonina. De hecho, la modula
revisó la experiencia existente, pero ninguna revisó la experiencia existente, pero ninguna Modelos Funcionalesrevisó la experiencia existente, pero ninguna
Fig. 11. Resonancia Magnética Cerebral y Cartografía Cerebral en fase sintomática (izquierda) y en fase de remisión (derecha) de una paciente de 40 años con Trastorno Bipolar Familiar de 15 años de evolución. Último episodio: Depresión Puerperal severa
Fig. 12. Resonancia Magnética Cerebral, EEG y Patrón Cartográfico Cerebral en fase sintomática (izquierda) y tras remisión de síntomas (derecha) en una mujer de 67 años con Trastorno Bipolar en Fase Maníaca y Encefalopatía Vascular tipo Binswanger
Fuente de Datos: R. Cacabelos, Base de Datos CIBE Fuente de Datos: R. Cacabelos, Base de Datos CIBE

Julio 2011 51
ciencia
ción de la melatonina con fármacos melato-nérgicos mejora las condiciones clínicas de los pacientes depresivos.
GenéticaDesde la antigüedad se ha visto que la depresión y los trastornos bipolares se acumulaban en determi-los trastornos bipolares se acumulaban en determi-los trastornos bipolares se acumulaban en determinadas familias, poniendo de manifiesto el compo-nente genético de la depresión. En los últimos 30 años se han realizado numerosos estudios de genéti-años se han realizado numerosos estudios de genéti-años se han realizado numerosos estudios de genética poblacional y epidemiología genética; y en la últi-ca poblacional y epidemiología genética; y en la últi-ca poblacional y epidemiología genética; y en la última década se han refinado los estudios de genética molecular y análisis genómicos a gran escala. Los estudios realizados entre 1980 y 1995 mostraban un riesgo genético del 3.5-8.0% para el trastorno bipo-lar y del 5.7-23.0% para la depresión unipolar. Los estudios en gemelos monozigóticos reflejaban una concordancia del 58.5% al 92.6%; y en gemelos dizi-concordancia del 58.5% al 92.6%; y en gemelos dizi-concordancia del 58.5% al 92.6%; y en gemelos dizigóticos la concordancia era del 16.4% al 34.9%. En cuanto al modelo de transmisión, se habló de una herencia autosómica recesiva, de una posible heren-cia ligada al sexo, e incluso de assorting mating, un assorting mating, un assorting matingfenómeno según el cual los pacientes depresivos tie-nen tendencia a casarse con otros pacientes depresi-nen tendencia a casarse con otros pacientes depresi-nen tendencia a casarse con otros pacientes depresivos. La percepción genética de la depresión cambió cuando fue posible realizar estudios de ligamiento y estudios de asociación. Igual que con otras enferme-dades complejas del sistema nervioso central, se lle-gó a la conclusión de que la depresión y el trastorno bipolar son síndromes poligénicos y multifactoriales que se caracterizan por la presencia de múltiples defectos genómicos cuya expresión dependerá de la interacción con factores externos (educación, stress, eventos vitales, condiciones médicas, microlesiones cerebrales) y fenómenos epigenéticos. Glatt et al
también sugieren que el splicing alternativo de pre-RNAm puede contribuir a la disregulación genética presente en muchos trastornos neuropsiquiátricos. La regla de oro establecida para este tipo de patolo-gías complejas es que a mayor número de defectos genómicos, más precoz es la enfermedad y peor es su respuesta terapéutica; y a menor número de de-fectos genómicos, más tardía es la expresión de la enfermedad y mejor es la respuesta al tratamiento.
Entre 1987 y 2000 se encontraron los primeros de-fectos genómicos asociados a depresión y trastorno bipolar en diferentes poblaciones étnicas. En 1987 y 1989, Egeland y Kelsoe, respectivamente, halla-y 1989, Egeland y Kelsoe, respectivamente, halla-y 1989, Egeland y Kelsoe, respectivamente, hallaron ligamiento genético entre la región 11p15 y el
trastorno bipolar; y entre 1990 y 1995, los grupos de Leboyer y Gurling comprobaron que ese locus se correspondía con el gen de la tirosina hidroxilasa. Baron et al encontró ligamiento a Xq28 en 1987, y Pekkarinen y Nurnberger asociaron el trastorno bipolar a Xq26 en 1995 y 1997, respectivamente. Otros autores destacados de la época fueron Berret-Otros autores destacados de la época fueron Berret-Otros autores destacados de la época fueron Berrettini, Stine y Freimar, que encontraron asociación con el cromosoma 18 en 18p-q y 18q22-23; Straub, Gurling y Nurnberger, en trabajos independientes, asociaron el trastorno bipolar a defectos en 21q22.3; los grupos de Dawson y Baden vieron cierto liga-los grupos de Dawson y Baden vieron cierto liga-los grupos de Dawson y Baden vieron cierto liga
ción de la melatonina con fármacos melato también sugieren que el splicing alternativo de pre-también sugieren que el splicing alternativo de pre-también sugieren que el splicing alternativo de pre-
Fig. 13. Resonancia Magnética Cerebral, EEG y Patrón Cartográfico Cerebral en fase sintomática (izquierda) y tras remisión de síntomas (derecha) en un varón de 42 años con Depresión Psicótica
Fig. 14. Depresión post-ictal en mujer de 45 años que desarrolla crisis convulsivas secundarias a accidente cerebrovascular agudo. RMN. Cartografía cerebral en el momento del diagnóstico (superior), con un MMSE de 24 y un Hamilton-Depresión de 34; y cartografía cerebral un año después, con remisión total de síntomas (MMSE: 30; Hamilton-Depresión: 8)
Fuente de Datos: R. Cacabelos, Base de Datos CIBE Fuente de Datos: R. Cacabelos, Base de Datos CIBE
El futuro pasa por la implantación irremediable y progresiva de protocolos de intervención
farmacogenética para optimizar la seguridad y la eficacia de los psicofármacos en general y los
antidepresivos en particular

52
Estados Depresivos
miento con 12q23-24, el locus de la enfermedad de Darier; Lachman y Nurnberger, en 1996 y 1997, respectivamente, vieron una posible asociación con 22q11; Blackwood et al con 4q16; Ewald y Nurnber-22q11; Blackwood et al con 4q16; Ewald y Nurnber-22q11; Blackwood et al con 4q16; Ewald y Nurnberger, con 16p13; Kelsoe en 1996 asoció el trastorno bipolar a 5p15, el locus del transportador de dopa-bipolar a 5p15, el locus del transportador de dopa-bipolar a 5p15, el locus del transportador de dopamina; Coon et al hallaron asociación con 5q35; y el grupo de Ginns comprobó que los loci 6pter-p24, 13q13 y 15q11-qter podían estar relacionados con depresión y trastorno bipolar. Desde el año 2000 hasta el presente se han estudiado más de 1000 ge-nes potencialmente relacionados con la depresión y el trastorno bipolar (Fig. 6-7; Fig. 6-7; Fig. 6-7 Tabla 1). Algunos de los genes asociados a trastorno bipolar también están asociados a esquizofrenia y trastornos psicóti-están asociados a esquizofrenia y trastornos psicóti-están asociados a esquizofrenia y trastornos psicóticos. De todos los genes investigados, es interesante
comprobar que muchos loci relacionados con los sistemas de neurotransmisión potencialmente alte-rados en la depresión, como es el caso de la sero-tonina y la noradrenalina, muestran defectos a ni-tonina y la noradrenalina, muestran defectos a ni-tonina y la noradrenalina, muestran defectos a nivel de receptores y enzimas cuya afectación podría ser responsable, en parte, del fracaso del equilibrio emocional que se observa en los estados depresivos. Los polimorfismos rs10994336 del gen ANK3 y el ANK3 y el ANK3rs1006737 del gen CACNA1C están entre los más CACNA1C están entre los más CACNA1Cconsistentemente asociados a trastorno bipolar. Otros polimorfismos recientemente confirmados son el rs383902 de ADAM10 y el rs2053053 de ADAM10 y el rs2053053 de ADAM10 CA-MK2A. ANK3, ANK3, ANK3 CACNA1C, CACNA1C, CACNA1C CMTM8, CMTM8, CMTM8 DGKH, DGKH, DGKH EGFR y EGFR y EGFR
NPAS3 aparecen asociados a trastorno bipolar en NPAS3 aparecen asociados a trastorno bipolar en NPAS3varios estudios genómicos. De los 4 polimorfismos presentes en el gen del transportador de dopami-presentes en el gen del transportador de dopami-presentes en el gen del transportador de dopamina (DAT)(rs6347, intrón-8 5/6 VNTR, rs27072 y DAT)(rs6347, intrón-8 5/6 VNTR, rs27072 y DAT3’UTR 9/10 VNTR), sólo el rs27072 parece estar asociado a trastorno bipolar. Varios polimorfismos de DISC1 y DISC1 y DISC1 DISC2, relacionados con esquizofrenia, DISC2, relacionados con esquizofrenia, DISC2también se asocian a depresión y trastorno bipolar. Varios genes pleiotrópicos, como el APOE, han mosAPOE, han mosAPOE -trado una distribución de frecuencias genotípicas diferenciadas en la depresión con respecto a otras patologías del sistema nervioso. En general, de los más de 2000 estudios genéticos realizados en los úl-más de 2000 estudios genéticos realizados en los úl-más de 2000 estudios genéticos realizados en los últimos 20 años, hasta el momento existen unos 300 genes cuyas variantes polimórficas podrían tener influencia patogénica en los estados depresivos o en la diátesis de vulnerabilidad emocional (Fig. 7; Fig. 7; Fig. 7Tabla 1). El establecimiento de un clúster genético altamente informativo puede ser de utilidad como ayuda en el diagnóstico precoz y en la prevención de estados depresivos en la población a riesgo.
DiagnósticoEl diagnóstico de los estados depresivos es funda-El diagnóstico de los estados depresivos es funda-El diagnóstico de los estados depresivos es fundamentalmente clínico, con diversos matices que se explicitan en el DSM-IV y en el ICD-10. Para alcanzar un diagnóstico de alta precisión, que per-alcanzar un diagnóstico de alta precisión, que per-alcanzar un diagnóstico de alta precisión, que permita el establecimiento de una pauta terapéutica temprana, todo paciente debe ser sometido a un protocolo diagnóstico que incluya: (i) anamnesis e historia clínica; (ii) examen psiquiátrico y neuroló-gico; (iii) análisis de sangre y orina, para descartar cualquier patología bioquímica o metabólica que pueda enmascarar o causar un cuadro depresivo; (iv) pruebas radiológicas (neuroimagen estática: TAC, MRI)(Fig. 9-10); (v) pruebas de neuroimaFig. 9-10); (v) pruebas de neuroimaFig. 9-10 -); (v) pruebas de neuroima-); (v) pruebas de neuroimagen funcional (SPECT, PET, Cartografía Cerebral, Topografía Óptica Digital)(disponibles en centros
Fig. 15. Distimia asociada a enfermedad de Parkinson en mujer de 58 años. Cartografía cerebral basal y posterior a remisión sintomática con tratamiento antidepresivo. Patrón topográfico cerebral basal y tras estimulación visual
Fig. 16. Trastorno depresivo asociado a enfermedad de Alzheimer precoz con componente depresivo en mujer de 60 años. RMN. Cartografía cerebral basal y posterior a remisión de síntomas depresivos tras tratamiento con bajas dosis de antidepresivos
Fuente de Datos: R. Cacabelos, Base de Datos CIBE Fuente de Datos: R. Cacabelos, Base de Datos CIBE
En términos globales, la instauración de la farmacogenómica en el tratamiento de las enfermedades del sistema nervioso central podría suponer un ahorro del 18-32% en gasto farmacéutico en un horizonte de 2-3 años

Julio 2011 53
ciencia
superespecializados)(Fig. 11); (vi) pruebas psico-métricas, para evaluar el grado de depresión y las condiciones psicopatológicas del paciente, especial-condiciones psicopatológicas del paciente, especial-condiciones psicopatológicas del paciente, especialmente los componentes psicótico y ansiogénico; y (vii) pruebas genéticas, en aquellos casos en que las circunstancias lo aconsejen, sólo disponibles en cen-tros de excelencia.
Los criterios clínicos esenciales para el diagnóstico de los diferentes trastornos depresivos incluyen una serie de características que definen los episodios de-presivos o maníacos.
Episodio Depresivo Mayor: humor deprimido co-tidiano, objetivo y subjetivo; pérdida de interés por las cosas cotidianas, anhedonía; pérdida de peso, sin hacer dieta; insomnio o hipersomnia habitual; agitación psicomotriz o enlentecimien-to funcional; fatiga y pérdida de energía; senti-to funcional; fatiga y pérdida de energía; senti-to funcional; fatiga y pérdida de energía; sentimientos de inutilidad o autoinculpación injus-tificada; dificultad para pensar o concentrarse, indecisión operativa; pensamientos recurrentes de muerte con o sin ideación suicida.
Episodio Maníaco: expansión emocional, irri-: expansión emocional, irri-: expansión emocional, irritabilidad; hipertrofia de la autoestima e ideas de grandiosidad personal; disminución de la necesidad de sueño; lenguaje apresurado, par-necesidad de sueño; lenguaje apresurado, par-necesidad de sueño; lenguaje apresurado, parlanchín y poco coherente; fuga de ideas o ex-lanchín y poco coherente; fuga de ideas o ex-lanchín y poco coherente; fuga de ideas o experiencia subjetiva de hechos irreales; distracti-periencia subjetiva de hechos irreales; distracti-periencia subjetiva de hechos irreales; distractibilidad e inatención; hiperactividad y agitación psicomotriz; propensión a meterse en activida-psicomotriz; propensión a meterse en activida-psicomotriz; propensión a meterse en actividades de riesgo o placer ilógico.
Los episodios hipomaníacos se parecen a los manía-Los episodios hipomaníacos se parecen a los manía-Los episodios hipomaníacos se parecen a los maníacos pero son menos intensos y duraderos, con cierta preservación de la racionalidad y el autocontrol ante presiones externas. Los episodios mixtos son una mezcla de fases depresivas y maníacas alternantes en el tiempo, sin un patrón lógico definido.
En el proceso diagnóstico, el médico no debe igno-rar que determinadas condiciones médicas pueden causar episodios depresivos o maníacos. Diversas lesiones en regiones selectivas del cerebro pueden
causar trastornos conductuales que simulan una de-presión o un estado maniforme. Esto es frecuente en personas con traumatismos craneoencefálicos y en pacientes que han sufrido ictus o accidentes cere-brovasculares de cualquier naturaleza. Algunos pa-brovasculares de cualquier naturaleza. Algunos pa-brovasculares de cualquier naturaleza. Algunos pacientes post-quirúrgicos, como consecuencia de la anestesia o del stress quirúrgico, pueden desarrollar trastornos depresivos y alteraciones conductuales. Diversos fármacos pueden causar estados de deli-Diversos fármacos pueden causar estados de deli-Diversos fármacos pueden causar estados de delirium y/o estados depresivos o hipomaníacos (alco-hol, anfetaminas, cocaína, sustancias alucinógenas, inhalantes, opiáceos, fenciclidina, ansiolíticos, hip-nóticos, sedantes, anticonceptivos, reserpina, metil-nóticos, sedantes, anticonceptivos, reserpina, metil-nóticos, sedantes, anticonceptivos, reserpina, metildopa, anticolinesterásicos, cimetidina, indometaci-dopa, anticolinesterásicos, cimetidina, indometaci-dopa, anticolinesterásicos, cimetidina, indometacina, fenotiazinas, cicloserina, vincristina, vinblastina). Algunos trastornos carenciales crónicos (ferropenia, déficit de fólico, hipovitaminosis, pelagra, anemia perniciosa) también pueden generar importantes alteraciones del humor. Diversas patologías pueden cursar con síndromes depresivos: trastornos endo-crinos (hipotirosidismo, hipertiroidismo, hiperpara-crinos (hipotirosidismo, hipertiroidismo, hiperpara-crinos (hipotirosidismo, hipertiroidismo, hiperparatiroidismo, hipopituitarismo, enfermedad de Addi-tiroidismo, hipopituitarismo, enfermedad de Addi-tiroidismo, hipopituitarismo, enfermedad de Addison, enfermedad de Cushing, diabetes); infecciones (sífilis terciaria, toxoplasmosis, influenza, neumonía vírica, hepatitis, mononucleosis infecciosa, SIDA); reacciones inflamatorias y colagenopatías (artiritis reumatoide, lupus eritematoso sistémico); trastor-reumatoide, lupus eritematoso sistémico); trastor-reumatoide, lupus eritematoso sistémico); trastornos neurológicos (esclerosis múltiple, enfermedad de Parkinson, traumatismo craneal, epilepsia par-de Parkinson, traumatismo craneal, epilepsia par-de Parkinson, traumatismo craneal, epilepsia parcial, apnea de sueño, tumores cerebrales, patología cerebrovascular); y neoplasias (tumores abdomina-cerebrovascular); y neoplasias (tumores abdomina-cerebrovascular); y neoplasias (tumores abdominales, carcinomatosis diseminada, metástasis cerebra-les, carcinomatosis diseminada, metástasis cerebra-les, carcinomatosis diseminada, metástasis cerebrales). Un estudio reciente de Bond en Canadá en-cuentra una clara correlación entre el aumento del índice de masa corporal y la disminución del volu-índice de masa corporal y la disminución del volu-índice de masa corporal y la disminución del volumen cerebral en pacientes con un primer episodio de manía. En más de un 30% de pacientes ancianos polimedicados pueden aparecer episodios compati-polimedicados pueden aparecer episodios compati-polimedicados pueden aparecer episodios compatibles con trastornos depresivos. En todos estos casos, la precisión diagnóstica es fundamental para optimi-la precisión diagnóstica es fundamental para optimi-la precisión diagnóstica es fundamental para optimizar la intervención terapéutica, centrada en la causa más que en el síntoma depresivo.
Fig. 17. Topografía óptica digital en mujer de 76 años con Trastorno Bipolar de más de 30 años de evolución
Fig. 18. RMN, topografía óptica digital y cartografía cerebral de mujer de 65 años con depresión unipolar recurrente. La cartografía muestra actividad bioeléctrica cerebral en fase depresiva (izquierda) y en fase asintomática (derecha)
Fuente de Datos: R. Cacabelos, Base de Datos CIBE Fuente de Datos: R. Cacabelos, Base de Datos CIBE

54
Estados Depresivos
NeuroimagenLa neuroimagen estática o estructural (TAC, MRI)(Fig. 9-10) muestra hallazgos bastante inespecífiFig. 9-10) muestra hallazgos bastante inespecífiFig. 9-10 -) muestra hallazgos bastante inespecífi-) muestra hallazgos bastante inespecíficos y comunes en diversas patologías del sistema nervioso: incremento del ratio ventrículo-cerebral, alargamiento de los ventrículos laterales, ensancha-alargamiento de los ventrículos laterales, ensancha-alargamiento de los ventrículos laterales, ensanchamiento de los surcos de las circunvoluciones cere-brales, atrofia vermiana en cerebelo, y anomalías en ganglios de la base y sustancia blanca subcortical. La presencia de focos isquémicos en regiones críticas de los circuitos córtico-límbicos es frecuente en pa-de los circuitos córtico-límbicos es frecuente en pa-de los circuitos córtico-límbicos es frecuente en pacientes depresivos.
La neuroimagen funcional (SPECT, PET)(Fig. 23) Fig. 23) Fig. 23refleja alteraciones regionales en el consumo de oxígeno, con especial compromiso de los ganglios basales, y anomalías en receptores serotonérgicos, noradrenérgicos e histaminérgicos. La carestía de estas pruebas y su relación coste-beneficio hace que
se reserven para circunstancias muy especiales, sobre todo en situaciones donde el diagnóstico diferencial lo requiere.
Con la cartografía cerebral (EEG computerizado)(Fig. 11-16,18-20) se puede ver un ligero enlenteciFig. 11-16,18-20) se puede ver un ligero enlenteciFig. 11-16,18-20 -) se puede ver un ligero enlenteci-) se puede ver un ligero enlentecimiento de la actividad cerebral de localización prefe-rente en regiones fronto-temporales y centroencefá-rente en regiones fronto-temporales y centroencefá-rente en regiones fronto-temporales y centroencefálicas. La Topografía Óptica Digital muestra patrones diferenciados en el consumo de oxígeno cortical en pacientes depresivos y maníacos (Fig. 17,21-22). Fig. 17,21-22). Fig. 17,21-22Aunque todas estas técnicas son esenciales en el diagnóstico diferencial de depresión y en el despis-diagnóstico diferencial de depresión y en el despis-diagnóstico diferencial de depresión y en el despistaje de lesiones cerebrales que puedan causar depre-sión, ninguna de ellas es suficientemente específica y carecen de valor diagnóstico primario, al nivel de resolución con el que trabajamos en la actualidad. A pesar de estas limitaciones, deben utilizarse en todos aquellos casos donde la destreza clínica sugiera un despistaje diagnóstico diferencial.
TratamientoEn 1995 vieron la luz dos grandes obras de psico-farmacología, editadas por Floyd E. Bloom y David J. Kupfer, en representación del American College of American College of American CollegeNeuropsychopharmacology, y Alan F. Schatzberg y Char-, y Alan F. Schatzberg y Char-, y Alan F. Schatzberg y Charles B. Nemeroff, de la American Psychiatric Press. AmAmerican Psychiatric Press. AmAmerican Psychiatric Press -. Am-. Ambos tratados recogen lo más relevante del tratamien-bos tratados recogen lo más relevante del tratamien-bos tratados recogen lo más relevante del tratamiento psicofarmacológico hasta mediados de la última década del siglo XX. Aunque desde entonces han sido aprobados nuevos fármacos para los trastornos depresivos, no ha habido grandes cambios concep-tuales a lo largo de los últimos 15 años desde una perspectiva estrictamente farmacéutica, a excepción de la entrada en el mercado de los agentes melato-nérgicos con acción antidepresiva. Un interesante trabajo de López-Muñoz y Álamo ilustra la evolución de la neurotransmisión monoaminérgica y el desa-de la neurotransmisión monoaminérgica y el desa-de la neurotransmisión monoaminérgica y el desarrollo histórico de los antidepresivos.
El tratamiento de los estados depresivos debe ser adaptado a la razón causal más que al síntoma. En los últimos años se ha producido una administración abusiva de antidepresivos, hasta el punto de que esta categoría de fármacos se encuentra entre los de máximo consumo a nivel mundial. Nada tiene que ver la intervención terapéutica de un episodio aisla-ver la intervención terapéutica de un episodio aisla-ver la intervención terapéutica de un episodio aislado de distimia o una depresión asociada a un proble-ma médico con el tratamiento que debe seguir un paciente con una depresión unipolar o con un tras-paciente con una depresión unipolar o con un tras-paciente con una depresión unipolar o con un trastorno bipolar. En el caso de la distimia hay que pres-torno bipolar. En el caso de la distimia hay que pres-torno bipolar. En el caso de la distimia hay que prestar atención a la causa desencadenante (en ocasio-nes de claro origen externo) e intentar neutralizarla. En los casos de depresión vinculada a una patología subyacente o concomitante hay que dar prioridad a la enfermedad de fondo. En casos de depresión a fármacos o tóxicos hay que eliminar el agente cau-fármacos o tóxicos hay que eliminar el agente cau-fármacos o tóxicos hay que eliminar el agente causante. En los casos genuinos de depresión, manía o trastorno bipolar hay que recurrir a estrategias tera-trastorno bipolar hay que recurrir a estrategias tera-trastorno bipolar hay que recurrir a estrategias terapéuticas concretas pautadas por médicos expertos en el manejo de fármacos antidepresivos u otros
Fig. 19. RMN y cartografía cerebral de mujer de 66 años con pseudodemencia depresiva en fase sintomática (izquierda) y 3 meses después (derecha), a tratamiento con antidepresivos
Fig. 20. RMN, topografía óptica digital y cartografía cerebral de mujer de 33 años con depresión asociada a esclerosis múltiple en fase sintomática (izquierda) y tras remisión de síntomas (derecha) a tratamiento durante 3 meses con antidepresivos
Fuente de Datos: R. Cacabelos, Base de Datos CIBE
Fuente de Datos: R. Cacabelos, Base de Datos CIBE

Julio 2011 55
ciencia
psicofármacos. Tradicionalmente, los estados depre-sivos han sido tratados con psicoterapia, fármacos o terapia electroconvulsiva en aquellos pacientes que no responden a tratamientos convencionales. La psicoterapia sigue teniendo su nicho de aplicación en casos de distimia o depresiones leves, así como adyuvante o complemento de otras formas terapéu-adyuvante o complemento de otras formas terapéu-adyuvante o complemento de otras formas terapéuticas. Las diferentes formas de psicoterapia incluyen: terapia interpersonal, terapia cognitivo-conductual, terapia conductual, terapia psicosocial, y terapia psi-terapia conductual, terapia psicosocial, y terapia psi-terapia conductual, terapia psicosocial, y terapia psicodinámica (acorde a las teorías de Sigmund Freud, Karl Abraham, Bertram Lewin, Melanie Klein, Adolf Meyer, Karen Horney, Sandor Rado, John Bowlby y Harry Stack Sullivan).
El tratamiento con antidepresivos responde a varias categorías farmacológicas o mecanismos de acción: (i) Inhibidores de la recaptación de noradrenalina (desipramina, protriptilina, nortriptilina, maprotili-(desipramina, protriptilina, nortriptilina, maprotili-(desipramina, protriptilina, nortriptilina, maprotilina); (ii) inhibidores de la recaptación de serotonina (citalopram, fluoxetina, fluvoxamina, paroxetina, sertralina); (iii) inhibidores de la recaptación de se-rotonina y noradrenalina (amitriptilina, doxepina, imipramina, trimipramina, venlafaxina); (iv) agen-imipramina, trimipramina, venlafaxina); (iv) agen-imipramina, trimipramina, venlafaxina); (iv) agentes activos a nivel presináptico y post-sináptico (ne-fazodona, mirtazapina); (v) inhibidores de la recap-tación de dopamina (bupropión); (vi) agentes de
acción mixta (clomipramina, trazodona); (vii) inhi-acción mixta (clomipramina, trazodona); (vii) inhi-acción mixta (clomipramina, trazodona); (vii) inhibidores de MAO (tranilcipromina, fenelzina, selegi-bidores de MAO (tranilcipromina, fenelzina, selegi-bidores de MAO (tranilcipromina, fenelzina, selegilina, moclobemida); y (viii) agentes melatonérgicos (agomelatina)(Tabla 2). Los primeros fármacos anTabla 2). Los primeros fármacos anTabla 2 -). Los primeros fármacos an-). Los primeros fármacos antidepresivos fueron las aminas cíclicas, que se clasifi-tidepresivos fueron las aminas cíclicas, que se clasifi-tidepresivos fueron las aminas cíclicas, que se clasifican en 3 categorías: (i) fármacos tricíclicos terciarios (amitriptilina, clomipramina, doxepina, imiprami-(amitriptilina, clomipramina, doxepina, imiprami-(amitriptilina, clomipramina, doxepina, imipramina, trimipramina), (ii) fármacos tricíclicos secunda-na, trimipramina), (ii) fármacos tricíclicos secunda-na, trimipramina), (ii) fármacos tricíclicos secundarios (desipramina, nortriptilina, protriptilina), y (iii) fármacos tetracíclicos (amoxapina, maprotilina). Estos fármacos han sido la base del tratamiento anti-Estos fármacos han sido la base del tratamiento anti-Estos fármacos han sido la base del tratamiento antidepresivo durante más de 30 años hasta la introduc-depresivo durante más de 30 años hasta la introduc-depresivo durante más de 30 años hasta la introducción de los inhibidores de la recaptación de serotoni-ción de los inhibidores de la recaptación de serotoni-ción de los inhibidores de la recaptación de serotonina. Tricíclicos y teracíclicos tienen un mecanismo de acción compleja sobre receptores adrenérgicos α
1 y α2, receptores histaminérgicos H1, receptores musca-, receptores musca-, receptores muscarínicos M1, y receptores serotonérgicos 5HT1 y 5HT2, con la consecuente modulación de noradrenalina, serotonina, histamina, acetilcolina y, en menor me-dida, dopamina. Esta complejidad farmacológica les hace proclives a desarrollar efectos secundarios rele-vantes, que van desde alteraciones cardiovasculares a aumento de peso y síndrome serotonérgico; este último, especialmente relevante en los inhibidores de la recaptación de serotonina.
Fig. 21. Topografía óptica digital de una paciente de 40 años con Depresión Mayor Familiar, Síndrome de Ansiedad Generalizada y Síndrome Estrogénico Secundario a uso crónico de anticonceptivos
Fig. 22. Topografía óptica digital de un paciente de 64 años con Trastorno Bipolar
Fuente de Datos: Iván Tellado y Ramón Cacabelos. Base de Datos CIBE
Fuente de Datos: Iván Tellado y Ramón Cacabelos. Base de Datos CIBE

56
Estados Depresivos
La terapia con IMAOs (iproniazida, isoniazida, fe-nelzina, isocarboxacida, tranilcipromina, clorgilina, pargilina, selegilina, moclobemida, brofaromina, lazabemida, mofegilina, milacemida, toloxatona, befloxatona), en franca decadencia, tiene restric-befloxatona), en franca decadencia, tiene restric-befloxatona), en franca decadencia, tiene restricciones e incompatibilidades; su aplicación exige restricciones dietéticas de tiramina, y es incompa-restricciones dietéticas de tiramina, y es incompa-restricciones dietéticas de tiramina, y es incompatible con estimulantes (anfetaminas, cocaína, ano-rexígenos), descongestionantes (efedrina, fenile-fedrina, fenilpropanolamina), antihipertensivos (metildopa, guanetidina, reserpina), antidepresivos tricíclicos (imipramina, desipramina, clomiprami-tricíclicos (imipramina, desipramina, clomiprami-tricíclicos (imipramina, desipramina, clomipramina), otros IMAOs, simpaticomiméticos (dopamina, metaraminol), precursores aminérgicos (L-dopa, L-triptófano) y narcóticos (meperidina). La admi-L-triptófano) y narcóticos (meperidina). La admi-L-triptófano) y narcóticos (meperidina). La administración de IMAOs también requiere precaución con opiáceos (morfina, codeína), sedantes (alcohol, barbitúricos, benzodiacepinas), anestésicos locales
que contengan vasoconstrictores, diversos agentes simpaticomiméticos y los anestésicos generales. A pesar de su complejidad, existen claras diferencias en términos de seguridad y eficacia dependiendo de que los IMAOs sean no-selectivos e irreversibles, selectivos y reversibles (RIMAs) o que actúen sobre la monoaminaoxidasa (MAO) A o B.
El uso de antidepresivos específicos se ajusta a: (i) la experiencia del prescriptor, (ii) las propiedades far-experiencia del prescriptor, (ii) las propiedades far-experiencia del prescriptor, (ii) las propiedades farmacocinéticas y farmacodinámicas de cada agente antidepresivo, (iii) los efectos secundarios y reaccio-nes adversas potenciales, (iv) las patologías concomi-nes adversas potenciales, (iv) las patologías concomi-nes adversas potenciales, (iv) las patologías concomitantes que requieren tratamiento farmacológico y pueden causar interacciones medicamentosas, y (v) el perfil farmacogenético de cada paciente. En un futuro próximo, la personalización del tratamiento mediante la farmacogenética permitirá obviar mu-mediante la farmacogenética permitirá obviar mu-mediante la farmacogenética permitirá obviar muchos de los problemas de manejo que hoy plantean los antidepresivos. Las dosis deben ser individualiza-los antidepresivos. Las dosis deben ser individualiza-los antidepresivos. Las dosis deben ser individualizadas en base a las condiciones del paciente y a la seve-ridad del cuadro depresivo. En niños debe ajustarse la dosis al peso corporal; y en ancianos debe aplicar-la dosis al peso corporal; y en ancianos debe aplicar-la dosis al peso corporal; y en ancianos debe aplicarse un 50% de la dosis recomendada en adultos.
El planteamiento terapéutico en casos de manía y trastorno bipolar es completamente diferente y al-trastorno bipolar es completamente diferente y al-trastorno bipolar es completamente diferente y altamente complejo en la mayoría de los casos. En pacientes maniformes los fármacos de elección, de-pendiendo de cada caso, son el ácido valproico, la carbamazepina y el litio. Otras alternativas podrían ser gabapentina, tiagabina, lamotrigina, vigabatrina, felbamato y topiramato. En cuadros de manía aguda, el litio es la primera opción, seguido de valproico, car-el litio es la primera opción, seguido de valproico, car-el litio es la primera opción, seguido de valproico, carbamazepina, clonazepam, lorazepam, antagonistas de receptores dopaminérgicos, antagonistas de sero-tonina y dopamina, antagonistas de los canales L del calcio iónico y reguladores GABAérgicos. Cuando se precisa administrar neurolépticos, pueden usarse los antipsicóticos típicos (clorpromazina, haloperi-los antipsicóticos típicos (clorpromazina, haloperi-los antipsicóticos típicos (clorpromazina, haloperidol, pimozida) o los atípicos (risperidona, clozapina, trimipramina, olanzapina, quetiapina, ziprasidona). El manejo de todos estos fármacos es complejo, re-quiere experiencia y un estricto seguimiento de sus efectos, tanto terapéuticos como indeseables a nivel cerebral y sistémico. El uso de carbamazepina debe ser extremadamente prudente cuando el paciente necesite medicación concomitante. Por ejemplo, la carbamazepina disminuye los niveles de alprazolam, amitriptilina, anticonceptivos, antifúngicos, bupro-pión, clobazam, clonazepam, clozapina, ciclospori-pión, clobazam, clonazepam, clozapina, ciclospori-pión, clobazam, clonazepam, clozapina, ciclosporina, dexametasona, dicumarol, doxacurium, doxepi-na, dexametasona, dicumarol, doxacurium, doxepi-na, dexametasona, dicumarol, doxacurium, doxepina, doxiciclina, etoxusimida, felbamato, fentanilo, flufenazina, haloperidol, imipramina, lamotrigina, metadona, metilprednisolona, mianserina, nimo-dipino, oxiracetam, pancuronium, fenitoína, pred-dipino, oxiracetam, pancuronium, fenitoína, pred-dipino, oxiracetam, pancuronium, fenitoína, prednisolona, primidona, teofilina, tiotixeno, valproato, vecuronium y warfarina. Hay una serie de fármacos que pueden aumentar las concentraciones de car-que pueden aumentar las concentraciones de car-que pueden aumentar las concentraciones de carbamazepina, como acetazolamida, baclofeno, cime-tidina, claritromicina, danazol, dextropropoxifeno, diltiazem, eritromicina, fluoxetina, fluritromicina, fluvoxamina, gemfibrozilo, isoniazida, josamicina,
La terapia con IMAOs (iproniazida, isoniazida, fe
barbitúricos, benzodiacepinas), anestésicos locales
Antidepresivos
24%
56%
40%47%
38% 38%
85%
5%0
0
CYP1A2 CYP2B6 CYP2C19
Antidepresivos Neurolépticos Benzodiazepinas
CYP2D6 CYP3A4
20
10
20
30
40
50
60
%
40
60
80
100
24%
CYP1A2 CYP2B6 CYP2C19
5%
CYP1A2 CYP2B6 CYP2C19CYP1A2 CYP2B6 CYP2C19CYP1A2 CYP2B6 CYP2C19CYP1A2 CYP2B6 CYP2C19
85%
CYP2D6 CYP3A4CYP2D6 CYP3A4CYP2D6 CYP3A4
%
CYP1A2CYP2B6CYP2C19CYP2D6CYP3A4
Fig. 23. PET scan en un caso control y un paciente con depresión
Fig. 24. Porcentaje de antidepresivos que actúan como sustratos mayores de diversas enzimas CYP
Fuente de Datos: Mayo Foundation for Medical Education and Research. (Consultado 23 Junio, 2011, en http://www.mayoclinic.com/health/medical/IM00356)
Fuente de Datos: R. Cacabelos, 2008

Julio 2011 57
ciencia
lamotrigina, nicotinamida, ponsinomicina, propoxi-lamotrigina, nicotinamida, ponsinomicina, propoxi-lamotrigina, nicotinamida, ponsinomicina, propoxifeno, terfenadina, triacetiloleandomicina, valproato, verapamilo y viloxazina. Algunos fármacos disminu-verapamilo y viloxazina. Algunos fármacos disminu-verapamilo y viloxazina. Algunos fármacos disminuyen la concentración de carbamazepina, tales como adriamicina, cisplatino, felbamato, fenobarbital, pri-adriamicina, cisplatino, felbamato, fenobarbital, pri-adriamicina, cisplatino, felbamato, fenobarbital, primidona y valproato; la propia carbamazepina, por autoinducción, puede reducir los niveles de carba-autoinducción, puede reducir los niveles de carba-autoinducción, puede reducir los niveles de carbamazepina. Todos estos fenómenos interactivos tie-nen un componente idiosincrático relacionado con el perfil farmacogenético de cada paciente.
FarmacogenómicaLa farmacogenómica persigue la personalización del tratamiento farmacológico en función del perfil genómico de cada paciente. La farmacocinética de los medicamentos sigue 4 fases que son exclusivas de cada persona: (i) absorción, (ii) distribución, (iii) metabolismo, y (iv) eliminación. A lo largo de este proceso de transformación farmacológica, hasta que el fármaco ejerce su acción terapéutica, se metaboli-el fármaco ejerce su acción terapéutica, se metaboli-el fármaco ejerce su acción terapéutica, se metaboliza y elimina, pueden ocurrir fenómenos que causan problemas de diversa índole. Por ejemplo, los agen-tes antiácidos y los anticolinérgicos pueden alterar la absorción de los antidepresivos y los antimaníacos. Aquellos fármacos que se unen fuertemente a pro-teínas plasmáticas pueden desplazar a otros medica-teínas plasmáticas pueden desplazar a otros medica-teínas plasmáticas pueden desplazar a otros medicamentos e incrementar su fracción libre, alterando con ello la distribución del fármaco. El metabolismo de la inmensa mayoría de los fármacos sigue reaccio-nes de fase I y fase II a nivel hepático, dependientes de enzimas del citocromo P-450 (monooxigenasas) o enzimas de fase II, responsables de otros procesos de biotransformación catabólica de los medicamen-tos. Alteraciones en estas enzimas, debido a defectos en los genes que las codifican, son responsables de gran parte de los efectos secundarios y las interaccio-nes medicamentosas que se observan en la clínica. Muchos casos de depresión resistente se deben a al-Muchos casos de depresión resistente se deben a al-Muchos casos de depresión resistente se deben a alteraciones en el metabolismo de los medicamentos asociados a variantes polimórficas en los genes que codifican a las enzimas que catalizan las reacciones de fase I y fase II. La eliminación de los fármacos también se puede ver afectada por la administración concomitante de otros agentes terapéuticos. El bicar-concomitante de otros agentes terapéuticos. El bicar-concomitante de otros agentes terapéuticos. El bicarbonato sódico y el ácido ascórbico pueden aumen-tar la excreción de algunos fármacos. Los IECAs (in-hibidores de la enzima conversora de angiotensina, utilizados para el tratamiento de la hipertensión) pueden aumentar la concentración de litio.
Para optimizar el rendimiento terapéutico de los agentes antidepresivos y antimaníacos es necesario definir el perfil farmacogenómico de los pacientes. Los genes que regulan la respuesta farmacogenómi-Los genes que regulan la respuesta farmacogenómi-Los genes que regulan la respuesta farmacogenómica y a la postre van a ser responsables de que logre-mos o no el efecto terapéutico deseado se clasifican en 5 categorías: (i) genes patogénicos relacionados con la causa de la depresión, la manía o el trastorno bipolar; (ii) genes relacionados con el mecanismo de acción del fármaco, a nivel de receptores y enzi-de acción del fármaco, a nivel de receptores y enzi-de acción del fármaco, a nivel de receptores y enzimas; (iii) genes relacionados con el metabolismo de
los fármacos a través de las reacciones enzimáticas de fase I y fase II; (iv) genes relacionados con las pro-teínas transportadoras que permiten el acceso de los medicamentos a las células diana (por ejemplo, la accesibilidad al cerebro mediante el paso a través de la barrera hematoencefálica); y (v) genes pleiotró-picos que participan en múltiples rutas metabólicas o procesos fisiológicos, como puede ser el caso del APOE en el metabolismo lipídico o las interleukinas APOE en el metabolismo lipídico o las interleukinas APOEy el TNF en las reacciones inflamatorias y procesos TNF en las reacciones inflamatorias y procesos TNFinmunológicos (Tablas 2-5). Tablas 2-5). Tablas 2-5
los fármacos a través de las reacciones enzimáticas
Fig. 25. Distribución y frecuencia de fenotipos metabolizador normal (EM), metabolizador intermedio (IM), metabolizador lento (PM) y metabolizador ultra-rápido (UM) asociados a variantes polimórficas del gen CYP2D6 en pacientes con enfermedades del sistema nervioso
Fig. 26. Distribución y frecuencia de fenotipos metabolizador normal (EM), metabolizador intermedio (IM) y metabolizador lento (PM) asociados a variantes polimórficas del gen CYP2C19 en pacientes con enfermedades del sistema nervioso
Fuente de Datos: R. Cacabelos, 2010
Fuente de Datos: R. Cacabelos, 2010
En las condiciones actuales, la tasa de error promedio en la administración de psicótropos
en España es del 50%

58
Estados Depresivos
Prácticamente, todos los antidepresivos sufren un proceso de biotransformación metabólica en híga-proceso de biotransformación metabólica en híga-proceso de biotransformación metabólica en hígado por acción de las monooxigenasas del citocromo P-450 (CYPs). Existen más de 200 enzimas de la su-P-450 (CYPs). Existen más de 200 enzimas de la su-P-450 (CYPs). Existen más de 200 enzimas de la superfamilia CYP. En el ser humano, los CYPs más im-perfamilia CYP. En el ser humano, los CYPs más im-perfamilia CYP. En el ser humano, los CYPs más importantes, que metabolizan cerca del 90% de todos los fármacos de uso común son CYP2D6, CYP2D6, CYP2D6 CYP2C19, CYP2C19, CYP2C19CYP2C9, CYP2C9, CYP2C9 CYP3A4/5, y otros de menor relevancia CYP3A4/5, y otros de menor relevancia CYP3A4/5genérica. Estas enzimas se codifican en genes de la familia CYP que presentan importantes diferencias étnicas en diversas latitudes geográficas, por lo que un mismo fármaco sufre transformaciones metabóli-un mismo fármaco sufre transformaciones metabóli-un mismo fármaco sufre transformaciones metabólicas distintas en función de las variantes polimórficas que presentan estos genes y que van a dar lugar al fenotipo de normometabolizadores (EMs, Extensive Metabolizers), metabolizadores intermedios (IMs, Intermediate Metabolizers), metabolizadores lentos (PMs, Poor Metabolizers), y metabolizadores ultra-rápidos (UMs, Ultra-Rapid Metabolizers). Los EMs
presentan una función enzimática normal y eficien-te. Los IMs tienen un déficit enzimático causado por mutaciones en uno de los alelos del gen codificante. Los PMs son metabolizadores lentos, carentes de ac-Los PMs son metabolizadores lentos, carentes de ac-Los PMs son metabolizadores lentos, carentes de actividad enzimática por mutaciones en ambos alelos o por deleciones que anulan el gen. Los UMs son metabolizadores rápidos que resultan de la duplica-metabolizadores rápidos que resultan de la duplica-metabolizadores rápidos que resultan de la duplicación, triplicación o multiplicación del gen CYP co-rrespondiente. A dosis convencionales, los PMs no metabolizan el fármaco, éste se acumula en sangre y aparecen efectos secundarios, en ocasiones severos y altamente tóxicos. Los IMs metabolizan el fármaco entre un 25% y un 50% por debajo de lo normal, por lo cual requieren una reducción de la dosis del 30-50%. Los UMs destruyen rápidamente el fárma-30-50%. Los UMs destruyen rápidamente el fárma-30-50%. Los UMs destruyen rápidamente el fármaco y éste no hace el efecto terapéutico deseado. El incremento de la dosis en los UMs no siempre con-duce a un efecto terapéutico y lo más probable es que aparezcan efectos tóxicos con anterioridad a la acción deseada. En PMs y UMs lo ideal es evitar los medicamentos que se metabolizan por la vía CYP deficiente y elegir fármacos cuyo metabolismo siga rutas enzimáticas alternativas eficientes. Los antide-presivos pueden actuar como sustratos, inhibidores o inductores de las enzimas CYP. Un 24% de los an-Un 24% de los an-Un 24% de los antidepresivos son sustratos mayores del CYP1A2, un 5% del CYP2B6, un 38% del CYP2C19, un 85% del CYP2D6, y un 38% del CYP3A4/5 (Fig. 24).Fig. 24).Fig. 24
Aunque no existen grandes diferencias en la distri-Aunque no existen grandes diferencias en la distri-Aunque no existen grandes diferencias en la distribución de frecuencias alélicas y fenotipos CYP en-tre pacientes con diferentes estados depresivos y la población general, no es infrecuente encontrar va-población general, no es infrecuente encontrar va-población general, no es infrecuente encontrar variaciones del 1-5% en nuestro medio y por encima del 10% en otras etnias. En España, un 55.71% de la población es EM para CYP2D6, un 34.7% es IM, CYP2D6, un 34.7% es IM, CYP2D6un 2.28% es PM, y un 7.31% es UM. En pacientes con depresión, la distribución de EMs, IMs, PMs y UMs es del 64.42%, 27.31%, 4.85% y 3.08%, respec-UMs es del 64.42%, 27.31%, 4.85% y 3.08%, respec-UMs es del 64.42%, 27.31%, 4.85% y 3.08%, respectivamente, con diferencias significativas respecto a la población normal, y a pacientes con psicosis, enfer-población normal, y a pacientes con psicosis, enfer-población normal, y a pacientes con psicosis, enfermedad de Parkinson y tumores cerebrales (Fig. 25). Fig. 25). Fig. 25Para CYP2C19, un 68.54% de la población es EM, CYP2C19, un 68.54% de la población es EM, CYP2C19un 30.05% es IM, y un 1.41% es PM. En pacientes depresivos, la frecuencia de CYP2C19 EMs, IMs y CYP2C19 EMs, IMs y CYP2C19PMs es del 77.14%, 22.45% y 0.41%, respectivamen-te (Fig. 26). Para CYP2C9, un 60.56% de la población CYP2C9, un 60.56% de la población CYP2C9española es EM, un 32.39% es IM y un 7.04% es PM. Los pacientes depresivos en España muestran una distribución de CYP2C9 EM, IM y PM del 65.75%, CYP2C9 EM, IM y PM del 65.75%, CYP2C927.4% y 6.85%, respectivamente. Existen claras dife-rencias en la distribución de frecuencias en depresi-rencias en la distribución de frecuencias en depresi-rencias en la distribución de frecuencias en depresivos con respecto a pacientes con déficit de atención e hipercinesia, epilepsia y retraso mental (Fig. 27). Fig. 27). Fig. 27Cuando integramos en clústers trigénicos las varian-tes polimórficas de los genes CYP2D6, CYP2D6, CYP2D6 CYP2C19 y CYP2C19 y CYP2C19CYP2C9, para definir el perfil genómico de nuestra CYP2C9, para definir el perfil genómico de nuestra CYP2C9población, comprobamos que sólo un 26.51% de la población española es normometabolizadora para estos 3 genes, indicando ello que más del 70% de la población es deficiente para la metabolización de fármacos comunes que se catabolizan a través de
Prácticamente, todos los antidepresivos sufren un
Fig. 27. Distribución y frecuencia de fenotipos metabolizador normal (EM), metabolizador intermedio (IM), y metabolizador lento (PM) asociados a variantes polimórficas del gen CYP2C9 en pacientes con enfermedades del sistema nervioso
Fig. 28. Distribución de fenotipos asociados a variantes del clúster trigénico integrado por los genes CYP2D6, CYP2C19 y CYP2C9 en la población española
Fuente de Datos: R. Cacabelos, 2010
Fuente de Datos: R. Cacabelos, 2009

Julio 2011 59
ciencia
estas rutas enzimáticas (Fig. 28).Fig. 28).Fig. 28 El análisis genómico de pacientes psiquiátricos que reciben psicofárma-de pacientes psiquiátricos que reciben psicofárma-de pacientes psiquiátricos que reciben psicofármacos por ensayo y error, según criterio médico, reve-cos por ensayo y error, según criterio médico, reve-cos por ensayo y error, según criterio médico, reveló que cuando prescribimos estos medicamentos desconociendo el perfil farmacogenómico de los pacientes, la tasa de error (y riesgo) es del 56% al dar antidepresivos, del 40% cuando damos neuro-lépticos y del 47% cuando damos benzodiazepinas (Fig. 29). Estos datos reflejan claramente la necesiFig. 29). Estos datos reflejan claramente la necesiFig. 29 -). Estos datos reflejan claramente la necesi-). Estos datos reflejan claramente la necesidad de disponer de un perfil farmacogenómico de los pacientes que requieren psicofármacos. En las condiciones actuales, la tasa de error promedio en la administración de psicótropos en España es del 50%.
FuturoParece evidente que el futuro pasa por la implan-tación irremediable y progresiva de protocolos de intervención farmacogenética para optimizar la se-guridad y la eficacia de los psicofármacos en general y los antidepresivos en particular. Hay una serie de normas esenciales que deberían ser tenidas en cuen-ta a la hora de tratar a los pacientes psiquiátricos y a cualquier paciente que requiere tratamiento cróni-cualquier paciente que requiere tratamiento cróni-cualquier paciente que requiere tratamiento crónico. Algunas normas básicas podrían resumirse de la siguiente manera: (i) antes de iniciar un tratamiento psiquiátrico (antidepresivos, antimaníacos, neuro-lépticos, bendodiazepinas), solicitar un perfil farma-lépticos, bendodiazepinas), solicitar un perfil farma-lépticos, bendodiazepinas), solicitar un perfil farmacogenético del paciente para saber el fármaco que le conviene y las categorías de fármacos que debe evitar de por vida. (ii) Reducir la dosis de fármacos entre un 25% y un 30% en pacientes con perfil IM. (iii) Evitar fármacos que se metabolicen por una ruta CYP determinada en PMs y UMs; buscar soluciones alternativas de medicamentos cuyo metabolismo se realice por otras vías diferentes a las deficitarias o nulas. (iv) Evitar simultanear fármacos con acción inhibidora sobre enzimas CYP porque podrían blo-quear el metabolismo enzimático convirtiendo a pacientes EMs en potenciales PMs. (v) Cuando se requiera la administración de varios psicofármacos para tratar una depresión, un trastorno bipolar, una crisis maníaca o cualquier otra patología psiquiátrica o neurológica que requiera la administración de psi-o neurológica que requiera la administración de psi-o neurológica que requiera la administración de psicofármacos (psicosis, ansiedad, epilepsia, Parkinson, demencia, etc), elegir fármacos que se metabolicen por rutas diferentes, que no sean inhibidores ni in-ductores de rutas CYP. (vi) Prestar especial atención a pacientes polimedicados con patologías crónicas (cardiovasculares, hipertensos, hipercolesterolémi-(cardiovasculares, hipertensos, hipercolesterolémi-(cardiovasculares, hipertensos, hipercolesterolémicos, cerebrovasculares, diabéticos, cancerosos, etc.) y establecer estrategias terapéuticas que minimicen la interacción farmacológica. Especial interés merecen los pacientes seniles, que por la complejidad de sus dolencias pueden tomar de 6 a 10 o más fármacos diarios. Los pacientes con demencia que son por-diarios. Los pacientes con demencia que son por-diarios. Los pacientes con demencia que son portadores del genotipo APOE-4/4, principal factor de APOE-4/4, principal factor de APOE-4/4riesgo para enfermedad de Alzheimer y demencia vascular (Fig. 8), responden peor que los portadores Fig. 8), responden peor que los portadores Fig. 8de otros genotipos a tratamientos convencionales
anti-demencia y a estrategias terapéuticas diversas para reducir el componente depresivo que suele afectar a más de un 40% de los pacientes en algún momento de su enfermedad (Fig. 30). (vii) Antes de Fig. 30). (vii) Antes de Fig. 30etiquetar a un paciente como “resistente”, verificar
Antidepresivos
24%
56%
40%47%
38% 38%
85%
5%0
0
CYP1A2 CYP2B6 CYP2C19
Antidepresivos Neurolépticos Benzodiazepinas
CYP2D6 CYP3A4
20
10
20
30
40
50
60
%
40
60
80
100
%56%
Antidepresivos Neurolépticos Benzodiazepinas
40%
Antidepresivos Neurolépticos BenzodiazepinasAntidepresivos Neurolépticos Benzodiazepinas
47%
Antidepresivos Neurolépticos BenzodiazepinasAntidepresivos Neurolépticos Benzodiazepinas
CYP1A2CYP2B6CYP2C19CYP2D6CYP3A4
anti-demencia y a estrategias terapéuticas diversas
Fig. 29. Tasa de error en la prescripción de psicofármacos cuando se ignora el perfil farmacogenómico de los pacientes psiquiátricos que requieren neurolépticos, antidepresivos o benzodiazepinas
Fig. 30. Efecto antidepresivo de un plan terapéutico multifactorial en pacientes con enfermedad de Alzheimer portadores de diferentes genotipos APOE. Nótese la mejora en la escala de Hamilton para Depresión en todas las variantes polimórficas, excepto en los pacientes con APOE-4/4, a lo largo de un año de tratamiento
Fuente de Datos: R. Cacabelos, 2008
Fuente de Datos: R. Cacabelos, 2009
La personalización del tratamiento antidepresivo, mediante el conocimiento de las características genómicas de cada paciente, permite optimizar
los recursos terapéuticos disponibles, mejorando el nivel de eficacia de los fármacos, reduciendo
efectos secundarios y racionalizando el coste que supone un tratamiento crónico con antidepresivos

60
Estados Depresivos
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition, Washington DC, 1994.
Berrios G, Porter R. A History of Clinical Psychiatry. Athlone Press, London, 1995.
Sullivan, JP. Psychopharmacology: The fourth generation of progress. In: Bloom FE. Kupfer DJ (Eds). Drug Development Research. Raven Press, Ltd., New York, 1995, 185-188.
Bond DJ, Lang DJ, Noronha MM, et al. The Association of Elevated Body Mass Index with Reduced Brain Volumes in First-Episode Mania. Biol Psychiatry 2011. doi: 10.1016/j.biops-ych.2011.02.025.
Brown AD, Barton DA, Lambert GW. Cardiovascular abnormalities in patients with major depres-sive disorder: autonomic mechanisms and implications for treatment. CNS Drugs 2009; 23:583-602.
Cacabelos, R. Molecular pathology and pharmacogenomics in Alzheimer’s disease: Polygenic-related effects of multifactorial treatments on cognition, anxiety, and depression. Meth. Find. Exper. Clin. Pharmacol 2007; 29(Suppl. A):1-91.
Cacabelos R. Pharmacogenomics in Alzheimer’s disease. Meth Mol Biol 2008; 448:213-357.
Cacabelos R. Pharmacogenomic biomarkers in neuropsychiatry: The path to personalized me-dicine in mental disorders. In: Ritsner MS (Eds). The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes. Springer, Netherlands, 2009:3-63.
Cacabelos R. Pharmacogenomics and therapeutic strategies for dementia. Expert Rev Mol Diag 2009; 9:567-611.
Cacabelos R (Ed). World Guide for Drug Use and Pharmacogenomics. EuroEspes Publishing, Coruña, (2011) (In press).
Cacabelos R, Fernández-Novoa L, Lombardi V et al. Genomics of schizophrenia and psychotic disorders. EuroEspes J, Gen-T International, 2011; 3:6-86.
Cacabelos R, Hashimoto R, Takeda M. Pharmacogenomics of antipsychotics efficacy for schizo-phrenia. Psychiat Clin Neurosci 2011; 65:3-19.
Cacabelos, R. and Martínez-Bouza, R. Genomics and Pharmacogenomics of Dementia. CNS Neu-roscience & Therapeutics, 2011. doi: 10.1111/j.1755-5949.2010.00189.x.
Cacabelos R, Martínez-Bouza R, Fernández-Novoa L et al. Personalized medicine. Pharmacoge-nomics of Alzheimer’s disease. Gen-T 2009; 4:18-48.
Drevets WC. Functional neuroimaging studies of depression. Annu Rev Med 1998; 49:341-61.
Gao SF, Bao AM. Corticotropin-releasing hormone, glutamate, and γ-aminobutyric acid in depression. Neuroscientist 2011; 17:124-44.
Glatt SJ, Cohen OS, Faraone SV, Tsuang MT. Dysfunctional gene splicing as a potential contributor to neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet 2011. doi: 10.1002/ajmg.b.31181.
Hashimoto K. Emerging role of glutamate in the pathophysiology of major depressive disor-der. Brain Res Rev 2009; 61:105-23.
Hughes ZA, Liu F, Marquis K et al. Estrogen receptor neurobiology and its potential for translation into broad spectrum therapeutics for CNS disorders. Curr Mol Pharmacol 2009; 2:215-36.
López-Muñoz F, Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des 2009; 15:1563-86.
Mahon K, Burdick KE, Szeszko PR. A role for white matter abnormalities in the pathophysiology of bipolar disorder. Neurosci Biobehav Rev 2010; 34:533-54.
Martinowich K, Schloesser RJ, Manji HK. Bipolar disorder: from genes to behavior pathways. J Clin Invest 2009; 119:726-36. doi: 10.1172/JCI37703.
Roussos P, Giakoumaki SG, Georgakopoulos A et al. The CACNA1C and ANK3 risk alleles impact on affective personality traits and startle reactivity but not on cognition or gating in healthy males. Bipolar Disord 2011. doi: 10.1111/j.1399-5618.2011.00924.x.
Peter CJ, Akbarian S. Balancing histone methylation activities in psychiatric disorders. Trends Mol Med 2011. doi: 10.1016/j.molmed.2011.02.003.
Sadock BJ, Sadock VA. Comprehensive Textbook of Psychiatry. 7th Ed. Lippincott Williams & Wilkins, Philadelphia, 2000; 1284-1440.
Sales AJ, Biojone C, Terceti MS et al. Antidepressant-like effect induced by systemic and intra-hippocampal administration of DNA methylation inhibitors. Br J Pharmacol 2011. doi: 10.1111/j.1476-5381.2011.01489.x.
Schatzberg AF, Nemeroff CB, Textbook of Psychopharmacology. The American Psychiatric Press, Washington DC, 1995.
Weber H, Kittel-Schneider S, Gessner A et al. Cross-Disorder Analysis of Bipolar Risk Genes: Further Evidence of DGKH as a Risk Gene for Bipolar Disorder, but also Unipolar Depression and Adult ADHD. Neuropsychopharmacology 2011. doi: 10.1038/npp.2011.98.
Referencias Bibliográficas:
su perfil genómico y farmacogenómico, potencial responsable del fracaso terapéutico en más de un 60% de los casos. (viii) Evitar la coadministración de fármacos con reconocido potencial interactivo. Anti-inflamatorios no esteroideos, antihipertensivos, esta-inflamatorios no esteroideos, antihipertensivos, esta-inflamatorios no esteroideos, antihipertensivos, estatinas, antifúngicos, antineoplásicos, anticoagulantes y anticonceptivos suelen dar complicaciones serias cuando se administran conjuntamente con algunos psicofármacos. (ix) Evitar antiácidos y “protectores gástricos”, salvo en casos excepcionales, porque pueden alterar la absorción de los psicofármacos. (x) Evitar medicinas alternativas de origen vegetal, infusiones y productos de herbolario. Un 30% de estos productos pueden causar interacciones graves al actuar como inhibidores o inductores de las enzi-al actuar como inhibidores o inductores de las enzi-al actuar como inhibidores o inductores de las enzimas CYP.
Con la implementación gradual de estos procesos basados en el conocimiento de la farmacogenómica podrían reducirse en más de un 40% los problemas de seguridad farmacológica (efectos secundarios,
toxicidad, reacciones catastróficas) e incrementar, en un 20% de los casos, la eficacia terapéutica. Los costes derivados de la administración de tratamien-tos para neutralizar efectos secundarios a psicofár-tos para neutralizar efectos secundarios a psicofár-tos para neutralizar efectos secundarios a psicofármacos puede alcanzar un 10-20% de los costes di-macos puede alcanzar un 10-20% de los costes di-macos puede alcanzar un 10-20% de los costes directos del tratamiento farmacológico en pacientes psiquiátricos. En términos globales, la instauración de la farmacogenómica en el tratamiento de las en-fermedades del sistema nervioso central podría su-fermedades del sistema nervioso central podría su-fermedades del sistema nervioso central podría suponer un ahorro del 18-32% en gasto farmacéutico en un horizonte de 2-3 años.
Ramón Cacabelos [email protected]

Julio 2011 61
ciencia
Tabla 1. Genes potencialmente asociados a estados depresivos y trastorno bipolar
Locus Símbolo Nombre
1p22 CMM4 melanoma, cutaneous malignant, 4
1p31 PDE4B phosphodiesterase 4B, cAMP-specific
1p32-p31 DAB1 disabled homolog 1 (Drosophila)
1p33-p32 ARTN artemin
1p34 HDAC1 histone deacetylase 1
1p34-p33 GRIK3 glutamate receptor, ionotropic, kainate 3
1p35-p34 FAAH fatty acid amide hydrolase
1p36 CMM cutaneous malignant melanoma/dysplastic nevus
1p36.23 PER3 period homolog 3 (Drosophila)
1p36.3 MTHFR methylenetetrahydrofolate reductase (NAD(P)H)
1p36.3-p34.3 HTR1D 5-hydroxytryptamine (serotonin) receptor 1D
1p36-p35 HTR6 5-hydroxytryptamine (serotonin) receptor 6
1q21 BCL9 B-cell CLL/lymphoma 9
1q21 GBA glucosidase, beta, acid
1q21-q22 NTRK1 neurotrophic tyrosine kinase, receptor, type 1
1q21-q23 CRP C-reactive protein, pentraxin-related
1q23.3 NOS1AP nitric oxide synthase 1 (neuronal) adaptor protein
1q23.3 RGS4 regulator of G-protein signaling 4
1q24.3 FMO1 flavin containing monooxygenase 1
1q25 PLA2G4A phospholipase A2, group IVA (cytosolic, calcium-dependent)
1q25.2-q25.3 PTGS2 prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase)
1q31-q32 IL10 interleukin 10
1q31-q32 PTPRC protein tyrosine phosphatase, receptor type, C
1q32.1 CHI3L1 chitinase 3-like 1 (cartilage glycoprotein-39)
1q42.1 DISC1 disrupted in schizophrenia 1
1q42.1 DISC2 disrupted in schizophrenia 2 (non-protein coding)
1q42.1 TSNAX translin-associated factor X
1q43 RYR2 ryanodine receptor 2 (skeletal)
2p11.2 TGOLN2 trans-golgi network protein 2
2p15-p13 SLC1A4 solute carrier family 1 (glutamate/neutral amino acid transporter), member 4
2p16.3RTN4, NOGO-A, Nogo-B, Nogo-CNogo-B, Nogo-C
reticulon 4
2p22-p21 CAD carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase
2p23.3 ADCY3 adenylate cyclase 3
2q11.2 NPAS2 neuronal PAS domain protein 2
2q14 IL1B interleukin 1, beta
2q14.2 IL1RN interleukin 1 receptor antagonist
2q22-q23 NR4A2, NURR1 nuclear receptor subfamily 4, group A, member 2
2q24 TBR1 T-box, brain, 1
2q31 GAD1, Gad67 glutamate decarboxylase 1 (brain, 67kDa)
2q32 DLX1 distal-less homeobox 1
2q32 INPP1 inositol polyphosphate-1-phosphatase
2q33 CTLA4 cytotoxic T-lymphocyte-associated protein 4
2q33.3-q34 ERBB4 v-erb-a erythroblastic leukemia viral oncogene homolog 4 (avian)
62
Estados Depresivos
Locus Símbolo Nombre
2q34 CREB1 cAMP responsive element binding protein 1
2q34-q35 MAP2 microtubule-associated protein 2
2q37.3 HDAC4 histone deacetylase 4
2q37.3 PER2 period homolog 2 (Drosophila)
3p21.2 GRM2 glutamate receptor, metabotropic 2
3p21.3 LARS2 leucyl-tRNA synthetase 2, mitochondrial
3p22.1 MOBP myelin-associated oligodendrocyte basic protein
3p22-p21.3 CCK cholecystokinin
3p25 HRH1 histamine receptor H1
3p25 OXTR oxytocin receptor
3p25 SYN2 synapsin II
3p26BHLHE40, BHLHB2BHLHB2
basic helix-loop-helix family, member e40
3p26.1-p25.1 GRM7 glutamate receptor, metabotropic 7
3q12 ABI3BP ABI family, member 3 (NESH) binding protein
3q12-q13.3 NR1I2 nuclear receptor subfamily 1, group I, member 2
3q13.1-q13.2 GAP43 growth associated protein 43
3q13.2-q21 ADCY5 adenylate cyclase 5
3q13.3 DRD3 dopamine receptor D3
3q13.3 GSK3B glycogen synthase kinase 3 beta
3q22.1 TF transferrin
4p12 GABRA2 gamma-aminobutyric acid (GABA) A receptor, alpha 2
4p15.2 PI4K2B phosphatidylinositol 4-kinase type 2 beta
4p16 ADRA2C adrenergic, alpha-2C-, receptor
4p16 WFS1 Wolfram syndrome 1 (wolframin)
4p16.1 DRD5 dopamine receptor D5
4p16.3 CPLX1 complexin 1
4q12 CLOCK clock homolog (mouse)
4q12 REST RE1-silencing transcription factor
4q22 PDLIM5 PDZ and LIM domain 5
4q23 ADH1B alcohol dehydrogenase 1B (class I), beta polypeptide
4q31 NPY2R neuropeptide Y receptor Y2
4q31.1 NR3C2 nuclear receptor subfamily 3, group C, member 2
4q31.3-q32 NPY1R neuropeptide Y receptor Y1
4q35.1 MTNR1A melatonin receptor 1A
5p13 SLC1A3 solute carrier family 1 (glial high affinity glutamate transporter), member 3
5p13 IL7R interleukin 7 receptor
5p13.1-p12 GDNF glial cell derived neurotrophic factor
5p15.3 SLC6A3, DAT solute carrier family 6 (neurotransmitter transporter, dopamine), member 3
5q11.2-q13 HTR1A 5-hydroxytryptamine (serotonin) receptor 1A
5q12 PDE4D phosphodiesterase 4D, cAMP-specific
5q23 HBEGF heparin-binding EGF-like growth factor
5q31.1 GRIA1 glutamate receptor, ionotropic, AMPA 1
5q31.1 HSPA4, Hsp70 heat shock 70kDa protein 4
5q31.1 TRPC7 transient receptor potential cation channel, subfamily C, member 7
Julio 2011 63
ciencia
Locus Símbolo Nombre
5q31.2 HINT1 HINT1 histidine triad nucleotide binding protein 1
5q31.3 NR3C1 nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor)
5q31-q33 HTR4 5-hydroxytryptamine (serotonin) receptor 4
5q32 CAMK2A calcium/calmodulin-dependent protein kinase II alpha
5q33.3 ADRA1B adrenergic, alpha-1B-, receptor
5q34 GABRA1 gamma-aminobutyric acid (GABA) A receptor, alpha 1
5q35.1 DRD1 dopamine receptor D1
6p21 MDGA1 MAM domain containing glycosylphosphatidylinositol anchor 1
6p21.3 GRM4 glutamate receptor, metabotropic 4
6p21.3 TNF tumor necrosis factor
6p21.3 HLA-DQB1 major histocompatibility complex, class II, DQ beta 1
6p21.3 HLA-DRB1 major histocompatibility complex, class II, DR beta 1
6p21.31 FKBP5 FK506 binding protein 5
6p21.31 GABBR1 gamma-aminobutyric acid (GABA) B receptor, 1
6p22.1 MOG myelin oligodendrocyte glycoprotein
6p22.3 DTNBP1 dystrobrevin binding protein 1
6p24-p22 SCZD3 schizophrenia disorder 3
6q13 HTR1B 5-hydroxytryptamine (serotonin) receptor 1B
6q14-q15 CNR1 cannabinoid receptor 1 (brain)
6q16.3-q21 GRIK2 glutamate receptor, ionotropic, kainate 2
6q21 HDAC2 histone deacetylase 2
6q21 CD24 CD24 molecule
6q23 SGK1 serum/glucocorticoid regulated kinase 1
6q23.2 TAAR6 trace amine associated receptor 6
6q24-q25 OPRM1 opioid receptor, mu 1
6q25 VIP vasoactive intestinal peptide
6q25.1 ESR1 estrogen receptor 1
6q25.3 SOD2 superoxide dismutase 2, mitochondrial
7p12 EGFR epidermal growth factor receptor
7p12.2 DDC dopa decarboxylase (aromatic L-amino acid decarboxylase)
7p12.3 ABCA13 ATP-binding cassette, sub-family A (ABC1), member 13
7p14.3 CRHR2 corticotropin releasing hormone receptor 2
7p15.1 NPY neuropeptide Y
7p21.2 DGKB diacylglycerol kinase, beta 90kDa
7q21.1 CYP3A4/5 cytochrome P450, family 3, subfamily A, polypeptide 4/5
7q21.12 ABCB1 ATP-binding cassette, sub-family B (MDR/TAP), member 1
7q21.1-q21.2 GRM3 glutamate receptor, metabotropic 3
7q21-q22 TAC1 tachykinin, precursor 1
7q22 RELN reelin
7q22.1 VGF VGF nerve growth factor inducible
7q31 KCND2 potassium voltage-gated channel, Shal-related subfamily, member 2
7q31.3 CADPS2 Ca++-dependent secretion activator 2
7q34-q35 TBXAS1 thromboxane A synthase 1 (platelet)
7q36.1 HTR5A 5-hydroxytryptamine (serotonin) receptor 5A

64
Estados Depresivos
Locus Símbolo Nombre
7q36.3 VIPR2 vasoactive intestinal peptide receptor 2
8p11.2 CHRNB3 cholinergic receptor, nicotinic, beta 3
8p11.21 CHRNA6 cholinergic receptor, nicotinic, alpha 6
8p11.21 SFRP1 secreted frizzled-related protein 1
8p11.2-p11.1 DKK4 dickkopf homolog 4 (Xenopus laevis)
8p12 ADRB3 adrenergic, beta-3-, receptor
8p12 FGFR1 fibroblast growth factor receptor 1
8p12 NRG1 neuregulin 1
8p12 PLAT plasminogen activator, tissue
8p21 CHRNA2 cholinergic receptor, nicotinic, alpha 2 (neuronal)
8p21 FGF17 fibroblast growth factor 17
8p21 FZD3 frizzled family receptor 3
8p21 FZD3 frizzled family receptor 3
8p21 NEFM, NEF3 neurofilament, medium polypeptide
8p21 SCZD6 schizophrenia disorder 6
8p21.1 FBXO16 F-box protein 16
8p21.2 ADRA1A adrenergic, alpha-1A-, receptor
8p21.3 PPP3CC protein phosphatase 3, catalytic subunit, gamma isozyme
8p21.3 SLC18A1 solute carrier family 18 (vesicular monoamine), member 1
8p22 FGF20 fibroblast growth factor 20
8p22 NAT2 N-acetyltransferase 2 (arylamine N-acetyltransferase)
8p22-p21 DPYSL2 dihydropyrimidinase-like 2
8p22-p21.3 PCM1 pericentriolar material 1
8p23 ARHGEF10 Rho guanine nucleotide exchange factor (GEF) 10
8p23-p21 EGR3 early growth response 3
8q11.2 OPRK1, KOR opioid receptor, kappa 1
8q13 CRH corticotropin releasing hormone
8q21.13-q21.3 IMPA1 inositol(myo)-1(or 4)-monophosphatase 1
8q23-q24 PENK proenkephalin
9p11.1 ALDH1B1 aldehyde dehydrogenase 1 family, member B1
9p12 BAG1 BCL2-associated athanogene
9p13.1 CNTNAP3 contactin associated protein-like 3
9p13.3 CREB3 cAMP responsive element binding protein 3
9p24 VLDLR very low density lipoprotein receptor
9q22.1 NTRK2 neurotrophic tyrosine kinase, receptor, type 2
9q31.1 GRIN3A glutamate receptor, ionotropic, N-methyl-D-aspartate 3A
9q34 DBH dopamine beta-hydroxylase (dopamine beta-monooxygenase)
9q34 TOR1A torsin family 1, member A (torsin A)
9q34.3 GRIN1 glutamate receptor, ionotropic, N-methyl D-aspartate 1
10p11.23 GAD2, Gad65 glutamate decarboxylase 2 (pancreatic islets and brain, 65kDa)
10p12.2PIP4K2A, PIP5K2APIP5K2A
phosphatidylinositol-5-phosphate 4-kinase, type II, alpha
10q21 ANK3 ankyrin 3, node of Ranvier (ankyrin G)
10q22.3 SCZD11 schizophrenia susceptibility locus, chromosome 10q-related
10q24 CYP2C9 cytochrome P450, family 2, subfamily C, polypeptide 9

Julio 2011 65
ciencia
Locus Símbolo Nombre
10q24 CYP2C19 cytochrome P450, family 2, subfamily C, polypeptide 19
10q24 C10orf2 chromosome 10 open reading frame 2
10q24-q26 ADRA2A adrenergic, alpha-2A-, receptor
10q24-q26 ADRB1 adrenergic, beta-1-, receptor
10q25 SLC18A2 solute carrier family 18 (vesicular monoamine), member 2
11p11.2 CRY2 cryptochrome 2 (photolyase-like)
11p13 BDNF brain-derived neurotrophic factor
11p13-p12 SLC1A2 solute carrier family 1 (glial high affinity glutamate transporter), member 2
11p15 ARNTL, BMAL1 aryl hydrocarbon receptor nuclear translocator-like
11p15.2 CALCA calcitonin-related polypeptide alpha
11p15.3-p14 TPH1 tryptophan hydroxylase 1
11p15.4 ADM adrenomedullin
11p15.5 DRD4 dopamine receptor D4
11q13 MEN1 multiple endocrine neoplasia I
11q13.3 GAL galanin prepropeptide
11q14-q21 SCZD2 schizophrenia disorder 2
11q21-q22 MTNR1B melatonin receptor 1B
11q22.2-q22.3 IL18 interleukin 18 (interferon-gamma-inducing factor)
11q22.3 GRIK4 glutamate receptor, ionotropic, kainate 4
11q23 ALG9, DIBD1 asparagine-linked glycosylation 9, alpha-1,2-mannosyltransferase homolog (S. cerevisiae)
11q23 DRD2 dopamine receptor D2
11q23.1 HTR3B 5-hydroxytryptamine (serotonin) receptor 3B
11q23.1 NCAM1 neural cell adhesion molecule 1
12p12 GRIN2B glutamate receptor, ionotropic, N-methyl D-aspartate 2B
12p12.1BHLHE41, BHLHB3BHLHB3
basic helix-loop-helix family, member e41
12p12.2 SLCO1C1 solute carrier organic anion transporter family, member 1C1
12p12.2-p11.2 ARNTL2 aryl hydrocarbon receptor nuclear translocator-like 2
12p13 NTF3 nurotrophin 3
12p13 GNB3 guanine nucleotide binding protein (G protein), beta polypeptide 3
12p13.3 CACNA1C calcium channel, voltage-dependent, L type, alpha 1C subunit
12q12-q13 TIMELESS timeless homolog (Drosophila)
12q13 SCN8A sodium channel, voltage gated, type VIII, alpha subunit
12q13.13 RNF41 ring finger protein 41
12q14 IFNG interferon, gamma
12q21 PAWR PRKC, apoptosis, WT1, regulator
12q21.1 TPH2 tryptophan hydroxylase 2
12q22-q23 DUSP6 dual specificity phosphatase 6
12q22-q23.2 MDD1 major depressive disorder 1
12q23-q24.1 CRY1 cryptochrome 1 (photolyase-like)
12q24 CIT citron (rho-interacting, serine/threonine kinase 21)
12q24 DAO D-amino-acid oxidase
12q24 P2RX7 purinergic receptor P2X, ligand-gated ion channel, 7
12q24.11 ATP2A2 ATPase, Ca++ transporting, cardiac muscle, slow twitch 2
12q24.12 CUX2 cut-like homeobox 2

66
Estados Depresivos
Locus Símbolo Nombre
12q24.12FAM109A, FLJ32356FLJ32356
family with sequence similarity 109, member A
12q24.2 CAMKK2 calcium/calmodulin-dependent protein kinase kinase 2, beta
12q24.2-q24.31 NOS1 nitric oxide synthase 1 (neuronal)
13q14.11 DGKH diacylglycerol kinase, eta
13q14-q21 HTR2A 5-hydroxytryptamine (serotonin) receptor 2A
13q32 SCZD7 schizophrenia disorder 7
13q32-33 DAOA/G30 (D-amino acid oxidase activator) gene complex
13q34 DAOA, G72 D-amino acid oxidase activator
13q34 DAOA-AS1, G30 DAOA antisense RNA 1 (non-protein coding)
14q22.1-q22.2 GCH1 GTP cyclohydrolase 1
14q22.3 OTX2 orthodenticle homeobox 2
14q23.2 ESR2 estrogen receptor 2 (ER beta)
14q24.2-q24.3 DIO2 deiodinase, iodothyronine, type II
14q24.3 FOS FBJ murine osteosarcoma viral oncogene homolog
14q31 GALC galactosylceramidase
14q32.1-q32.2 BDKRB2 bradykinin receptor B2
14q32.32 AKT1 v-akt murine thymoma viral oncogene homolog 1
15q11.2-q12 GABRA5 gamma-aminobutyric acid (GABA) A receptor, alpha 5
15q11.2-q12 GABRB3 gamma-aminobutyric acid (GABA) A receptor, beta 3
15q13 SLC12A6 solute carrier family 12 (potassium/chloride transporters), member 6
15q14-q15 RYR3 ryanodine receptor 3
15q15 SCZD10 schizophrenia disorder 10 (periodic catatonia)
15q22 ADAM10 ADAM metallopeptidase domain 10
15q22.2 RORA RAR-related orphan receptor A
15q24.1 CYP1A2 cytochrome P450, family 1, subfamily A, polypeptide 2
15q25 NTRK3 neurotrophic tyrosine kinase, receptor, type 3
15q25 POLG polymerase (DNA directed), gamma
15q25.3-q26.2 MDD2 major depressive disorder 2
16p13.2 GRIN2A glutamate receptor, ionotropic, N-methyl D-aspartate 2A
16p13.3 ADCY9 adenylate cyclase 9
16p13.3 PGP phosphoglycolate phosphatase
16q12.2 SLC6A2, NET solute carrier family 6 (neurotransmitter transporter, noradrenalin), member 2
16q22.1 HP haptoglobin
16q22.1 NQO1 NAD(P)H dehydrogenase, quinone 1
16q24.3 MC1R melanocortin 1 receptor (alpha melanocyte stimulating hormone receptor)
17p13 ARRB2 arrestin, beta 2
17p13.1 ALOX12 arachidonate 12-lipoxygenase
17p13.1 DLG4, PSD95 discs, large homolog 4 (Drosophila)
17p13.1 PER1 period homolog 1 (Drosophila)
17p13.3 YWHAE tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, epsilon polypeptide
17q11.2 NR1D1 nuclear receptor subfamily 1, group D, member 1
17q11.2SLC6A4, 5-HTTLPR, SERTPRSERTPR
solute carrier family 6 (neurotransmitter transporter, dopamine), member 4
17q11.2-q12 CCL2, MCP-1 chemokine (C-C motif) ligand 2
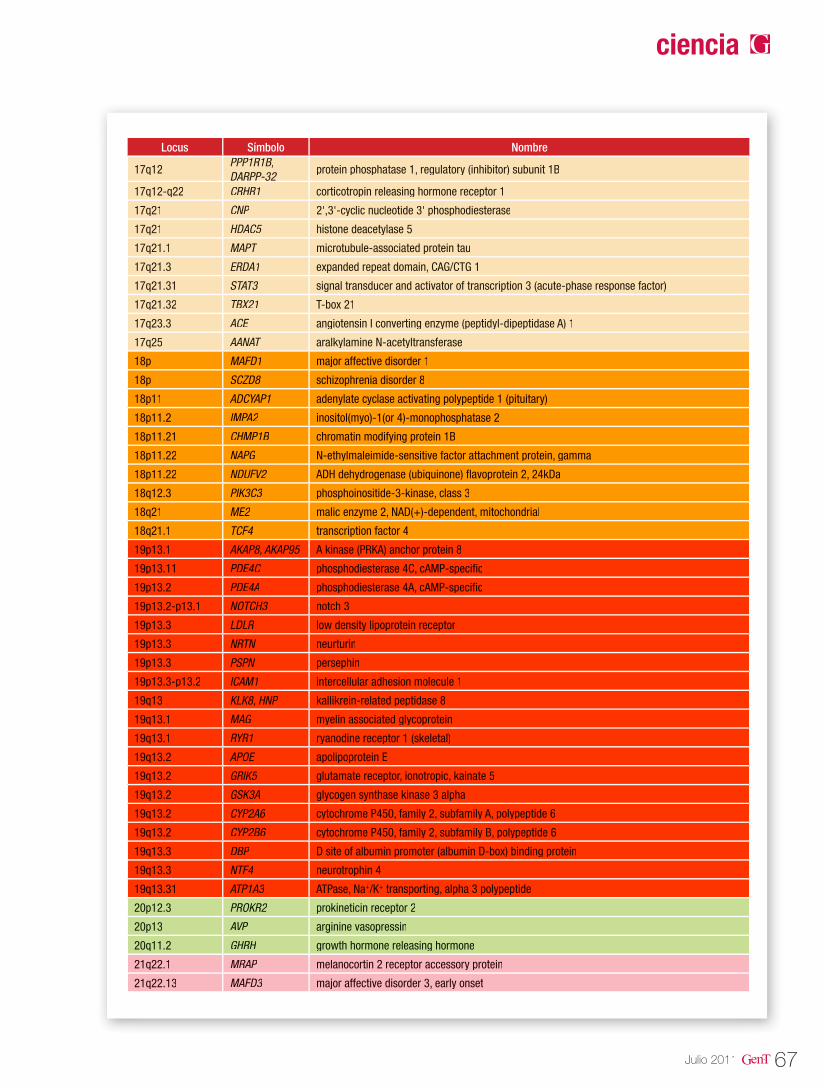
Julio 2011 67
ciencia
Locus Símbolo Nombre
17q12PPP1R1B, DARPP-32DARPP-32
protein phosphatase 1, regulatory (inhibitor) subunit 1B
17q12-q22 CRHR1 corticotropin releasing hormone receptor 1
17q21 CNP 2',3'-cyclic nucleotide 3' phosphodiesterase
17q21 HDAC5 histone deacetylase 5
17q21.1 MAPT microtubule-associated protein tau
17q21.3 ERDA1 expanded repeat domain, CAG/CTG 1
17q21.31 STAT3 signal transducer and activator of transcription 3 (acute-phase response factor)
17q21.32 TBX21 T-box 21
17q23.3 ACE angiotensin I converting enzyme (peptidyl-dipeptidase A) 1
17q25 AANAT aralkylamine N-acetyltransferase
18p MAFD1 major affective disorder 1
18p SCZD8 schizophrenia disorder 8
18p11 ADCYAP1 adenylate cyclase activating polypeptide 1 (pituitary)
18p11.2 IMPA2 inositol(myo)-1(or 4)-monophosphatase 2
18p11.21 CHMP1B chromatin modifying protein 1B
18p11.22 NAPG N-ethylmaleimide-sensitive factor attachment protein, gamma
18p11.22 NDUFV2 ADH dehydrogenase (ubiquinone) flavoprotein 2, 24kDa
18q12.3 PIK3C3 phosphoinositide-3-kinase, class 3
18q21 ME2 malic enzyme 2, NAD(+)-dependent, mitochondrial
18q21.1 TCF4 transcription factor 4
19p13.1 AKAP8, AKAP95 A kinase (PRKA) anchor protein 8
19p13.11 PDE4C phosphodiesterase 4C, cAMP-specific
19p13.2 PDE4A phosphodiesterase 4A, cAMP-specific
19p13.2-p13.1 NOTCH3 notch 3
19p13.3 LDLR low density lipoprotein receptor
19p13.3 NRTN neurturin
19p13.3 PSPN persephin
19p13.3-p13.2 ICAM1 intercellular adhesion molecule 1
19q13 KLK8, HNP kallikrein-related peptidase 8
19q13.1 MAG myelin associated glycoprotein
19q13.1 RYR1 ryanodine receptor 1 (skeletal)
19q13.2 APOE apolipoprotein E
19q13.2 GRIK5 glutamate receptor, ionotropic, kainate 5
19q13.2 GSK3A glycogen synthase kinase 3 alpha
19q13.2 CYP2A6 cytochrome P450, family 2, subfamily A, polypeptide 6
19q13.2 CYP2B6 cytochrome P450, family 2, subfamily B, polypeptide 6
19q13.3 DBP D site of albumin promoter (albumin D-box) binding protein
19q13.3 NTF4 neurotrophin 4
19q13.31 ATP1A3 ATPase, Na+/K+ transporting, alpha 3 polypeptide
20p12.3 PROKR2 prokineticin receptor 2
20p13 AVP arginine vasopressin
20q11.2 GHRH growth hormone releasing hormone
21q22.1 MRAP melanocortin 2 receptor accessory protein
21q22.13 MAFD3 major affective disorder 3, early onset
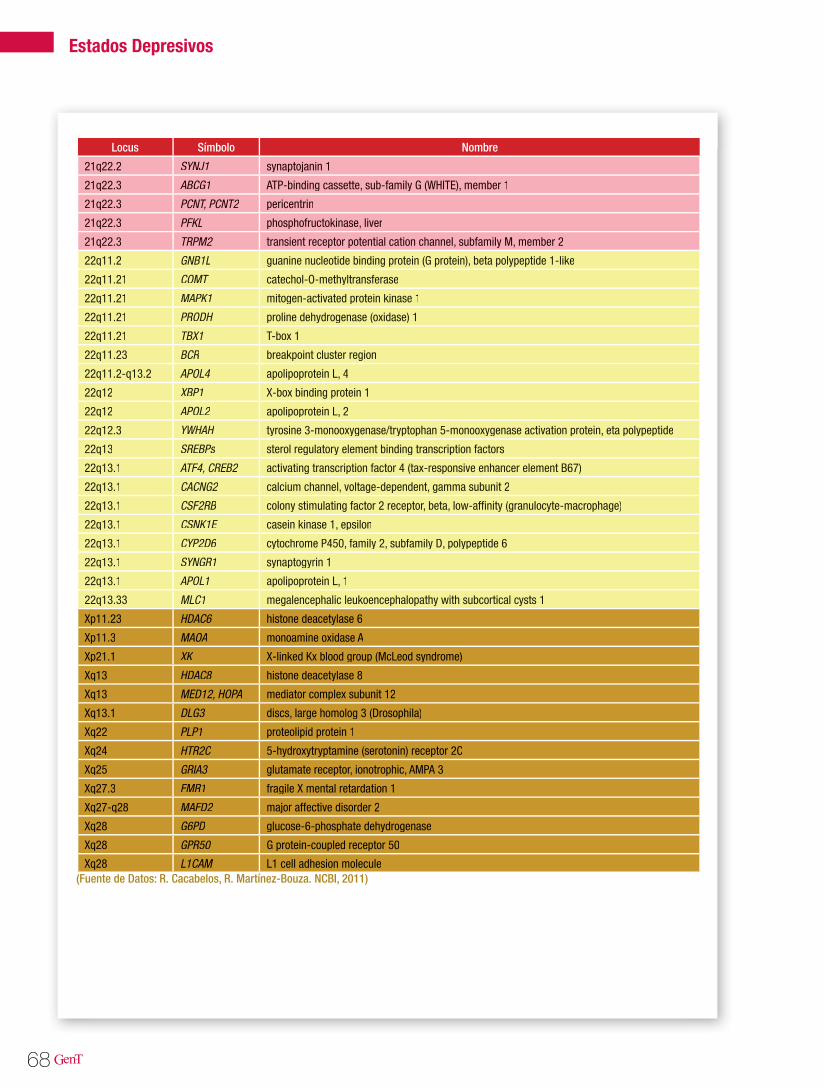
68
Estados Depresivos
Locus Símbolo Nombre
21q22.2 SYNJ1 synaptojanin 1
21q22.3 ABCG1 ATP-binding cassette, sub-family G (WHITE), member 1
21q22.3 PCNT, PCNT2 pericentrin
21q22.3 PFKL phosphofructokinase, liver
21q22.3 TRPM2 transient receptor potential cation channel, subfamily M, member 2
22q11.2 GNB1L guanine nucleotide binding protein (G protein), beta polypeptide 1-like
22q11.21 COMT catechol-O-methyltransferase
22q11.21 MAPK1 mitogen-activated protein kinase 1
22q11.21 PRODH proline dehydrogenase (oxidase) 1
22q11.21 TBX1 T-box 1
22q11.23 BCR breakpoint cluster region
22q11.2-q13.2 APOL4 apolipoprotein L, 4
22q12 XBP1 X-box binding protein 1
22q12 APOL2 apolipoprotein L, 2
22q12.3 YWHAH tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, eta polypeptide
22q13 SREBPs sterol regulatory element binding transcription factors
22q13.1 ATF4, CREB2 activating transcription factor 4 (tax-responsive enhancer element B67)
22q13.1 CACNG2 calcium channel, voltage-dependent, gamma subunit 2
22q13.1 CSF2RB colony stimulating factor 2 receptor, beta, low-affinity (granulocyte-macrophage)
22q13.1 CSNK1E casein kinase 1, epsilon
22q13.1 CYP2D6 cytochrome P450, family 2, subfamily D, polypeptide 6
22q13.1 SYNGR1 synaptogyrin 1
22q13.1 APOL1 apolipoprotein L, 1
22q13.33 MLC1 megalencephalic leukoencephalopathy with subcortical cysts 1
Xp11.23 HDAC6 histone deacetylase 6
Xp11.3 MAOA monoamine oxidase A
Xp21.1 XK X-linked Kx blood group (McLeod syndrome)
Xq13 HDAC8 histone deacetylase 8
Xq13 MED12, HOPA mediator complex subunit 12
Xq13.1 DLG3 discs, large homolog 3 (Drosophila)
Xq22 PLP1 proteolipid protein 1
Xq24 HTR2C 5-hydroxytryptamine (serotonin) receptor 2C
Xq25 GRIA3 glutamate receptor, ionotrophic, AMPA 3
Xq27.3 FMR1 fragile X mental retardation 1
Xq27-q28 MAFD2 major affective disorder 2
Xq28 G6PD glucose-6-phosphate dehydrogenase
Xq28 GPR50 G protein-coupled receptor 50
Xq28 L1CAM L1 cell adhesion molecule(Fuente de Datos: R. Cacabelos, R. Martínez-Bouza. NCBI, 2011)
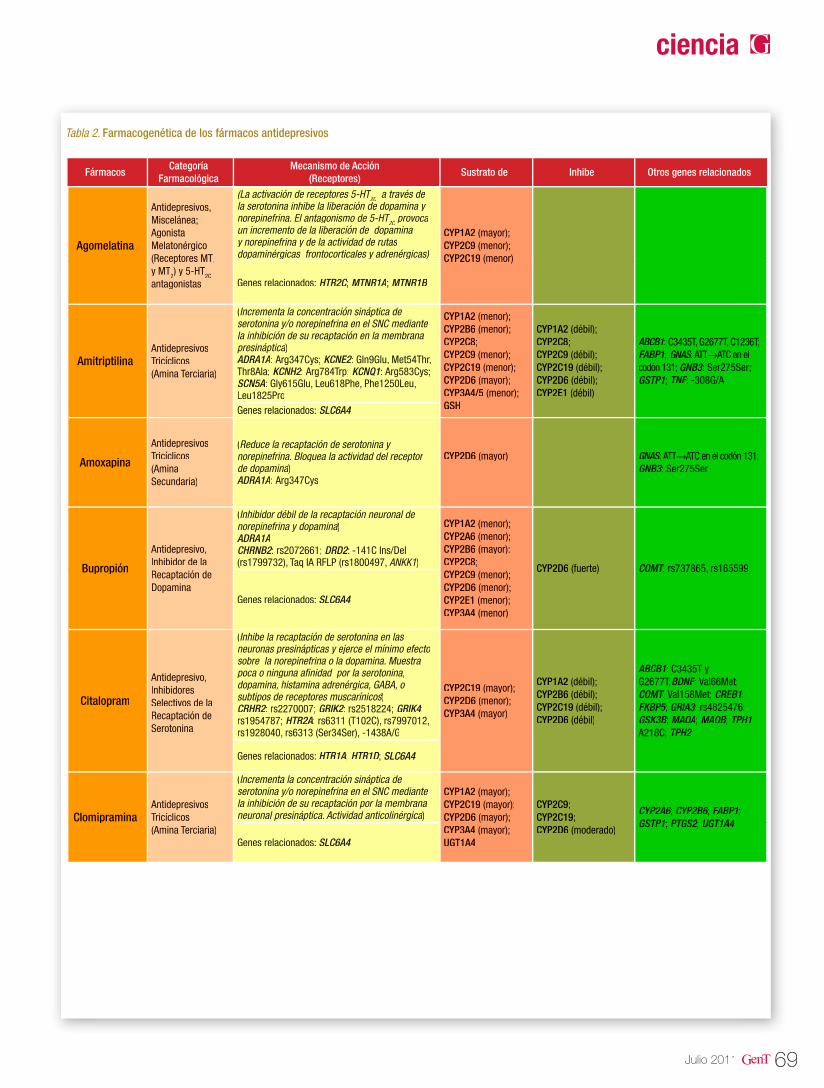
Julio 2011 69
ciencia
Tabla 2. Farmacogenética de los fármacos antidepresivos
FármacosCategoría
FarmacológicaMecanismo de Acción
(Receptores)Sustrato de Inhibe Otros genes relacionados
Agomelatina
Antidepresivos, Miscelánea; Agonista Melatonérgico (Receptores MT1
y MT2) y 5-HT2C
antagonistas
(La activación de receptores 5-HT2C(La activación de receptores 5-HT2C(La activación de receptores 5-HT a través de 2C a través de 2Cla serotonina inhibe la liberación de dopamina y norepinefrina. El antagonismo de 5-HT2Cnorepinefrina. El antagonismo de 5-HT2Cnorepinefrina. El antagonismo de 5-HT provoca 2C provoca 2Cun incremento de la liberación de dopamina y norepinefrina y de la actividad de rutas dopaminérgicas frontocorticales y adrenérgicas)
CYP1A2 (mayor); CYP2C9 (menor); CYP2C19 (menor)
Genes relacionados: HTR2C; HTR2C; HTR2C MTNR1A; MTNR1B
AmitriptilinaAntidepresivos Tricíclicos (Amina Terciaria)
(Incrementa la concentración sináptica de serotonina y/o norepinefrina en el SNC mediante la inhibición de su recaptación en la membrana presináptica)presináptica)presinápticaADRA1A: Arg347Cys; KCNE2:KCNE2:KCNE2 Gln9Glu, Met54Thr, Thr8Ala; KCNH2:KCNH2:KCNH2 Arg784Trp; KCNQ1: Arg583Cys;SCN5A: Gly615Glu, Leu618Phe, Phe1250Leu, Leu1825Pro
CYP1A2 (menor); CYP2B6 (menor); CYP2C8; CYP2C9 (menor); CYP2C19 (menor); CYP2D6 (mayor); CYP3A4/5 (menor); GSH
CYP1A2 (débil); CYP2C8; CYP2C9 (débil); CYP2C19 (débil); CYP2D6 (débil); CYP2E1 (débil)
ABCB1: C3435T, G2677T, C1236T;FABP1; GNAS:GNAS:GNAS ATT→ATC en el →ATC en el →codón 131; GNB3: Ser275Ser; GSTP1; TNF: -308G/A TNF: -308G/A TNF
Genes relacionados: SLC6A4
Amoxapina
Antidepresivos Tricíclicos (Amina Secundaria)
(Reduce la recaptación de serotonina y norepinefrina. Bloquea la actividad del receptor de dopamina)de dopamina)de dopaminaADRA1A: Arg347Cys
CYP2D6 (mayor) GNAS:GNAS:GNAS ATT→ATC en el codón 131;→ATC en el codón 131;→GNB3: Ser275Ser
Bupropión
Antidepresivo, Inhibidor de la Recaptación de Dopamina
(Inhibidor débil de la recaptación neuronal de norepinefrina y dopamina)norepinefrina y dopamina)norepinefrina y dopaminaADRA1A CHRNB2:CHRNB2:CHRNB2 rs2072661; DRD2: -141C Ins/Del DRD2: -141C Ins/Del DRD2(rs1799732), Taq IA RFLP (rs1800497, ANKK1)
CYP1A2 (menor); CYP2A6 (menor); CYP2B6 (mayor): CYP2C8; CYP2C9 (menor); CYP2D6 (menor); CYP2E1 (menor); CYP3A4 (menor)
CYP2D6 (fuerte) COMT:COMT:COMT rs737865, rs165599
Genes relacionados: SLC6A4
Citalopram
Antidepresivo,Inhibidores Selectivos de la Recaptación de Serotonina
(Inhibe la recaptación de serotonina en las neuronas presinápticas y ejerce el mínimo efecto sobre la norepinefrina o la dopamina. Muestra poca o ninguna afinidad por la serotonina, dopamina, histamina adrenérgica, GABA, o subtipos de receptores muscarínicos)subtipos de receptores muscarínicos)subtipos de receptores muscarínicosCRHR2:CRHR2:CRHR2 rs2270007; GRIK2:GRIK2:GRIK2 rs2518224; GRIK4:rs1954787; HTR2A: rs6311 (T102C), rs7997012, rs1928040, rs6313 (Ser34Ser), -1438A/G
CYP2C19 (mayor); CYP2D6 (menor); CYP3A4 (mayor)
CYP1A2 (débil); CYP2B6 (débil); CYP2C19 (débil); CYP2D6 (débil)
ABCB1: C3435T y G2677T;BDNF:BDNF:BDNF Val66Met;COMT:COMT:COMT Val158Met; CREB1;FKBP5;FKBP5;FKBP5 GRIA3: rs4825476;GSK3B; MAOA; MAOB; TPH1:A218C; TPH2
Genes relacionados: HTR1A; HTR1D; SLC6A4
ClomipraminaAntidepresivos Tricíclicos (Amina Terciaria)
(Incrementa la concentración sináptica de serotonina y/o norepinefrina en el SNC mediante la inhibición de su recaptación por la membrana neuronal presináptica. Actividad anticolinérgica)neuronal presináptica. Actividad anticolinérgica)neuronal presináptica. Actividad anticolinérgica
CYP1A2 (mayor); CYP2C19 (mayor);CYP2D6 (mayor); CYP3A4 (mayor); UGT1A4
CYP2C9; CYP2C19; CYP2D6 (moderado)
CYP2A6; CYP2A6; CYP2A6 CYP2B6; CYP2B6; CYP2B6 FABP1;GSTP1; PTGS2;PTGS2;PTGS2 UGT1A4
Genes relacionados: SLC6A4

70
Estados Depresivos
FármacosCategoría
FarmacológicaMecanismo de Acción
(Receptores)Sustrato de Inhibe Otros genes relacionados
Desipramina
Antidepresivos Tricíclicos (Amina Secundaria)
(Incrementa la concentración sináptica de norepinefrina en el SNC mediante la inhibición de su recaptación por la membrana neuronal presináptica. Reducción de la sensibilidad de la adenilato ciclasa, inhibición de receptores β-adrenérgicos y de serotonina)-adrenérgicos y de serotonina)-adrenérgicos y de serotoninaCRHR1: rs1876828, rs242939, rs2422941;CRHR2:CRHR2:CRHR2 rs917195; NTRK2: NTRK2: NTRK2 rs2289657, rs56142442, rs11140793, rs2289658; SLC6A2: SLC6A2: SLC6A2Phe528Cys
CYP1A2 (menor); CYP2D6 (mayor)
CYP2A6 (moderado); CYP2B6 (moderado); CYP2C19 (moderado); CYP2D6 (moderado); CYP2E1 (débil); CYP3A4 (moderado)
ABCB1: rs1202186, rs17064;CYP2C9;CYP2C9;CYP2C9 FKBP5:FKBP5:FKBP5 rs3800373, rs1360780; FOS; IFNA1; IL1B;NR3C1; PDE1C:PDE1C:PDE1C rs30585, rs992185; PDE5A; PSMD9:PSMD9:PSMD9rs1043307; PRKCSH:PRKCSH:PRKCSHrs34095; STAT3:STAT3:STAT3 rs3744483, rs3809758; TBX21
Genes relacionados: HTR1A; SLC6A3; SLC6A4
Desvenlafaxina
Antidepresivo, Inhibidor de la Recaptación de Serotonina/ Norepinefrina
(Un potente inhibidor selectivo de serotonina y norepinefrina) CYP3A4 (mayor);
UGT isoformasCYP2D6 ABCB1; UGTs
Genes relacionados: SLC6A2;SLC6A2;SLC6A2 SLC6A4
DoxepinaAntidepresivos Tricíclicos (Amina Terciaria)
(Incrementa la concentración sináptica de serotonina y norepinefrina en el SNC mediante la inhibición de su recaptación por la membrana neuronal presináptica)neuronal presináptica)neuronal presináptica
CYP1A1 (menor); CYP1A2 (menor); CYP2C9 (menor); CYP2C19 (mayor); CYP2D6 (mayor); CYP3A4/5 (menor); UGT1A3; UGT1A4
ABCB1: C3435T, G2677T, C1236T; GSTP1
Genes relacionados: KCNH2; KCNH2; KCNH2 HRH1; SLC6A2; SLC6A2; SLC6A2SLC6A4
Duloxetina
Antidepresivo, Inhibidor de la Recaptación de Serotonina/ Norepinefrina
(Inhibidor sInhibidor sInhibidor electivo de serotonina y norepinefrina e inhibidor débil de la recaptación de dopamina)e inhibidor débil de la recaptación de dopamina)e inhibidor débil de la recaptación de dopamina
CYP1A2 (mayor); CYP2D6 (mayor)
CYP1A2 (moderado); CYP2B6 (moderado); CYP2C19 (moderado); CYP2D6 (moderado);CYP3A4/5 (moderado)
SLC6A2;SLC6A2;SLC6A2 SLC6A4Genes relacionados: SLC6A2;SLC6A2;SLC6A2 SLC6A4
Escitalopram
Antidepresivo, Inhibidores Selectivos de la Recaptación de Serotonina
(Inhibe la recaptación de serotonina con poco o ningún efecto sobre la recaptación de la norepinefrina o dopamina. Posee ninguna o muy poca afinidad por los receptores 5-HT1-7poca afinidad por los receptores 5-HT1-7poca afinidad por los receptores 5-HT , 1-7, 1-7α- y β-adrenérgicos, D1-5, H1-5, H1-5 1-3, M1-5, y 1-5, y 1-5benzodiazepínicos. No se une a, o posee baja afinidad por, los canales iónicos de Na+, K+, K+, K , Cl-, Cl-, Cl y - y -
Ca2+Ca2+Ca )2+)2+
GRIK2:GRIK2:GRIK2 rs2518224; GRIK4: rs1954787; HTR2A:rs9316233
ABCB1; CYP2C19 (mayor); CYP2D6 (mayor); CYP3A4 (mayor)
ABCB1; CYP1A2 (débil); CYP2C9 (débil); CYP2C19 (débil); CYP2D6 (débil); CYP2E1 (débil); CYP3A4 (débil)
ABCB1; CREB1; FKBP5:FKBP5:FKBP5rs1360780, rs4713916; GRIA3:rs4825476
Genes relacionados: NR3C1; SLC6A4
Fluoxetina
Inhibidores Selectivos de la Recaptación de Serotonina
(Potenciación de la actividad serotonérgica en el SNC como resultado de su inhibición de la recaptación neuronal de serotonina. Gran inhibidor selectivo de la recaptación de serotonina. Incrementa las concentraciones sinápticas de serotonina en el SNC pero posee poco o ningún efecto sobre otros neurotransmisores)neurotransmisores)neurotransmisoresHTR1A: C-1019G
CYP1A2 (menor); CYP2B6 (menor); CYP2C8 (mayor); CYP2C9 (mayor); CY2C19 (mayor); CYP2D6 (mayor); CYP2E1 (menor); CYP3A4 (menor);POR
CYP1A2 (moderado);CYP2B6 (débil); CYP2C8 (moderado);CYP2C9 (débil);CYP2C19 (moderado);CYP2D6 (fuerte); CYP3A4 (moderado)
ABCB1; BDNF:BDNF:BDNF Val66Met;CREB1; CYP1A2;CYP1A2;CYP1A2 CYP2B6;CYP2B6;CYP2B6CYP2E1; FABP1; FKBP5:FKBP5:FKBP5 rs3800373, rs1360780;GSK3B; IFNA1; MAOA; PDE5A;TBX21; TPH1; TPH2
Genes relacionados: DRD3; HTR1B; HTR1D;HTR2A; HTR2C;HTR2C;HTR2C HTR3A; HTR6;HTR6;HTR6 KCNH2; KCNH2; KCNH2 NR3C1;NTRK2;NTRK2;NTRK2 SLC6A4
Fluvoxamina
Antidepresivo, Inhibidores Selectivos de la Recaptación de Serotonina
(Inhibe la captación neuronal de serotonina en el CNS)el CNS)el CNSHTR1A: rs6295; HTR2A: G-1438A
CYP1A2 (mayor); CYP2D6 (mayor); CYP2C19 (mayor); CYP3A4 (mayor)
CYP1A2 (fuerte); CYP2B6 (débil); CYP2C9 (moderado); CYP2C19 (moderado); CYP2D6 (débil); CYP3A4 (débil)
ABCB1: C3435T, G2677T, C1236T;BDNF:BDNF:BDNF G196A; CREB1;CYP2B6;CYP2B6;CYP2B6 CYP2C9;CYP2C9;CYP2C9 MAOA;TPH1
Genes relacionados: HTR3A; KCNH2;KCNH2;KCNH2 SLC6A4

Julio 2011 71
ciencia
FármacosCategoría
FarmacológicaMecanismo de Acción
(Receptores)Sustrato de Inhibe Otros genes relacionados
ImipraminaAntidepresivo Tricíclico (Amina Terciaria)
(Puede implicar la inhibición de la recaptación de norepinefrina y/o serotonina. Actividad anticolinérgica)anticolinérgica)anticolinérgicaADRB2:ADRB2:ADRB2 Arg16Gly, Gln27Glu, Val34Met, Thr164Ile;SLC6A4: Phe586Val
CYP1A2 (menor); CYP2B6 (menor); CYP2C9 (menor); CYP2C19 (mayor); CYP2D6 (mayor); CYP3A4 (menor); UGT1A3; UGT1A4
CYP1A2 (débil); CYP2C19 (débil); CYP2D6 (moderado); CYP2E1 (débil)
ABCB1; BDNF;BDNF;BDNF FABP1;FMO1; FOS; GSTP1;UGT1A3; UGT1A4: Leu48Val, Leu150Leu, 43fcX22Genes relacionados: HTR
Isocarboxazida
Antidepresivo, Inhibidor Monoamino Oxidasa
(Se cree que actúa incrementando las concentraciones endógenas de la epinefrina, norepinefrina, dopamina, y serotonina a través de la inhibición de la enzima MAO)la inhibición de la enzima MAO)la inhibición de la enzima MAO
COMT;COMT;COMT MAOA; MAOB
Maprotilina Antidepresivo Tetracíclico
(Puede implicar la inhibición de la recaptación de norepinefrina en el manejo de desórdenes depresivos y de ansiedad. Actividad anticolinérgica y propiedades sedativas)anticolinérgica y propiedades sedativas)anticolinérgica y propiedades sedativas
CYP1A2 (menor); CYP2D6 (mayor)
ABCB1; CYP2C19;CYP2C19;CYP2C19 CYP3A4;MAOB
Genes relacionados: ADRA1A; HRH1; SLC6A2
Milnacipran
Antidepresivo, Inhibidor de la Recaptación de Serotonina/ Norepinefrina
(Es un potente inhibidor neuronal de la recaptación de norepinefrina y serotonina; inhibe la recaptación de norepinefrina con aproximadamente el triple de potencia in vitro que la serotonina, sin afectar directamente a la recaptación de dopamina u otros neurotransmisores. No posee afinidad significativa por los receptores serotonérgicos (5-HT1-75-HT1-75-HT )1-7)1-7 , α- y β- y β- y -adrenérgicos, muscarínicos (M1-5)1-5)1-5 , histaminérgicos (H1-4)1-4)1-4 , dopaminérgicos (D1-5)1-5)1-5 , opiáceos, benzodiazepínicos, y GABA in vitro. No posee afinidad significativa por los canales de Ca2+canales de Ca2+canales de Ca , K+, K+, K , Na+ y Cl+ y Cl+ – y Cl– y Cl y no inhibe la actividad de las monoamino oxidasas humanas (MAO-A y MAO-B) o la acetilcolinesterasa)(MAO-A y MAO-B) o la acetilcolinesterasa)(MAO-A y MAO-B) o la acetilcolinesterasaADRA2A: C-1297G; SLC6A2: T-182C, G1287A; SLC6A2: T-182C, G1287A; SLC6A2SLC6A4: 5-HTTLPR, 5-HTTVNTR
CYP1A2; CYP2A6; CYP2B6; CYP2C9; CYP2C19; CYP2D6; CYP2E1; CYP3A4 isoenzimas (bajo)
BDNF: G196A; BDNF: G196A; BDNF COMT: COMT: COMTVal158Met
MirtazapinaAntidepresivo, Antagonista α2-Adrenérgico
(Incrementa la liberación de norepinefrina y serotonina. Potente antagonista de los receptores serotonérgicos 5-HT2serotonérgicos 5-HT2serotonérgicos 5-HT y 5-HT2 y 5-HT2 3 y 5-HT3 y 5-HT , H1 histaminérgicos y antagonista moderado de receptores α1-adrenérgicos y muscarínicos. No inhibe la recaptación de norepinefrina o serotonina)recaptación de norepinefrina o serotonina)recaptación de norepinefrina o serotoninaHTR2A: A-1438G; SLC6A3: 3' UTR 40-bp VNTR; SLC6A4: 5-HTTLPR
CYP1A2 (mayor); CYP2C9 (menor); CYP2D6 (mayor); CYP3A4 (mayor)
CYP1A2 (débil); CYP3A4 (débil)
ABCB1; FKBP5;FKBP5;FKBP5 MAOA: T941G (rs6323);MAOB; TPH2
Genes relacionados: ADRA2A; HRH1; HTR3A
Moclobemida
Antidepresivo, Inhibidor Reversible Monoamino Oxidasa
(Inhibe el metabolismo (deaminación)deaminación)deaminación de la serotonina, norepinefrina, y dopamina, incrementando su concentración)incrementando su concentración)incrementando su concentración
CYP2C19 (mayor);CYP2D6 (mayor)
CYP1A2 (débil); CYP2C19 (débil); CYP2D6 (débil)
MAOA; MAOB
Nefazodona
Antidepresivo, Inhibidores Selectivos de la Recaptación de Serotonina/Antagonista
(Inhibe la recaptación neuronal de serotonina y norepinefrina. Bloquea los receptores 5-HT2norepinefrina. Bloquea los receptores 5-HT2norepinefrina. Bloquea los receptores 5-HT y2 y2 α y α y 1)HTR1A: C-1019G CYP2D6 (mayor);
CYP3A4 (mayor)
CYP1A2 (débil); CYP2B6 (débil); CYP2C8 (débil); CYP2D6 (débil); CYP3A4 (fuerte)
ABCB1; ABCB11
Genes relacionados: ADRA1A; HTR2C
Nortriptilina
Antidepresivo Tricíclico (Amina Secundaria)
(Se cree que incrementa la concentración sináptica de serotonina y/sináptica de serotonina y/sináptica de serotonina y o norepinefrina en el SNC mediante la inhibición de su recaptación por la membrana neuronal presináptica. Otros efectos: reducción de la sensibilidad de la adenilato ciclasa, inhibición de receptores β-adrenérgicos e inhibición de receptores serotonérgicos)serotonérgicos)serotonérgicosSLC6A2:SLC6A2:SLC6A2 G1287A
CYP1A2 (menor); CYP2C19 (menor);CYP2D6 (mayor); CYP3A4 (menor)
CYP2C8 (moderado); CYP2C9 (moderado); CYP2D6 (débil); CYP2E1 (débil); CYP3A4 (moderado)
ABCB1: C3435T; GNB3:Ser275Ser
Genes relacionados: HTR1B; NR3C1; SLC6A4
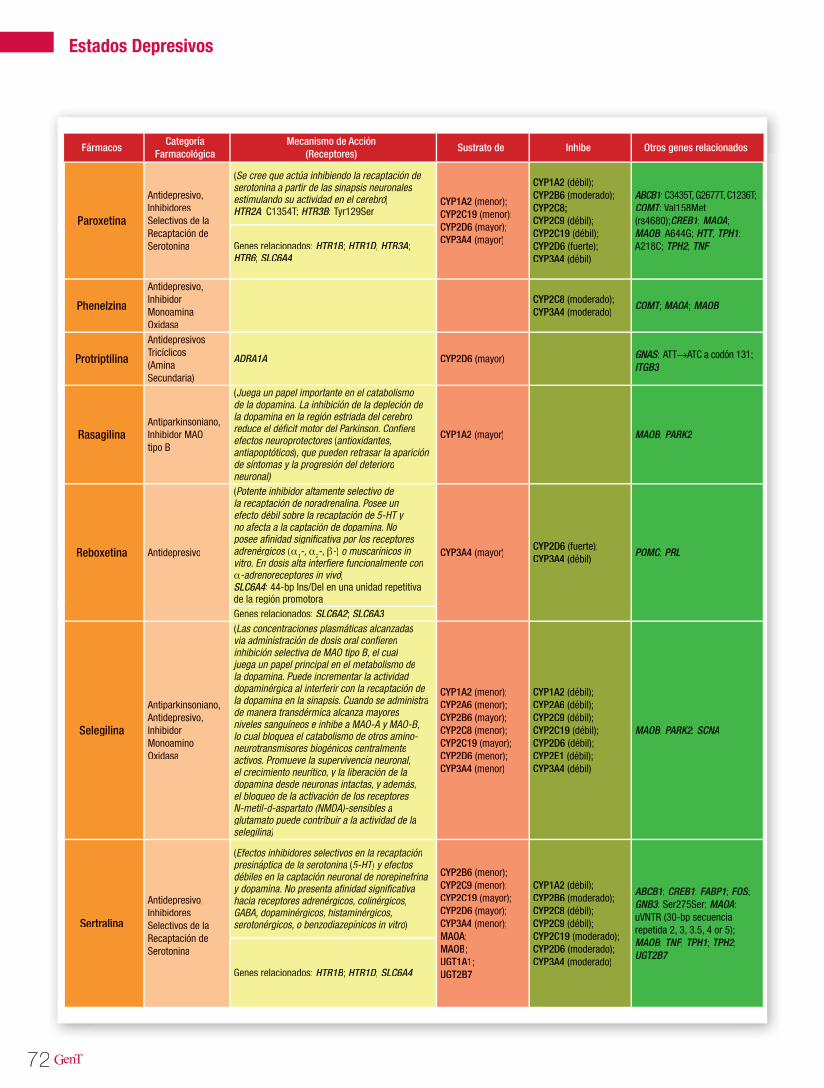
72
Estados Depresivos
FármacosCategoría
FarmacológicaMecanismo de Acción
(Receptores)Sustrato de Inhibe Otros genes relacionados
Paroxetina
Antidepresivo, Inhibidores Selectivos de la Recaptación de Serotonina
(Se cree que actúa inhibiendo la recaptación de serotonina a partir de las sinapsis neuronales estimulando su actividad en el cerebro)estimulando su actividad en el cerebro)estimulando su actividad en el cerebroHTR2A: C1354T; HTR3B: Tyr129Ser
CYP1A2 (menor); CYP2C19 (menor);CYP2D6 (mayor); CYP3A4 (mayor)
CYP1A2 (débil); CYP2B6 (moderado); CYP2C8; CYP2C9 (débil); CYP2C19 (débil); CYP2D6 (fuerte); CYP3A4 (débil)
ABCB1: C3435T, G2677T, C1236T;COMT:COMT:COMT Val158Met (rs4680);CREB1; MAOA; MAOB: A644G; HTT;HTT;HTT TPH1:A218C; TPH2;TPH2;TPH2 TNFGenes relacionados: HTR1B; HTR1D; HTR3A;
HTR6;HTR6;HTR6 SLC6A4
Phenelzina
Antidepresivo, Inhibidor Monoamina Oxidasa
CYP2C8 (moderado); CYP3A4 (moderado)
COMT;COMT;COMT MAOA; MAOB
Protriptilina
Antidepresivos Tricíclicos (Amina Secundaria)
ADRA1A CYP2D6 (mayor) GNAS:GNAS:GNAS ATT→ATC a codón 131; ITGB3
RasagilinaAntiparkinsoniano, Inhibidor MAO tipo B
(Juega un papel importante en el catabolismo de la dopamina. La inhibición de la depleción de la dopamina en la región estriada del cerebro reduce el déficit motor del Parkinson. Confiere efectos neuroprotectores (antioxidantes, antiapoptóticos)antiapoptóticos)antiapoptóticos , que pueden retrasar la aparición de síntomas y la progresión del deterioro neuronal)
CYP1A2 (mayor) MAOB; PARK2
Reboxetina Antidepresivo
(Potente inhibidor altamente selectivo de la recaptación de noradrenalina. Posee un efecto débil sobre la recaptación de 5-HT y no afecta a la captación de dopamina. No posee afinidad significativa por los receptores adrenérgicos (α1-, α2-, β-)-)- o muscarínicos in vitro. En dosis alta interfiere funcionalmente con α-adrenoreceptores in vivo)-adrenoreceptores in vivo)-adrenoreceptores in vivoSLC6A4: 44-bp Ins/Del en una unidad repetitiva de la región promotora
CYP3A4 (mayor)CYP2D6 (fuerte);CYP3A4 (débil)
POMC;POMC;POMC PRL
Genes relacionados: SLC6A2; SLC6A2; SLC6A2 SLC6A3
Selegilina
Antiparkinsoniano, Antidepresivo, Inhibidor Monoamino Oxidasa
(Las concentraciones plasmáticas alcanzadas via administración de dosis oral confieren inhibición selectiva de MAO tipo B, el cual juega un papel principal en el metabolismo de la dopamina. Puede incrementar la actividad dopaminérgica al interferir con la recaptación de la dopamina en la sinapsis. Cuando se administra de manera transdérmica alcanza mayores niveles sanguíneos e inhibe a MAO-A y MAO-B, lo cual bloquea el catabolismo de otros amino-neurotransmisores biogénicos centralmente activos. Promueve la supervivencia neuronal, el crecimiento neurítico, y la liberación de la dopamina desde neuronas intactas, y además, el bloqueo de la activación de los receptores N-metil-d-aspartato (NMDA)-sensibles a glutamato puede contribuir a la actividad de la selegilina)
CYP1A2 (menor); CYP2A6 (menor); CYP2B6 (mayor); CYP2C8 (menor); CYP2C19 (mayor);CYP2D6 (menor); CYP3A4 (menor)
CYP1A2 (débil); CYP2A6 (débil); CYP2C9 (débil); CYP2C19 (débil); CYP2D6 (débil); CYP2E1 (débil); CYP3A4 (débil)
MAOB; PARK2;PARK2;PARK2 SCNA
Sertralina
Antidepresivo,Inhibidores Selectivos de la Recaptación de Serotonina
(Efectos inhibidores selectivos en la recaptación presináptica de la serotonina (5-HT)5-HT)5-HT y efectos débiles en la captación neuronal de norepinefrina y dopamina. No presenta afinidad significativa hacia receptores adrenérgicos, colinérgicos, GABA, dopaminérgicos, histaminérgicos, serotonérgicos, o benzodiazepínicos in vitro)serotonérgicos, o benzodiazepínicos in vitro)serotonérgicos, o benzodiazepínicos in vitro
CYP2B6 (menor); CYP2C9 (menor);CYP2C19 (mayor);CYP2D6 (mayor); CYP3A4 (menor); MAOA; MAOB; UGT1A1; UGT2B7
CYP1A2 (débil); CYP2B6 (moderado);CYP2C8 (débil); CYP2C9 (débil); CYP2C19 (moderado); CYP2D6 (moderado); CYP3A4 (moderado)
ABCB1; CREB1; FABP1; FOS; GNB3: Ser275Ser; MAOA: uVNTR (30-bp secuencia repetida 2, 3, 3.5, 4 or 5); MAOB; TNF;TNF;TNF TPH1; TPH2;TPH2;TPH2UGT2B7
Genes relacionados: HTR1B; HTR1D; SLC6A4
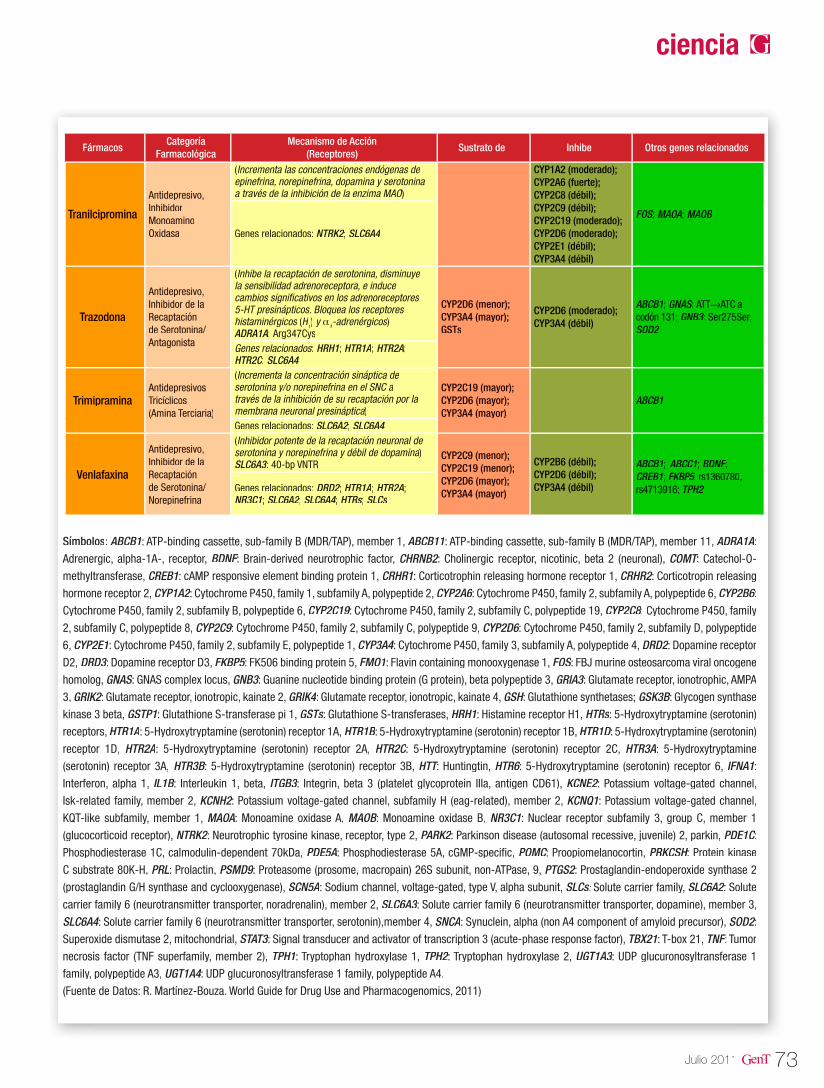
Julio 2011 73
ciencia
FármacosCategoría
FarmacológicaMecanismo de Acción
(Receptores)Sustrato de Inhibe Otros genes relacionados
Tranilcipromina
Antidepresivo, Inhibidor Monoamino Oxidasa
(Incrementa las concentraciones endógenas de epinefrina, norepinefrina, dopamina y serotonina a través de la inhibición de la enzima MAO)a través de la inhibición de la enzima MAO)a través de la inhibición de la enzima MAO
CYP1A2 (moderado); CYP2A6 (fuerte); CYP2C8 (débil); CYP2C9 (débil); CYP2C19 (moderado); CYP2D6 (moderado); CYP2E1 (débil); CYP3A4 (débil)
FOS; MAOA; MAOB
Genes relacionados: NTRK2;NTRK2;NTRK2 SLC6A4
Trazodona
Antidepresivo, Inhibidor de la Recaptación de Serotonina/ Antagonista
(Inhibe la recaptación de serotonina, disminuye la sensibilidad adrenoreceptora, e induce cambios significativos en los adrenoreceptores 5-HT presinápticos. Bloquea los receptores histaminérgicos (H1) y α1-adrenérgicos)-adrenérgicos)-adrenérgicosADRA1A: Arg347Cys
CYP2D6 (menor); CYP3A4 (mayor); GSTs
CYP2D6 (moderado); CYP3A4 (débil)
ABCB1; GNAS: ATT→ATC a codón 131; GNB3: Ser275Ser;SOD2
Genes relacionados: HRH1; HTR1A; HTR2A;HTR2C;HTR2C;HTR2C SLC6A4
TrimipraminaAntidepresivos Tricíclicos (Amina Terciaria)
(Incrementa la concentración sináptica de serotonina y/o norepinefrina en el SNC a través de la inhibición de su recaptación por la membrana neuronal presináptica)membrana neuronal presináptica)membrana neuronal presináptica
CYP2C19 (mayor); CYP2D6 (mayor); CYP3A4 (mayor)
ABCB1
Genes relacionados: SLC6A2;SLC6A2;SLC6A2 SLC6A4
Venlafaxina
Antidepresivo, Inhibidor de la Recaptación de Serotonina/ Norepinefrina
(Inhibidor potente de la recaptación neuronal de serotonina y norepinefrina y débil de dopamina)serotonina y norepinefrina y débil de dopamina)serotonina y norepinefrina y débil de dopaminaSLC6A3: 40-bp VNTR
CYP2C9 (menor); CYP2C19 (menor); CYP2D6 (mayor); CYP3A4 (mayor)
CYP2B6 (débil); CYP2D6 (débil); CYP3A4 (débil)
ABCB1; ABCC1; BDNF;BDNF;BDNFCREB1; FKBP5:FKBP5:FKBP5 rs1360780, rs4713916; TPH2Genes relacionados: DRD2; DRD2; DRD2 HTR1A; HTR2A;
NR3C1; SLC6A2;SLC6A2;SLC6A2 SLC6A4; HTRs; SLCs
Símbolos: ABCB1: ATP-binding cassette, sub-family B (MDR/TAP), member 1, ABCB11: ATP-binding cassette, sub-family B (MDR/TAP), member 11, ADRA1A:
Adrenergic, alpha-1A-, receptor, BDNF: Brain-derived neurotrophic factor, BDNF: Brain-derived neurotrophic factor, BDNF CHRNB2: Cholinergic receptor, nicotinic, beta 2 (neuronal), CHRNB2: Cholinergic receptor, nicotinic, beta 2 (neuronal), CHRNB2 COMT: Catechol-O-COMT: Catechol-O-COMTmethyltransferase, CREB1: cAMP responsive element binding protein 1, CRHR1: Corticotrophin releasing hormone receptor 1, CRHR2: Corticotropin releasing CRHR2: Corticotropin releasing CRHR2hormone receptor 2, CYP1A2: Cytochrome P450, family 1, subfamily A, polypeptide 2, CYP1A2: Cytochrome P450, family 1, subfamily A, polypeptide 2, CYP1A2 CYP2A6: Cytochrome P450, family 2, subfamily A, polypeptide 6, CYP2A6: Cytochrome P450, family 2, subfamily A, polypeptide 6, CYP2A6 CYP2B6: CYP2B6: CYP2B6Cytochrome P450, family 2, subfamily B, polypeptide 6, CYP2C19: Cytochrome P450, family 2, subfamily C, polypeptide 19, CYP2C19: Cytochrome P450, family 2, subfamily C, polypeptide 19, CYP2C19 CYP2C8: Cytochrome P450, family
2, subfamily C, polypeptide 8, CYP2C9: Cytochrome P450, family 2, subfamily C, polypeptide 9, CYP2C9: Cytochrome P450, family 2, subfamily C, polypeptide 9, CYP2C9 CYP2D6: Cytochrome P450, family 2, subfamily D, polypeptide CYP2D6: Cytochrome P450, family 2, subfamily D, polypeptide CYP2D66, CYP2E1: Cytochrome P450, family 2, subfamily E, polypeptide 1, CYP3A4: Cytochrome P450, family 3, subfamily A, polypeptide 4, DRD2: Dopamine receptor DRD2: Dopamine receptor DRD2D2, DRD3: Dopamine receptor D3, FKBP5: FK506 binding protein 5, FKBP5: FK506 binding protein 5, FKBP5 FMO1: Flavin containing monooxygenase 1, FOS: FBJ murine osteosarcoma viral oncogene
homolog, GNAS: GNAS complex locus, GNB3: Guanine nucleotide binding protein (G protein), beta polypeptide 3, GRIA3: Glutamate receptor, ionotrophic, AMPA
3, GRIK2: Glutamate receptor, ionotropic, kainate 2, GRIK2: Glutamate receptor, ionotropic, kainate 2, GRIK2 GRIK4: Glutamate receptor, ionotropic, kainate 4, GSH: Glutathione synthetases; GSH: Glutathione synthetases; GSH GSK3B: Glycogen synthase
kinase 3 beta, GSTP1: Glutathione S-transferase pi 1, GSTs: Glutathione S-transferases, HRH1: Histamine receptor H1, HTRs: 5-Hydroxytryptamine (serotonin)
receptors, HTR1A: 5-Hydroxytryptamine (serotonin) receptor 1A, HTR1B: 5-Hydroxytryptamine (serotonin) receptor 1B, HTR1D: 5-Hydroxytryptamine (serotonin)
receptor 1D, HTR2A: 5-Hydroxytryptamine (serotonin) receptor 2A, HTR2C: 5-Hydroxytryptamine (serotonin) receptor 2C, HTR2C: 5-Hydroxytryptamine (serotonin) receptor 2C, HTR2C HTR3A: 5-Hydroxytryptamine
(serotonin) receptor 3A, HTR3B: 5-Hydroxytryptamine (serotonin) receptor 3B, HTT: Huntingtin, HTT: Huntingtin, HTT HTR6: 5-Hydroxytryptamine (serotonin) receptor 6, HTR6: 5-Hydroxytryptamine (serotonin) receptor 6, HTR6 IFNA1:
Interferon, alpha 1, IL1B: Interleukin 1, beta, ITGB3: Integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61), KCNE2: Potassium voltage-gated channel, KCNE2: Potassium voltage-gated channel, KCNE2Isk-related family, member 2, KCNH2: Potassium voltage-gated channel, subfamily H (eag-related), member 2, KCNH2: Potassium voltage-gated channel, subfamily H (eag-related), member 2, KCNH2 KCNQ1: Potassium voltage-gated channel,
KQT-like subfamily, member 1, MAOA: Monoamine oxidase A, MAOB: Monoamine oxidase B, NR3C1: Nuclear receptor subfamily 3, group C, member 1
(glucocorticoid receptor), NTRK2: Neurotrophic tyrosine kinase, receptor, type 2, NTRK2: Neurotrophic tyrosine kinase, receptor, type 2, NTRK2 PARK2: Parkinson disease (autosomal recessive, juvenile) 2, parkin, PARK2: Parkinson disease (autosomal recessive, juvenile) 2, parkin, PARK2 PDE1C: PDE1C: PDE1CPhosphodiesterase 1C, calmodulin-dependent 70kDa, PDE5A: Phosphodiesterase 5A, cGMP-specific, POMC: Proopiomelanocortin, POMC: Proopiomelanocortin, POMC PRKCSH: Protein kinase PRKCSH: Protein kinase PRKCSHC substrate 80K-H, PRL: Prolactin, PSMD9: Proteasome (prosome, macropain) 26S subunit, non-ATPase, 9, PSMD9: Proteasome (prosome, macropain) 26S subunit, non-ATPase, 9, PSMD9 PTGS2: Prostaglandin-endoperoxide synthase 2 PTGS2: Prostaglandin-endoperoxide synthase 2 PTGS2(prostaglandin G/H synthase and cyclooxygenase), SCN5A: Sodium channel, voltage-gated, type V, alpha subunit, SLCs: Solute carrier family, SLC6A2: Solute SLC6A2: Solute SLC6A2carrier family 6 (neurotransmitter transporter, noradrenalin), member 2, SLC6A3: Solute carrier family 6 (neurotransmitter transporter, dopamine), member 3,
SLC6A4: Solute carrier family 6 (neurotransmitter transporter, serotonin),member 4, SNCA: Synuclein, alpha (non A4 component of amyloid precursor), SOD2: SOD2: SOD2Superoxide dismutase 2, mitochondrial, STAT3: Signal transducer and activator of transcription 3 (acute-phase response factor), TBX21: T-box 21, TNF: Tumor TNF: Tumor TNFnecrosis factor (TNF superfamily, member 2), TPH1: Tryptophan hydroxylase 1, TPH2: Tryptophan hydroxylase 2, TPH2: Tryptophan hydroxylase 2, TPH2 UGT1A3: UDP glucuronosyltransferase 1
family, polypeptide A3, UGT1A4: UDP glucuronosyltransferase 1 family, polypeptide A4.
(Fuente de Datos: R. Martínez-Bouza. World Guide for Drug Use and Pharmacogenomics, 2011)

74
Estados Depresivos
Tabla 3. Genes relacionados con la farmacogenética de las Sales de Litio
Farmacogenética primaria:BCR: Asn796SerBDNF: C/G (rs988748) and G/A (Val66Met)BDNF: C/G (rs988748) and G/A (Val66Met)BDNFCACNG2: rs2284017, rs2284018, rs5750285GSK3B: T-50C (GSK3-beta*C) P1: C973A (rs1882891)MT-DNA: 10398ANTRK2: rs1387923, rs1565445
Otros genes potencialmente relacionados con seguridad y eficacia:ABCG2; CCND1; CREB1; DRD1; ESR1; FMR1; GRIK2; NR3C1; PTGES; SLC6A4; VEGFA
(Fuente de Datos: R. Martínez-Bouza. World Guide for Drug Use and Pharmacogenomics, 2011)
Tabla 4. Genes relacionados con la farmacogenética de Carbamazepina
Farmacogenética primaria:ABCB1: C3435T, G2677T, C1236TABCC2: Val417IleCYP3A4 and CYP3A5: CYP3A5: CYP3A5 CYP3A4*1, CYP3A4*1B, CYP3A4*2, CYP3A4*3, CYP3A4*4, CYP3A4*5, CYP3A4*5, CYP3A4*5 CYP3A4*6, CYP3A4*8, CYP3A4*11,CYP3A4*12, CYP3A4*13, CYP3A4*15, CYP3A4*15, CYP3A4*15 CYP3A4*17, CYP3A4*17, CYP3A4*17 CYP3A4*18, CYP3A4*19, CYP3A5*3 EPHX1: Tyr113His, His139ArgGSTM1: A304G, Null (Del)GSTT1: Null (Del)HLA-A: HLA-A*3101HLA-B: HLA-B*1502HSPA1L: Thr493Met (C2437T)
Otros genes potencialmente relacionados con seguridad y eficacia:ABCB4; ABCG2; CASR; CSTB; CYP1A2; CYP2A6; CYP2B6; CYP2C8; CYP2C9; CYP2C19; CYP2D6; CYP2E1; FOS; GRIK2; GSTA1;HLA-A; IL6; MTHFR; NR1I2; NR3C1; RFC1; SCN1A; SCN2A; SCN3A; SULT1A1; UGT1A4; UGT2B7
Sustrato de:CYP1A2 (menor); CYP2A6 (menor); CYP2B6 (mayor); CYP2C8 (menor); CYP2C19 (menor); CYP2E1 (menor); CYP3A4 (mayor); CYP3A5 (menor); CYP3A7 (menor); HLA-A; HLA-B; UGT2B7
Inductor de: ABCB1; ABCB4; ABCC2; ABCG2; CASR; CYP1A2; CYP2B6; CYP2C8; CYP2C9; CYP2C19; CYP2D6; CYP3A4; GSTA1; NR1I2; NR3C1; SULT1A1; UGT1A4
(Fuente de Datos: R. Martínez-Bouza. World Guide for Drug Use and Pharmacogenomics, 2011)
Tabla 5. Genes relacionados con la farmacogenética del Ácido Valproico
Farmacogenética primaria:ABCB1: C3435T, G2677T/A
Otros genes potencialmente relacionados con seguridad y eficacia:ABAT;ABAT;ABAT ABCC2; ABCG2; AGPAT2; AKR1C4; CHRNA1; COL1A1; CREB1; CYP2A6; CYP2B6; CYP2C9; CYP2C19; CYP2D6; CYP2E1;CYP3A4; CYP4B1; FMR1; FOS; GSK3B; HBB; HDAC9; HFE; HFE; HFE HLA-A; HLA-B; IL6; IL10; MAOA; MT-TK; MT-TK; MT-TK NR3C1; PTGES; SCN2A; SLC5A5;SLC5A5;SLC5A5SLC6A2; SLC12A3; SLC22A16; TNF; TNF; TNF TP53; UGT1A6; UGT1A9; UGT2B7; UGT2B7; UGT2B7 UGT2B15
Sustrato de:ABCB1; CYP2A6 (mayor); CYP2B6 (menor); CYP2C9 (mayor); CYP2C19 (menor); CYP2E1 (menor); CYP3A4 (menor); UGT1A6; UGT1A9; UGT2B7
Inhibidor de:CYP2A6 (moderado); CYP2C9 (fuerte); CYP2C19 (moderado); CYP2D6 (débil); CYP3A4 (moderado); UGT1A9; UGT2B15; UGT2B7
Inductor de: ABCB1; CASR; CYP2A6; CYP3A4; NR1I2
(Fuente de Datos: R. Martínez-Bouza. World Guide for Drug Use and Pharmacogenomics, 2011)

75Julio 2011
Introducción
a revolución genómica está cambiando cómo entendemos nuestra identidad como especie y nuestro lugar en el mundo natural. No sólo compartimos el 99,9% de nuestros genes con los demás seres humanos, sino que también compartimos el 98% de nuestros genes con los chimpancés, el 90% con los ratones, el 21% de gusanos, y 7%, con una simple bac-teria como E. coli, una sorprendente demos-
tración de la continuidad de la vida en la Tierra. Debemos ser capaces de entender cómo funcionan los genes, cómo la natura-leza (herencia genética) interactúa con el desarrollo (medio ambiente) para influir en lo que llegamos a ser, y cómo podemos estar cambiando el equilibrio, ahora que tenemos la capacidad de alterar el código del genoma de la vida.
a revolución genómica está cambiando cómo entendemos nuestra identidad como especie y nuestro lugar en el mundo natural. No sólo compartimos el 99,9% de nuestros genes con los demás seres humanos, sino que también compartimos el 98% de nuestros genes con
L
La revolución genómica: del proyecto Genoma Humano a la megasecuenciación como herramienta diagnóstica
Juan C. CarrilDepartamento de Genómica, EuroEspes Biotecnología, Bergondo, Coruña

La revolución genómica está cambiando cómo entendemos nuestra identidad como especie y nuestro lugar en el mundo natural
76
La revolución genómica
El Proyecto Genoma Humano
En junio de 2000, el equipo de Craig Venter anunció triunfalmente que había descifrado el genoma humano, el plan codificado que per-mite desarrollar la vida humana. Al poner en orden los 3,2 millones de unidades de nuestro ADN, los investigadores desataron una tormen-ta de descubrimientos que han marcado el co-mienzo de una nueva era.
El Proyecto Genoma Humano es un programa de investigación colaborativo, cuyo objetivo principal fue descifrar por completo la informa-ción genética contenida en cada célula y escri-ta en el lenguaje del ADN: el código genético. La secuenciación completa del ADN finalizó en mayo de 2000 y arrojó interesantes resultados. Se estima que sólo el 2% del ADN es codifican-te, es decir, da lugar a productos de expresión o proteínas, mientras que el 50% está compuesto por secuencias repetitivas de diferente tipo cuya función está aún poco clara. La secuencia com-pleta tiene aproximadamente 3200 millones de pares de bases que codifican 30000 genes y sólo el 50% de éstos tiene secuencias de “ADN pa-trón” que sugieren su posible función. Se han identificado mutaciones responsables de dife-rentes patologías en alrededor de 1000 genes.
El dogma “un gen – una proteína” ha sido des-plazado por la evidencia de que estos 30000 ge-nes codifican alrededor de 100000 proteínas, es decir, el mismo gen da lugar a diferentes pro-ductos de expresión. Esto es posible a través de mecanismos como el denominado “splicing alternativo” (procesos alternativos de elimina-ción de intrones para la formación de distintas variantes de ARN mensajero maduro) y fenóme-nos epigenéticos (mecanismos que modifican
la expresión, pero no la información genética), como la metilación. Se ha identificado que los cromosomas 17, 19 y 22 son los que poseen ma-yor densidad génica y que los cromosomas 13, 18 y 21 son los que tienen menor contenido gé-nico, siendo ésta, quizás, la causa de que concep-ciones trisómicas para los mismos sean viables dando lugar a aberraciones cromosómicas o aneuploidías (síndrome de Down, síndrome de Edwards, síndrome de Patau, etc.).
Una característica del genoma humano, de re-levancia médica y social, reside en que dos in-dividuos no emparentados comparten el 99,9% de su secuencia de ADN. De todos modos, dado que la secuencia completa es de 3200 millones de nucleótidos, las diferencias entre cada dos individuos son de varios millones de bases nu-cleotídicas. Se realizan esfuerzos para catalogar estas variantes referidas como polimorfismos de nucleótido único (“Single Nucleotide Polymor-phisms” o SNPs). Estos SNPs son sólo marcado-res de la diversidad biológica y pueden o no re-lacionarse con genes involucrados en patologías en función de dónde se ubiquen.
Para el ojo inexperto, el vasto código de A, T, G y C resulta desconcertante. Sin embargo, el mapa de nuestro genoma ofrece un potencial ilimitado. Uno de los hitos más importantes se refiere a las perspectivas para nuestra salud, que van desde descubrir la cura para el cáncer hasta la posibilidad de cambiar o elegir nuestros ge-nes. También podemos aplicar este conocimien-to para alimentar a la creciente población del mundo, para la resolución de misterios forenses y para salvar especies en peligro de extinción. Realmente, se trata de una información de una utilidad incalculable, si la usamos con respon-sabilidad y somos capaces de gestionar adecua-damente los riesgos que este conocimiento lleva implícitos.
Poner el genoma a nuestro servicio plantea pre-guntas y dilemas para nosotros como individuos, familias, naciones e incluso como especie. Tene-mos que tomar decisiones sobre nuestra salud, nuestra comida, nuestra responsabilidad con la naturaleza y nuestras responsabilidades con la siguiente generación. La revolución genómica está aquí, pero, ¿estamos preparados?
El ADN: la molécula de la vida
Los científicos han sabido del ADN desde 1871, cuando Miescher describió la “nucleína” de los leucocitos. Pero fue un documento sin preten-siones científicas publicado por James Watson y Francis Crick en 1953 lo que revolucionó el concepto de la vida misma con la presentación

Poner el genoma a nuestro servicio plantea preguntas y dilemas para nosotros como
individuos, familias, naciones e incluso como especie
Julio 2011 77
ciencia
de la estructura del ADN: la doble hélice. “Esta estructura tiene nuevas características que son de considerable interés biológico”, escribieron. Junto con Maurice Wilkins, y gracias, en parte, al trabajo en la sombra de la nunca suficiente-mente reconocida, Rosalind Franklin Watson y Crick ganaron el Premio Nobel en 1962 por su descubrimiento.
Todos los seres vivos en la Tierra, cada bacteria, planta o animal, comparten la estructura más fundamental de la vida, el ácido desoxirribo-nucleico, o ADN. En total, el genoma humano contiene 3200 millones de unidades de ADN, dispuestos en una secuencia fija que define la especie humana. Cuatro componentes quími-cos, adenina, timina, guanina y citosina, común-mente abreviados como A, T, G y C, constituyen las bases del código genético, los peldaños de la escalera de doble hélice.
Los seres humanos no parecen tener mucho en común con los ratones y los gusanos. Sin embar-go, dentro de las células de cada uno están los genes, las instrucciones de funcionamiento co-dificadas en el ADN. A pesar de las diferencias entre los organismos - alas, hojas, pies o aletas – todos compartimos un sorprendente número de genes. Los seres humanos y la lombriz de tierra, por ejemplo, comparten el 21% de sus genes, los seres humanos y los ratones compar-ten el 90%.
Al comparar los códigos genéticos de plantas y animales con la secuencia de ADN humano podemos determinar el porcentaje de genes que tienen en común. Los mismos genes en di-ferentes organismos no tienen precisamente la misma disposición de A, T, G y C, pero se esta-blecen porcentajes de homología que nos per-miten determinar que se trata del mismo gen o de un gen con una funcionalidad semejante.
Puesto que los genes se heredan de los padres, cada generación está vinculada a la anterior. Los genes que compartimos con los demás or-ganismos vivos se transmiten de una generación a otra, a partir de un ancestro universal que vi-vió hace unos 3.5 millones de años. A través de la evolución, los cambios accidentales en el có-digo genético han dado lugar a la aparición de diferentes especies. Hoy en día, los rastros de genes antiguos se pueden encontrar dentro de todos nosotros, los gusanos y los seres humanos por igual.
Prácticamente cualquier tipo de célula en el cuerpo humano contiene una copia completa de todos los genes necesarios para “codificar” una persona. Los genes explican las instruccio-nes biológicas que dirigen el desarrollo y man-tenimiento de todo, desde el color de los ojos
a la disposición de las partes del cuerpo. Todos los seres vivos, desde una bacteria a una ballena, tiene genes. Las células del cuerpo contienen una copia del juego completo de instrucciones necesarias para el desarrollo y mantenimiento de un organismo. Este conjunto se denomina genoma.
La ampliación de una célula humana revela una masa de ADN contenida en el interior del nú-cleo. Durante la reproducción el ADN se orga-niza en paquetes llamados cromosomas, dentro de los cuales hay segmentos que codifican pro-teínas necesarias para la función celular. Cada uno de estos segmentos constituye un gen.
La Huella Genética
Si miramos a nuestro alrededor, no hay dos per-sonas exactamente iguales. Pero todos compar-timos el 99.9% del mismo ADN. Genéticamen-te hablando, nuestras diferencias se derivan de las variaciones ocasionales en la secuencia de ADN: en promedio, tan sólo nos diferen-ciamos en unas 1250 posiciones nucleotídicas. En esta pequeñísima fracción del ADN que nos diferencia, se contiene la explicación de todos nuestros rasgos morfológicos, color de ojos, estatura, etc., además de las diferencias en la predisposición a ciertas enfermedades.
Los nuevos datos del mapa del genoma huma-no revelan que todos los seres humanos son increíblemente similares, de hecho, genética-mente idénticos al 99.9%. Todos somos miem-bros de una especie, Homo sapiens. La Genética ha confirmado que no existe una base genética o biológica para la raza. La variación genética entre las personas dentro del mismo grupo “ra-cial” puede ser mayor que la variación entre las personas de dos grupos diferentes. Las diferen-cias genéticas entre los asiáticos y los europeos son menos pronunciadas que las diferencias entre los grupos de, por ejemplo, partes de África oriental y occidental.
La huella genética, la identificación de un ser vivo, sea humano o no, por las características singulares de su genoma, se ha convertido en un método de identificación universal en to-das las aplicaciones en que es factible, con una aceptación que hace 20 años apenas se vislum-braba.

Todos los seres vivos en la Tierra, cada bacteria, planta o animal, comparten la estructura más fundamental de la vida, el ácido desoxirribonucleico, o ADN
78
La revolución genómica
El primer caso de inmigración en el que se dis-puso de la ayuda inestimable de la huella gené-tica no tardó en llegar. Se trataba de un chico originario de Ghana al que, al volver de un viaje a su país, se le negó la residencia porque su do-cumentación parecía falsificada. Las pruebas de ADN demostraron con un 99.997% de probabi-lidad, que era hermano de los demás hijos de su madre, de nacionalidad británica, por lo que pudo quedarse en el Reino Unido.
Otro caso, de los primeros internacionales, fue el de Josef Mengele, criminal de guerra nazi, cu-yos supuestos restos fueron descubiertos en 1985 en un cementerio brasileño. En 1988 se compa-ró el ADN extraído de un hueso del esqueleto con el ADN de la sangre de la esposa y el hijo de Mengele. La conclusión, con un 99.94% de probabilidad fue positiva para la identificación de los restos encontrados como los pertenecien-tes a Mengele.
Aplicaciones de la Genética en la sociedad del bienestar
Pero donde las técnicas de ADN están abriendo un campo novedoso es en la autentificación de los alimentos. Para hacerse una idea de la situa-ción actual, basta con seguir los resultados de las inspecciones como las que realiza la Food Standards Agency (FSA), la agencia británica de alimentación. En 2004 dio a conocer los resul-tados del análisis realizado sobre las diferentes marcas de arroz Basmati que se comercializaban en aquel país. De las 363 muestras analizadas, 63 contenían más de un 20% de arroz que no era Basmati. Lo preocupante era que en 31 mues-tras, más del 60% del arroz no era Basmati, por lo que se trataba de un engaño deliberado. Para el ensayo se usó un test de ADN desarrollado por la FSA.
El Basmati, cuyo precio dobla el de las otras variedades, supone el 37% del volumen eco-nómico de las ventas en el mercado británico, moviendo anualmente alrededor de 75 millo-nes de euros. Especialmente apreciado por su aroma, es un producto en alza debido a la po-pularización de la comida india y pakistaní. La sustitución de arroz Basmati es claramente un fraude que beneficia al proveedor y perjudica al consumidor.
Fraudes similares se han descubierto en otros sectores. En 1998, la misma agencia británica halló en una inspección de salmón ahu-mado, que en varias muestras
Desde una hebra de cabello, o un cepillo de dientes, hasta los restos de sudor en el cuello de una camisa, pueden constituir muestras que ayuden a descifrar o conocer las características únicas de nuestro ADN.
El 22 de enero de 1988, Colin Pitchfork, un jo-ven inglés de 27 años, casado y con dos hijos, se convirtió en la primera persona en el mundo en ser condenado por el perfil de su ADN. El perfil genético delató al “asesino del sendero”, respon-sable de la violación y muerte de dos jóvenes, al comparar el ADN extraído del semen encontra-do en las víctimas con una muestra de sangre tomada por la policía de todos los hombres del pequeño pueblo de Narborough.
Muy cerca de allí, en la Universidad de Leices-ter, a tan sólo diez kilómetros de la escena del crimen, el Doctor Alec Jeffreys, connotado ge-netista, había descubierto accidentalmente una técnica que sería conocida popularmente como “Huella Genética”. Jeffreys investigaba por aquel entonces en la búsqueda de marcadores gené-ticos que ayudaran a identificar los genes im-plicados en la producción de mioglobina, y fue analizando las denominadas “regiones hiperva-riables” cuando observó que existían diferencias entre individuos en la longitud de dichas regio-nes. Estas regiones, denominadas minisatélites, tenían un potencial para identificación mayor que cualquier otro marcador genético con-vencional, como los grupos sanguíneos AB0 o HLA.
En casos recientes, se ha exonerado a perso-nas condenadas incluso a cadena perpetua y a la pena de muerte en su momento, sobre la base de su ADN. El primer caso fue el del es-tadounidense Kirk Bloodsworth, condenado a la pena de muerte en 1985 por el asesinato y violación de una niña de nueve años. La re-visión del caso se produjo en 1992 con el re-sultado de que Bloodsworth quedó en libertad en 1993.
La utilización del ADN en procesos criminales es lo que se denomina Criminalística Biológica, y consiste en el estudio de la variabilidad genéti-ca humana aplicada a la resolución de procesos criminales, mediante el análisis de vestigios bio-lógicos encontrados en el lugar de los hechos y su comparación con los perfiles genéticos de los posibles implicados.

Desde una hebra de cabello, o un cepillo de dientes, hasta los restos de sudor en el cuello
de una camisa, pueden constituir muestras que ayuden a descifrar o conocer las características
únicas de nuestro ADN
Julio 2011 79
ciencia
el pretendido salmón no era tal sino trucha. En la misma línea, en el año 2000 la FSA descubrió muestras de conservas en las que el atún había sido sustituido por bonito, mucho más económi-co. La identificación y autentificación de otros productos como el aceite de oliva virgen, las di-ferentes variedades de patata, los tipos de carne o la harina usada en la pasta también están entre los objetivos de los análisis de ADN.
Así por ejemplo, la pasta debería elaborarse con la harina derivada de la variedad de trigo Triti-cum durum. Su sustitución por la variedad co-mún (T. aestivum) da como resultado una pasta de peor calidad. Detectarlo ahora es mucho más fácil desde que se desarrolló un método que, tomando como referencia unos polimorfismos genéticos que están en un tipo de trigo pero no en el otro, permite identificar de forma fiable el contenido de una muestra.
En España también se trabaja para desarrollar técnicas que ayuden a la autentificación de los alimentos. El pescado, donde se vende mucho producto procesado y enlatado, es especial-mente sensible al fraude. La identificación de las especies biológicas mediante el empleo de fragmentos de ADN tiene como ventaja que se pueden seleccionar diferentes regiones de ADN dependiendo del nivel de resolución requerido: individuos, poblaciones, especies, géneros, fami-lias, etc.
La Medicina Genómica
En lo que se refiere a lo que se ha denominado Medicina Genómica, es necesario tener en cuen-ta que existe una brecha entre la aplicación de los conocimientos surgidos del Proyecto Geno-ma Humano y la práctica médica cotidiana. Para estrechar esta brecha, la investigación en Salud Pública tiene un papel importante, fundamen-talmente en tres áreas:
Investigación epidemiológica: Determinar la pre-valencia de las variantes génicas y la magnitud del riesgo de enfermar asociado a esas variantes, sus interacciones entre sí y con los factores am-bientales.
Investigación en políticas y comunicación: Cuando se va a utilizar la información genética para el beneficio de la salud es importante considerar la necesidad de información y comunicación de los individuos involucrados y desarrollar políti-cas eficientes para el uso adecuado de la infor-mación genética.
Investigación sobre los servicios de salud: Es impor-tante conocer cómo influirían en los distintos
servicios la realización de pruebas genéticas, el manejo de la información obtenida, la modifica-ción de las estrategias de intervención, etc.
Tanto la investigación médica como la investiga-ción en Salud Pública serán necesarias para ha-cer realidad lo enunciado por los doctores Co-llins y Mckusick, quienes aseguran que: “muchos proveedores de atención primaria se converti-rán en los practicantes de la medicina genómica y tendrán que explicar complejas informaciones estadísticas de riesgo genético a personas sanas que desean mejorar sus posibilidades de mante-nerse en buen estado de salud”.
La genética ha hecho grandes progresos en la comprensión de las causas de las relativamente raras enfermedades monogénicas, como la fi-brosis quística y la distrofia muscular. La búsque-da de factores genéticos en enfermedades más prevalentes, sin embargo, como el cáncer o la diabetes, resulta mucho más complejo, costoso y, hasta hace muy poco, prácticamente inútil.
Los estudios de asociación del genoma comple-to (o sus siglas en inglés, GWAS) centran el últi-mo de una serie de poderosos nuevos enfoques de investigación que combinan las tecnologías genómicas de alta resolución con los métodos rigurosos de la epidemiología, comenzando a dar sus frutos al identificar numerosos genes im-plicados en muchas enfermedades prevalentes. Para acelerar la aplicación de estas tecnologías y establecer una base sólida para traducir los resultados en la mejora de la atención médica, el National Human Genome Research Institute (NHGRI) recientemente creó la Oficina de Ge-nómica Poblacional.
Los estudios GWAS son altamente informativos ya que combinan las tecnologías de genómica de alta resolución con los métodos tradicionales de investigación epidemiológica. Los primeros resultados de estos estudios han identificado va-riantes genéticas que contribuyen al cáncer de próstata, cáncer de mama, diabetes, obesidad, etc. En el futuro, los médicos y los genetistas po-drán utilizar estas herramientas para dar a los pacientes información individualizada sobre sus riesgos de desarrollar ciertas enfermedades y, potencialmente, un plan de prevención adapta-do a reducir estos riesgos.

Existe una brecha entre la aplicación de los conocimientos surgidos del Proyecto Genoma Humano y la práctica médica cotidiana
80
La revolución genómica
Para llevar a cabo una exploración de todo el genoma, los investigadores recogen los datos de grupos de personas con la enfermedad y compa-ran su material genético con las personas sin la enfermedad o controles sanos. Luego, utilizando técnicas genómicas de última generación como los arrays de ADN, escanean todo el genoma de cada individuo buscando variaciones caracterís-ticas de la enfermedad en aproximadamente un millón de SNPs. El cambio detectado puede ser neutral y no cambiar el significado de la palabra – en este caso de la proteína – o se puede produ-cir un cambio aminoacídico o en los patrones de expresión del gen.
Estos estudios, con miles de participantes, pro-ducen millones de datos individuales, por lo que requieren el desarrollo de sofisticadas he-rramientas analíticas para distinguir variaciones reales de otras debidas al azar o la posibilidad de que los resultados no pueden ser replicados por otros investigadores. Para lograr esto, los ge-netistas y epidemiólogos tendrán que colaborar estrechamente para identificar factores de ries-go implicados en la salud y la enfermedad de las poblaciones humanas.
La secuenciación del genoma humano ha pro-porcionado al mundo de la medicina una nueva
visión de la relación existente entre salud y en-fermedad, ofreciendo la posibilidad de avanzar hacia una medicina más personalizada, teniendo en cuenta las diferencias genéticas individuales, las causas moleculares de las enfermedades y la influencia de factores ambientales en el desarro-llo de las mismas.
La genética predictiva o genética de riesgo ofre-ce oportunidades únicas de ayuda a los médicos en el control y el entendimiento en profundi-dad de una enfermedad en particular. Hoy en día, las pruebas genéticas pueden confirmar un diagnóstico probable, descartar enfermedades en un diagnóstico diferencial, predecir la apa-rición de futuras enfermedades en un individuo sano, y ayudar a parejas en su planificación fami-liar. El futuro es prometedor, ya que los avances en la tecnología genética podrán ayudar a los médicos en el diagnóstico y en el tratamiento de las enfermedades tanto a través de la farmaco-genética (fármacos confeccionados para indivi-duos con un determinado perfil genético) como a través de la terapia génica.
Muchos de los beneficios de la genética pre-dictiva son evidentes para el médico, por eso es importante que éstos reciban toda la infor-mación que necesiten para poder así ayudar a sus pacientes a entender los beneficios de una prueba genética. Por lo tanto, deberán conocer cómo y cuándo usar un test genético y cuál es la mejor manera de comunicar esta información a sus pacientes para garantizar que puedan tomar decisiones informadas. Ya que el diagnóstico ge-nético puede ir más allá del diagnóstico de un individuo ya enfermo, pues puede “predecir” si un individuo sano desarrollará síntomas en el futuro, este diagnóstico debe ser tratado con gran cuidado. Esto significa que hay que tomar en cuenta varias cuestiones como la confidencia-lidad de la información genética, de tal modo que los análisis genéticos no puedan ser utili-zados de manera impropia (ej. discriminación genética); el proporcionar la ayuda necesaria a los individuos para que conozcan y entiendan los beneficios y riesgos de una prueba genética, de tal modo que puedan tomar una decisión basada en el conocimiento; el cumplimiento de la legislación vigente sobre la realización de las pruebas genéticas, así como la legislación que se desarrolle en el futuro.
Se están desarrollando reglamentos que afectan al consentimiento informado, para asegurar que pacientes que se someten a test genéticos pre-dictivos (test para enfermedades que todavía no presentan síntomas) entiendan los beneficios y riesgos de esta información y sean libres de to-mar la decisión apropiada.
La Enfermedad Genética
Una enfermedad genética se produce por la alte-ración del ADN de un individuo. Está alteración puede afectar a un único gen, a varios genes, a

La genética predictiva o genética de riesgo ofrece oportunidades únicas de ayuda a los médicos en el control y el entendimiento en
profundidad de una enfermedad en particular
Julio 2011 81
ciencia
En cuanto a las enfermedades multifactoria-les o poligénicas, se trata de un grupo de pa-tologías determinadas por la herencia (mu-taciones en varios genes, generalmente en diferentes cromosomas) y por la interacción de múltiples factores ambientales (estilo de vida, nutrición, tipo de trabajo, exposiciones intrauterinas, etc.). Esta interacción entre ge-nes y ambiente conduce a la enfermedad.
La mayoría de los trastornos que afectan a un gran número de individuos a lo largo de su vida, en forma de procesos patológicos, no tienen un origen monogénico, sino que son debidos a múltiples causas, producto de influencias entre el componente genético del individuo, la acción particular de ciertos genes y los factores ambientales más diversos
ocurridos a lo largo de toda la vida, empe-zando por el período intrauterino. Este gru-po de enfermedades es extraordinariamente frecuente; aparecen generalmente en la edad adulta y suelen tener un carácter crónico. Ejemplos de estas patologías son: las enfer-medades neurodegenerativas, la enfermedad de Alzheimer, las enfermedades cardiovas-culares, la hipertensión arterial esencial, las enfermedades cerebrovasculares, el cáncer, la depresión, la esquizofrenia, la obesidad, la diabetes, el asma, la psoriasis, etc.
Las enfermedades poligénicas, también de-nominadas genéticamente complejas, están determinadas por la variación genética, es de-cir por los polimorfismos genéticos, que son variaciones comunes que ocurren en el ADN humano con una frecuencia aproximada de 1 por cada 1000 bases de ADN, y determinan la susceptibilidad o la resistencia a la enfer-medad y la respuesta al tratamiento. A raíz de la finalización del proyecto Genoma Huma-no se han encontrado aproximadamente 3.7 millones de variaciones en la secuencia de ba-ses de ADN humano. De estas variaciones las más comunes son los SNPs (“polimorfismo de un solo nucleótido”). Estas variaciones o polimorfismos genéticos identifican las dife-rencias de los individuos en cuanto a la sus-ceptibilidad a padecer ciertas enfermedades, la resistencia a las mismas o la respuesta a un tratamiento.
un cromosoma o a varios cromosomas. Todas las enfermedades en mayor o menor medida tienen un componente genético. Existen tres grandes grupos de enfermedades genéticas: cromosómicas, monogénicas y multifactoria-les o poligénicas.
Las alteraciones cromosómicas son frecuentes, constituyendo más del 30% de las causas de retraso mental, el 50% de los abortos espon-táneos y afectando a 6.2 de cada 1000 recién nacidos vivos.
Los cromosomas son los portadores del mate-rial genético y por lo tanto de las característi-cas hereditarias.
Las enfermedades cromosómicas se producen por la alteración de los cromosomas de un in-dividuo, pudiendo afectar a su número (pérdi-da y/o ganancia de uno o varios cromosomas completos) o a su estructura (las anomalías estructurales implican cambios en la estructu-ra de uno o varios cromosomas), tales como las deleciones, translocaciones, inversiones, inserciones y duplicaciones.
La citogenética se encarga del estudio de los cromosomas humanos y de sus anomalías, se trata por tanto de una prueba diagnóstica clave en el diagnóstico prenatal, en el retraso mental, en los casos de múltiples defectos de nacimiento, en las alteraciones del desarrollo sexual, en casos de infertilidad, en los abortos de repetición y en el estudio y el tratamiento del cáncer y de las enfermedades hematológi-cas malignas.
Las enfermedades monogénicas se producen por la alteración de la secuencia de ADN de un único gen, dando lugar a una alteración en la proteína codificada por ese gen. Se transmiten de generación en generación, en la descenden-cia, pudiendo manifestarse o no, siguiendo un patrón de herencia mendeliana. Se conocen más de 6000 enfermedades monogénicas, y su prevalencia es de 1 caso por cada 200 naci-mientos. Se cree que más de 10000 enferme-dades son de origen monogénico. Estas enfer-medades se producen por mutaciones en la secuencia nucleotídica de un gen, debido a la sustitución, a la pérdida o a la ganancia de nu-cleótidos. Una mutación puede causar la alte-ración de la secuencia de aminoácidos de una proteína y por lo tanto puede alterar su confor-mación y su función, dando lugar a un fenotipo de enfermedad. Por lo tanto, el efecto de una mutación depende de la naturaleza del cambio de la secuencia de ADN. Las enfermedades mo-nogénicas siguen patrones de herencia clásicos o mendelianos: herencia autosómica (domi-nante o recesiva) y herencia ligada al sexo.

El Riesgo Genético de la Enfermedad
Debido a la interacción entre genes y ambiente, las enfermedades complejas se pueden prevenir a través de la actuación sobre los factores am-bientales con un plan de prevención adecuado.
El conocimiento de los genes implicados en el desarrollo de estas enfermedades nos permite hacer ciertas predicciones sobre los riesgos, sus-ceptibilidades o resistencias a desarrollarlas.
El término susceptibilidad, predisposición ge-nética, o también riesgo genético a padecer una determinada enfermedad, se define como la presencia de determinadas variaciones en la secuencia de ADN y/o la combinación de una serie de ellas (haplotipos) en un individuo, que no son necesariamente anormales, pero que asociados pueden incrementar el riesgo de desarrollar una determinada enfermedad. En otras palabras, una persona ha heredado de sus padres una copia de un gen “problema” que aunque no es un agente causal por sí mismo, si hace más susceptible a la persona a desarrollar una enfermedad. Si las condiciones ambientales son adecuadas y se produce la interacción con el gen “problema” la enfermedad aparecerá. La historia familiar representa un factor importan-te para la estimación del riesgo o susceptibilidad de un individuo de desarrollar una determinada enfermedad. Si un padre tiene una determinada enfermedad, eso no significa que su hijo vaya a padecerla necesariamente, por lo tanto el riesgo no puede ser calculado sino estimado. ¿Qué he-chos de la historia familiar de un individuo ha-cen de él que sea más susceptible a una determi-nada patología? Tener dos o más familiares con una determinada enfermedad; tener un familiar con una enfermedad diagnosticada antes de los 55 años; tener un familiar al que se le diagnos-tica una enfermedad que no es frecuente que afecte a un sexo determinado (ej. el infarto de miocardio en la mujer); la presencia de dos o más enfermedades relacionadas en una familia (ej. diabetes y enfermedad cardiovascular).
La mayoría de las enfermedades de la vida adul-ta (cáncer, demencia, ictus, diabetes, hiperten-sión, hiperlipemias, osteoporosis, cardiopatías, etc.) se engloban dentro del concepto de enfer-medades poligénicas, caracterizadas por defec-
tos genéticos múltiples en diferentes regiones del genoma humano, que ante unas determi-nadas condiciones ambientales nos hacen más susceptibles a la enfermedad. Estas enfermeda-des constituyen una de las principales causas de enfermedad y muerte en casi todo el mundo, causando discapacidad y una gran inversión socio-sanitaria.
Paneles de Riesgo Genético
Las pruebas genéticas de enfermedades comple-jas determinan la susceptibilidad, el riesgo o la probabilidad de un individuo de padecer una en-fermedad. Por lo tanto, el resultado de las prue-bas indica que una persona puede tener mayor probabilidad, riesgo o susceptibilidad que la po-blación general de padecer una enfermedad en particular, pero no significa que la vaya a pade-cer, puesto que ese riesgo se ve influido por otras variables, como los factores externos condicio-nantes (dieta, tabaquismo, sedentarismo, etc.). Este tipo de pruebas genéticas están integradas por paneles multigénicos, ya que en el desarro-llo de estas enfermedades no interviene un solo gen sino que se producen por la interacción de una serie de genes. En estos paneles genéticos se estudian diferentes polimorfismos genéticos o variaciones en la secuencia de ADN que están involucrados en el desarrollo, el pronóstico y la evolución de estas patologías, constituyendo una herramienta clave para la práctica médica.
Así, por ejemplo, el panel genético de Neurode-generación estudia marcadores genéticos de sus-ceptibilidad que indican el riesgo de un indivi-duo para desarrollar demencia, particularmente demencia tipo Alzheimer; y genes implicados en formas familiares de la enfermedad de Alzhei-mer o de herencia autosómica dominante. Los genes que se analizan son marcadores genéticos de susceptibilidad (APOE, PSEN1, A2M, ACE, NOS3 y PRNP); y marcadores genéticos de for-mas familiares de la enfermedad (APP, PSEN1, TAU y PSEN2).
En el caso de la patología vascular concurren múltiples factores ambientales y mutaciones en diferentes genes. Estos polimorfismos genéticos determinan la susceptibilidad que se estudia en el panel genético Cerebrovascular o la resistencia a la enfermedad y la respuesta al tratamiento.
El panel aborda el estudio de genes implicados en los diferentes eventos que desencadenan el proce-so aterogénico, como son: metabolismo lipídico (modificación de LDL-colesterol), función endo-telial, respuesta inmunitaria (reclutamiento de macrófagos y formación de células espumosas) y estabilidad de la placa de ateroma (trombosis).
Las alteraciones cromosómicas son frecuentes, constituyendo más del 30% de las causas de retraso mental, el 50% de los abortos espontáneos y afectando a 6.2 de cada 1000 recién nacidos vivos
82
La revolución genómica

Una vez determinado el perfil genotípico vascu-lar, se pondera el riesgo relativo del paciente y se incide en los hábitos de vida modificables que mayor riesgo suponen para el paciente y que se recomiendan corregir.
El panel genético Cardiovascular está diseñado panel genético Cardiovascular está diseñado panel genético Cardiovascularpara conocer la predisposición de una persona a sufrir enfermedades cardiovasculares. La Hiper-tensión Arterial Esencial es una de las mayores causas de muerte hoy en día en el mundo. La sufren un 30% de la población adulta de las so-ciedades occidentales. La hipertensión arterial se asocia a un incremento del riesgo de padecer infarto de miocardio, fallo cardíaco, fallo renal y accidentes cerebrovasculares. Entre los facto-res genéticos conocemos que los hijos de padres hipertensos tienden a alcanzar mayor presión sanguínea que el resto de la población. Además, se ha demostrado que gemelos idénticos presen-tan una mayor correlación entre sus presiones arteriales sistólicas y diastólicas respecto a los gemelos no-idénticos. Por lo tanto la presión arterial tiene un componente hereditario, que puede ser condicionado por factores medioam-bientales.
Los paneles de Oncogenética estudian los mar-paneles de Oncogenética estudian los mar-paneles de Oncogenéticacadores genéticos involucrados en el desarrollo de distintas formas de cáncer. Los genetistas están continuamente trabajando en el conoci-miento de los mecanismos genéticos básicos que subyacen en el proceso del cáncer, y en cómo este conocimiento puede ser aplicado para per-mitir nuevos y más efectivos diagnósticos y trata-mientos de los pacientes con cáncer. La mayoría de los cánceres surgen a partir de una compleja interrelación entre múltiples genes (oncogenes
y genes supresores tumorales), y de estos genes con factores medioambientales. La información del historial familiar y del test genético aporta un medio para identificar personas con un ele-vado riesgo de padecer cáncer.
El panel genético de Obesidad estudia los tres procesos involucrados en el origen y la evolu-ción de esta condición física que puede conver-tirse en patológica: la Eficiencia Energética, el Control del Apetito y el Metabolismo Lipídico.
El panel genético de Eficiencia Energética anali-panel genético de Eficiencia Energética anali-panel genético de Eficiencia Energéticaza las causas potenciales de obesidad debidas a alteraciones en genes que se expresan en el teji-do adiposo. Se centra en genes reguladores del crecimiento y la diferenciación de los adipocitos y genes relacionados con el gasto energético.
El panel genético de Control del Apetito analiza las causas potenciales de obesidad por altera-ciones en genes relacionados con el centro de control del apetito. Son genes reguladores de proteínas que controlan las señales de hambre y saciedad.
El panel genético de Metabolismo Lipídico ana-liza las causas potenciales de obesidad debidas a alteraciones de genes que intervienen en el metabolismo de los lípidos. Los polimorfismos genéticos elegidos analizan la predisposición de un individuo a sufrir obesidad y/o riesgos adi-cionales relacionados con la misma.
Existen más de 10.000 enfermedades debidas a mutaciones en un único gen
Julio 2011 83
ciencia

Otro tipo de estudios genéticos, como el panel de Detoxificación, analiza la actividad de di-versas enzimas implicadas en los mecanismos fisiológicos reguladores de la carga tóxica del organismo. La actividad de dichas enzimas es di-ferente en cada individuo ya que son altamente polimórficas. Las variaciones genéticas elegidas analizan la predisposición de un individuo a su-frir un desequilibrio en la eliminación de estos compuestos tóxicos (toxinas, procarcinógenos, radicales libres, etc.).
El panel Farmacogenético de los CYPs analiza cuatro de los genes más importantes de la fami-lia del citocromo P450, también denominados CYPs, que son enzimas responsables del meta-bolismo oxidativo (fase I) de muchas drogas, esteroides y agentes carcinógenos. El objeto de este análisis es determinar la función del indivi-
duo a la hora de metabolizar dichos fármacos y valorar si presenta un perfil de metabolización rápido, lento o normal, y evitar en lo posible efectos indeseables como las reacciones adver-sas extremas.
El objetivo del panel Farmacogenético de los Anticoagulantes cumarínicos es valorar la seguridad y eficacia de la terapia anticoa-gulante. Analiza polimorfismos de los genes CYP2C9 y CYP2C9 y CYP2C9 VKORC1 implicados en el metabo-lismo y en la acción de dichos fármacos. Los polimorfismos genéticos elegidos son C430T, A1075C y G-1639A, relacionados con la ca-pacidad de un individuo para metabolizar y responder a un tratamiento con anticoagu-lantes.
Nuevas tecnologías al servicio de la Genética
Desde que la secuenciación del ADN fue-ra descrita en 1977, sucesivas mejoras en las técnicas y equipos de análisis, así como en la bioinformática necesaria para el análisis, han
La mayoría de las enfermedades de la vida adulta (cáncer, demencia, ictus, cardiopatías, diabetes, hipertensión) se engloban dentro de las denominadas enfermedades poligénicas
84
La revolución genómica

permitido la automatización y han mejorado la relación coste-beneficio de este tipo de análisis genéticos y su utilidad en la práctica médica.
En los últimos años han surgido en el panora-ma de la genómica médica diversos métodos de secuenciación masiva en paralelo denomi-nada genéricamente como “next-generation sequencing” (NGS) o secuenciación de segun-da generación, que permite la secuenciación de grandes extensiones de ADN de manera rápida y asequible.
La secuenciación de segunda generación aún no tiene un gran impacto en la clínica diag-nóstica, pero con la promesa del “genoma de 1000 dólares” de la mano del proyecto “1000 genomes”, parece cuestión de tiempo que esta nueva tecnología se implante de manera ruti-naria en todos los laboratorios de diagnóstico molecular.
La secuenciación de segunda generación per-mite buscar simultáneamente mutaciones en cientos de loci para desórdenes genéticamen-te heterogéneos, entre los que podemos citar la muerte súbita, así como las principales en-fermedades complejas: cáncer, enfermedades degenerativas y accidentes cardio- y cerebro-vasculares. Además, la secuenciación masiva en paralelo permitirá abordar de manera más comprensiva disciplinas tales como la farma-cogenética y la epigenética, integrando datos globales que permitan interpretar interaccio-nes génicas y mecanismos epigenéticos de re-gulación en la expresión.
El genoma humano comprende 6400 millo-nes de nucleótidos en dos dotaciones de 23 cromosomas. Las variaciones interindividua-les abarcan aproximadamente 6 millones de SNPs, unas 1000 variantes estructurales (SVs) de más de 3 kb y muchas más variaciones es-tructurales pequeñas, responsables de la varia-ción fenotípica entre individuos.
Las estrategias actualmente empleadas para detectar esta variabilidad pasan por la utiliza-ción de arrays de hibridación de SNPs, Real Time PCR y secuenciación cíclica de Sanger, con lo que la capacidad de detección de va-riaciones responsables de cambios fenotípicos es relativamente limitada. La llegada de la se-cuenciación NGS permitirá cambiar la escala en la longitud de la secuencia analizada, pa-sando de estudios de kilobases a megabases y, a medio plazo, estudios genómicos completos.
Conceptos tales como la PCR en emulsión (“emPCR”), la pirosecuenciación y la “profun-didad” de la secuenciación en paralelo, son
términos que pronto nos serán ciertamente familiares a los profesionales del diagnóstico molecular.
Actualmente, cuando hablamos de paneles ge-néticos predictivos que abarcan 30 o 40 genes, realmente estamos analizando unas decenas de SNPs, con lo que el esfuerzo de análisis se queda en la superficie de la variabilidad de es-tos genes y, por lo tanto, en la superficie de los cambios fenotípicos que explican el riesgo de padecer la enfermedad. La secuenciación masiva permitirá abordar en breve el análisis de estos 30 o 40 genes de manera global, toda la secuencia y toda la variabilidad que puedan entrañar, por lo que la capacidad predictiva de estos análisis se incrementará de manera exponencial acercando el cálculo de riesgo a niveles próximos al diagnóstico cierto.
La Medicina en la Era Post-Genómica
Ya durante el desarrollo fetal la interacción genotipo-ambiente condiciona el estableci-miento de las cualidades físicas, mentales, in-telectuales y, hasta cierto punto, la predispo-sición a padecer enfermedades más o menos graves durante el ciclo vital del individuo. Las condiciones nutricionales del feto y de la ma-dre condicionan la regulación epigenética en la expresión de determinados genes.
Hoy día conocemos que el ambiente fetal predispone al individuo a futuros cuadros de obesidad, hipertensión, dislipemias y, conse-cuentemente, patologías cerebro- y cardio-vasculares. También comenzamos a entender cómo mutaciones en genes implicados en la respuesta inmunitaria, como es el caso de factor del complemento H, predisponen al individuo a desarrollar enfermedades de tipo infeccioso como la meningitis que, a priori, no le presuponíamos componente genético alguno.
Los paneles de riesgo genético estudian diferentes variaciones en la secuencia de
ADN que están involucradas en el desarrollo, el pronóstico y la evolución de las patologías
complejas, constituyendo una herramienta clave para la práctica médica
Julio 2011 85
ciencia

La Medicina basada en el conocimiento exLa Medicina basada en el conocimiento ex-haustivo del genoma del individuo y de los haustivo del genoma del individuo y de los mecanismos que rigen la regulación en la exmecanismos que rigen la regulación en la ex-presión génica, así como la comprensión de presión génica, así como la comprensión de las interacciones entre los factores de riesgo las interacciones entre los factores de riesgo genéticos y ambientales dará lugar, no tan sólo genéticos y ambientales dará lugar, no tan sólo a un mejor tratamiento del individuo enfera un mejor tratamiento del individuo enfer-a un mejor tratamiento del individuo enfer-a un mejor tratamiento del individuo enfermo, sino más bien a la planificación de una mo, sino más bien a la planificación de una vida saludable de los individuos, aún sanos, vida saludable de los individuos, aún sanos, desde el momento de su nacimiento. Desde el desde el momento de su nacimiento. Desde el primer minuto de la vida del individuo, el méprimer minuto de la vida del individuo, el mé-dico conocerá el perfil farmacogenético del dico conocerá el perfil farmacogenético del recién nacido, no se arriesgará a la práctica recién nacido, no se arriesgará a la práctica irresponsable del ensayo-error, confiando en irresponsable del ensayo-error, confiando en los avances de la medicina de urgencias y en los avances de la medicina de urgencias y en la pericia de sus colegas para paliar los efectos la pericia de sus colegas para paliar los efectos de tan nociva praxis terapéutica.de tan nociva praxis terapéutica.
El médico, desde el mismo momento del naci-miento del individuo, conocerá las anomalías genéticas presentes en el genoma de dicho su-jeto, la concurrencia de polimorfismos de ries-go y la probabilidad “a priori” de desarrollar la patología. De este modo, el médico podrá anticiparse a la presentación de la misma, es-tableciendo pautas saludables en cuanto a la dieta y vigilando más estrechamente los facto-
8686
La revolución genómica
1. Bentley DR, Balasubramanian S, Swerdlow HP et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008; 456:53-9.
2. Glass CK, Witztum JL. Atherosclerosis: The road ahead. Cell 2001; 104:503-16.
3. Hindorff LA, Junkins HA, Hall PN, Mehta JP, Manolio TA. A catalog of published genome-wide association studies. National Human Genome Research Institute, 2010. (Accessed May 19, 2011, at http://www.genome.gov/gwastudies).
4. Jeffreys AJ, Wilson V, Thein SL. Individual-specific ‘fingerprints’ of human DNA. Nature 1985, 316:76-9.
5. Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA 1977; 74:560-4.
6. Paré G, Serre D, Brisson D et al. Genetic analysis of 103 candidate genes for coronary artery disease and associated phenotypes in a founder population reveals a new association between endothelin-1 and high-density lipoprotein cholesterol. Am J Hum Genet 2007; 80:673-82.
7. Sachidanandam R, Weissman D, Schmidt SC et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001; 409:928-33.
8. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad Sci USA 1977; 74:5463-7.
9. Siva N. 1000 Genomes project. Nat Biotechnol 2008; 26:256.
Referencias Bibliográficas:
Juan C. Carril [email protected]
res de riesgo modificables que podrían desen-cadenar la aparición de la patología para la que el individuo presenta una elevada predis-posición. Pero no sólo esto, sino que en el mo-mento de aparición de los primeros síntomas, el médico tendrá claro cuál es la mejor opción terapéutica para su paciente.
En definitiva, estaremos ante la práctica universal de una medici-na personalizada y fundamental-mente preventiva, dejando atrás los tiempos oscuros de la medici-na paliativa o reparadora.
La Tarjeta Farmacogenética le indica al paciente y a su médico qué medicamentos puede tomar y cuales debe evitar para no sufrir reacciones adversas

87Julio 2011
VConferencia Anual EuroEspes
II Reunión de la Asociación Mundialde Medicina Genómica (WAGEM)
Biotecnología y Genómica
sociedad
A las 09:00 horas, el Prof. Dr. Ramón Cacabelos, acompañado por el Profesor Masatoshi Takeda y por el Dr. Adolfo Sánchez, Vicerrector de Investi-gación de la Universidad Camilo José Cela de Ma-drid, inauguró el acto, dando la bienvenida a los asistentes y anticipándoles el formato que tendría la Conferencia: una primera parte dedicada al uso de la genómica como instrumento tanto predicti-vo de riesgo como diagnóstico y terapéutico; una segunda parte dedicada a los resultados de la in-vestigación básica y clínica llevada a cabo con bio-productos patentados por el Grupo EuroEspes y ya disponibles en muchos países; y un tercer bloque consistente en una muestra de la utilidad de la ge-nética como herramienta a usar en beneficio de la seguridad ciudadana, y cómo debe regularse el uso de la misma. El Dr. Cacabelos terminó el acto de bienvenida dándoles las gracias a los ponentes invitados, y deseando a los asistentes un día prove-choso en que todos pudieran aprender algo nue-vo. El Profesor Takeda y el Dr. Sánchez también pronunciaron unas palabras de bienvenida.
Adam McKayFundación EuroEspes,
15165-Bergondo, Coruña
l pasado día 17 de diciembre de 2.010, se celebró ante una numerosa audiencia en el auditorio del Centro de Investigación Biomédica EuroEspes, en Bergon-de Investigación Biomédica EuroEspes, en Bergon-de Investigación Biomédica EuroEspes, en Bergondo, A Coruña, la V Conferencia Anual EuroEspes, coincidiendo con la II Reunión de la Asociación Mundial de Medicina Genómica (WAGEM).WAGEM).WAGEM
Como tema central de esta Conferencia, se eli-gió la Biotecnología y Genómica, desde el con-texto de su aplicación en beneficio de la salud y del bienestar de la sociedad. Por este motivo, las ponencias tuvieron siempre un perfil práctico y documental.E

Efecto de E-CONGERINE-10423® (25 μL/pocillo)
sobre la permeabilidad de la monocapa de las líneas
tumorales SW-872, SW-982 y HT29-M6
88
V Conferencia Anual EuroEspes: Biotecnología y Genómica
A continuación, la Dra. Lucía FerA continuación, la Dra. Lucía Fer-A continuación, la Dra. Lucía Fer-A continuación, la Dra. Lucía Fernández-Novoa presentó su ponennández-Novoa presentó su ponen-cia “cia “Diagnóstico molecular y genética médica de patologías prevalentes: enfermédica de patologías prevalentes: enfer-médica de patologías prevalentes: enfer-médica de patologías prevalentes: enfermedades cardiovasculares, cáncer y trasmedades cardiovasculares, cáncer y tras-medades cardiovasculares, cáncer y tras-medades cardiovasculares, cáncer y trastornos neuropsiquiátricostornos neuropsiquiátricos”. En ella, hatornos neuropsiquiátricos”. En ella, hatornos neuropsiquiátricos -”. En ella, ha-”. En ella, ha
bló sobre la necesidad de conocer el perfil genético del individuo, con el objetivo de predecir la apari-del individuo, con el objetivo de predecir la apari-del individuo, con el objetivo de predecir la apari
en temas de vital importancia: el entendimiento de en temas de vital importancia: el entendimiento de la etiopatogenia de las enfermedades, la implanta-la etiopatogenia de las enfermedades, la implanta-la etiopatogenia de las enfermedades, la implantación de procedimientos de riesgo predictivo, y la optimización de los recursos terapéuticos. En su ponencia, el Profesor Cacabelos mostró datos ob-tenidos de la base de datos PubMed (NCBI) en di-ciembre, donde se mostraba el liderazgo mundial de EuroEspes en la Farmacogenómica del Sistema de EuroEspes en la Farmacogenómica del Sistema Nervioso Central.Nervioso Central.Nervioso Central.
La primera ponencia de la mañaLa primera ponencia de la mañaLa primera ponencia de la mañaLa primera ponencia de la maña--La primera ponencia de la maña-La primera ponencia de la mañaLa primera ponencia de la maña-La primera ponencia de la mañana, a cargo del Profesor Cacabelos, na, a cargo del Profesor Cacabelos, llevaba por título: “llevaba por título: “Biotecnología de la saludsalud”. El Profesor declaró que éste salud”. El Profesor declaró que éste saludha sido uno de los campos de máxiha sido uno de los campos de máxi-
ma expansión durante los últimos 20 años, y que ma expansión durante los últimos 20 años, y que ha permitido un avance espectacular en diferentes áreas de interés sanitario, entre ellas la de la Medi-cina Genómica. Ésta ha abierto nuevos horizontes cina Genómica. Ésta ha abierto nuevos horizontes
ción de las enfermedades. El conocimiento del geción de las enfermedades. El conocimiento del ge-noma es de gran utilidad en aquellos pacientes cuya historia familiar indica riesgo aumentado para una enfermedad en particular, como por ejemplo las enfermedades cardiovasculares, que representan la primera casusa de muerte en la Unión Europea, o los trastornos mentales, que representan el 20% del los trastornos mentales, que representan el 20% del total de enfermedades.
La tercera ponencia de la mañana La tercera ponencia de la mañana La tercera ponencia de la mañana La tercera ponencia de la mañana fue a cargo del Dr. Juan Carlos Cafue a cargo del Dr. Juan Carlos Ca-fue a cargo del Dr. Juan Carlos Ca-fue a cargo del Dr. Juan Carlos Carril, quien habló de la “rril, quien habló de la “Farmacoge-Farmacoge-Farmacogenética aplicada a la personalización del nética aplicada a la personalización del tratamiento: perfiles genéticos de fármacos tratamiento: perfiles genéticos de fármacos
de alto consumode alto consumo”. El Dr. Carril comparó la administra”. El Dr. Carril comparó la administrade alto consumo”. El Dr. Carril comparó la administrade alto consumode alto consumo”. El Dr. Carril comparó la administrade alto consumo -”. El Dr. Carril comparó la administra-”. El Dr. Carril comparó la administración tradicional de fármacos (de forma masiva, una ción tradicional de fármacos (de forma masiva, una misma dosis para todos los pacientes) con el uso de
la farmacogenética y farmacogenómica para lograr la farmacogenética y farmacogenómica para lograr una dosis individualizada y para eliminar el factor de ensayo-y-error, que puede dar lugar a costes innecesa-ensayo-y-error, que puede dar lugar a costes innecesa-ensayo-y-error, que puede dar lugar a costes innecesarios y a efectos adversos que hasta pueden conllevar la muerte del paciente. Mencionó, como dato pre-ocupante, que más del 70% de las prescripciones de medicamentos que se realizan no son las adecuadas medicamentos que se realizan no son las adecuadas para la capacidad metabolizadora de los pacientes.
Tras un breve descanso, el Dr. Pablo Tras un breve descanso, el Dr. Pablo Tras un breve descanso, el Dr. Pablo Tras un breve descanso, el Dr. Pablo Carnota ocupó el estrado para dar Carnota ocupó el estrado para dar su ponencia: “su ponencia: “Neuro-Oftalmologenó-Neuro-Oftalmologenó-Neuro-Oftalmologenómicamica”. Habló de la importancia de mica”. Habló de la importancia de micalos factores genéticos en el estudio los factores genéticos en el estudio
como ejemplos mencionó la neuropatía óptica hecomo ejemplos mencionó la neuropatía óptica he-reditaria de Leber (LHON), una enfermedad de origen mitocondrial, la degeneración macular aso-ciada a la edad (DMAE), una patología de origen multifactorial, y el glaucoma; todos dependientes
El Dr. Valter Lombardi inauguró la El Dr. Valter Lombardi inauguró la parte de la Conferencia dedicada a parte de la Conferencia dedicada a la investigación realizada con biola investigación realizada con bio-la investigación realizada con bio-la investigación realizada con bioproductos patentados por el Grupo productos patentados por el Grupo EuroEspes con su ponencia: “EuroEspes con su ponencia: “Biotec-Biotec-Biotec
nología y propiedades antitumorales de Congerina (Antinología y propiedades antitumorales de Congerina (Anti-nología y propiedades antitumorales de Congerina (Anti-nología y propiedades antitumorales de Congerina (AntiGan)”. Presentó resultados de ensayos realizados con Gan)”. Presentó resultados de ensayos realizados con Gan)un nuevo compuesto nutracéutico de origen marino, Congerina (E-Congerine-10423®, AntiGan®), obteni-), obteni-), obtenido a partir de la especie Conger conger, que ha demosConger conger, que ha demosConger conger -, que ha demos-, que ha demos
trado una significativa actividad antitumoral. En particular men--particular men-particular mencionó la activi-cionó la activi-cionó la actividad citotóxica in in vitro de Congevitro de Congevitro -rina utilizando diferentes líneas tumorales.
Efecto de E-CONGERINE-10423® (25 μL/pocillo)
sobre la permeabilidad de la monocapa de las líneas
los factores genéticos en el estudio los factores genéticos en el estudio de las enfermedades que afectan al sistema visual;
multifactorial, y el glaucoma; todos dependientes del factor genético.
El Dr. Valter Lombardi inauguró la El Dr. Valter Lombardi inauguró la trado una signi-trado una signi-trado una signi

Julio 2011 89
sociedad
12. Cacabelos et al. 2010.
A continuación, D. Pablo Bourkaib presentó su ponencia: “Nutrigenómica y actividad hipolipemiante y antiarteriosclerótica de Sardilipina (LipoEsar®y antiarteriosclerótica de Sardilipina (LipoEsar®y antiarteriosclerótica de Sardilipina (LipoEsar )”. E-)”. E-)SAR-94010®, o Sardilipina®, o Sardilipina®, o Sardilipina , es un extracto lipopro-teico no desnaturalizado obtenido a partir de la especie S. pilchardus. En el año 1999, los primeros S. pilchardus. En el año 1999, los primeros S. pilchardusestudios preclínicos mostraron un efecto hipolipe-miante y reductor de los niveles de colesterol y tri-miante y reductor de los niveles de colesterol y tri-miante y reductor de los niveles de colesterol y triglicéridos. En 2010, un tratamiento multifactorial, integrado por 750 mg de LipoEsar®integrado por 750 mg de LipoEsar®integrado por 750 mg de LipoEsar entre otros,
y LDL-Cho. La conclusión de estos estudios es que LipoEsar®estudios es que LipoEsar®estudios es que LipoEsar tiene un importante efecto hipolipemiante.
y LDL-Cho. La conclusión de estos tiene un
La última ponencia de la mañana, con el título: “Biopropiedades y efectos clínicos de Juritrofin (DefenVid®Biopropiedades y efectos clínicos de Juritrofin (DefenVid®Biopropiedades y efectos clínicos de Juritrofin (DefenVid )” )” )corrió a cargo de la Dra. Lola Corzo. Juritrofin® (E-JUR-94013®) es un complemento alimenticio a base de extracto lipoproteico de T. trachurus procedente T. trachurus procedente T. trachurus
del Océano Atlántico. En complejos del Océano Atlántico. En complejos análisis para la detección del princianálisis para la detección del princi-análisis para la detección del princi-análisis para la detección del principio activo responsable de su efecto pio activo responsable de su efecto inmunomodulador, se llegó a la inmunomodulador, se llegó a la conclusión de que el aminoácido conclusión de que el aminoácido espinacina podría ser el responsable de la actividad espinacina podría ser el responsable de la actividad inmunomoduladora observada en los ensayos in vitro realizados. Los resultados obtenidos mediante vitro realizados. Los resultados obtenidos mediante vitrola determinación de diferentes marcadores de acti-la determinación de diferentes marcadores de acti-la determinación de diferentes marcadores de activación linfocitaria por citometría de flujo demostra-vación linfocitaria por citometría de flujo demostra-vación linfocitaria por citometría de flujo demostraron una significativa inmunoactivación comparado con un grupo control y con otros conocidos induc-con un grupo control y con otros conocidos induc-con un grupo control y con otros conocidos induc
A continuación, y con el Profesor Cacabelos como moderador, se inició una discusión en la cual todos los ponentes de la sesión de la mañana respondie-
Tras el descanso para comer, el Profesor Dr. Masatoshi Takeda, quien había acudido desde el Japón para estar presente, tomó el estrado para pronun-ciar su ponencia: “Desafíos de la Medicina Genómica del futuro”. Habló de enfermedades neurodegenedel futuro”. Habló de enfermedades neurodegenedel futuro -rativas crónicas como el Alzheimer, el Parkinson y la esclerosis lateral amiotrófica entre otras, y afirmó que la mayoría de los casos de estas enfer-afirmó que la mayoría de los casos de estas enfer-afirmó que la mayoría de los casos de estas enfer
medades son esporádicos; los casos familiales representan solamente el 5-10% de los casos. Mencionó los genes causales y de riesgo para estas enfermedades, y los tests que se realizan, utilizando la terapia ge-nética, para investigar las enfermedades neuropsi-quiátricas.
medades son esporádicos; los casos familiales representan solamente el 5-10% de los casos. Mencionó los genes causales y de riesgo para estas enfermedades, y los tests que
-
bros de la audiencia, destacando la participación de varios miembros venidos de fuera de España. A las 14:00 horas aproximadamente, el Dr. Cacabelos dio
A continuación, D. Pablo Bourkaib presentó su y LDL-Cho. La conclusión de estos y LDL-Cho. La conclusión de estos
integrado por 750 mg de LipoEsarrealizado en 765 pacientes con demencia mostró una significativa reducción en los niveles de T-Cho
La última ponencia de la mañana, con el título: del Océano Atlántico. En complejos del Océano Atlántico. En complejos
tores linfocitarios, lo que lo convierte en un potente inmunoregulador.
los ponentes de la sesión de la mañana respondieron a numerosas preguntas formuladas por miem-ron a numerosas preguntas formuladas por miem-ron a numerosas preguntas formuladas por miem
Tras el descanso para comer, el Profesor Dr. Masa-Tras el descanso para comer, el Profesor Dr. Masa-Tras el descanso para comer, el Profesor Dr. Masa medades son esporádicos; los casos medades son esporádicos; los casos
14:00 horas aproximadamente, el Dr. Cacabelos dio término a la sesión.
Estudio in vitro en linfocitos humanos: 12 voluntarios sanos. Cultivo durante 48h. de linfocitos con 2 concentraciones de E-JUR-94013
CONTROL E-JUR-94013 10 L/w E-JUR-94013 25 L/w
Lombardi et al, 2004
T-Totales Linfocitos B Linfocitos T activados

90
V Conferencia Anual EuroEspes: Biotecnología y Genómica
La Conferencia de Clausura la pronunció D. Carlos Varela, Fiscal Superior de Galicia. Habló del as-pecto jurídico del uso de los avan-ces biotecnológicos y genéticos,
que todavía no gozan de una regulación deta-que todavía no gozan de una regulación deta-que todavía no gozan de una regulación detallada en nuestro ordenamiento jurídico interno, aunque esta cuestión ha sido abordada en el ám-bito internacional y sobre todo por la comunidad
La Conferencia de Clausura la pronunció D. Carlos Varela, Fiscal Superior de Galicia. Habló del aspecto jurídico del uso de los avances biotecnológicos y genéticos,
Al finalizar esta última intervención, se prosiguió con el Acto de Clausura, con el Profesor Cacabelos como moderador y la participación del Dr. Carril, D. Luis
Hombreiro y D. Carlos Varela, quienes respondieron a preguntas formuladas por miembros de la audien-a preguntas formuladas por miembros de la audien-a preguntas formuladas por miembros de la audiencia sobre temas relacionados con sus ponencias.
Tras una breve pausa, tomó la paTras una breve pausa, tomó la palabra D. Luis Hombreiro Noriega, labra D. Luis Hombreiro Noriega, Jefe del Laboratorio Territorial de Jefe del Laboratorio Territorial de Biología-ADN de la Brigada de PoliBiología-ADN de la Brigada de Poli-Biología-ADN de la Brigada de Poli-Biología-ADN de la Brigada de Policía Científica de la Jefatura Superior cía Científica de la Jefatura Superior
áreas técnico-científicas en las que trabaja el Cuerpo Nacional de Policía, haciendo especial énfasis en las técnicas de genética molecular y forense. Gracias a estas técnicas, se han identificado miles de autores de hechos delictivos que de otra forma hubiesen
Tras esta ponencia, el Dr. Cacabelos, Tras esta ponencia, el Dr. Cacabelos, acompañado por D. Javier Sánchez, acompañado por D. Javier Sánchez, se dirigió a los asistentes para inforse dirigió a los asistentes para infor-se dirigió a los asistentes para infor-se dirigió a los asistentes para informarles sobre la Asociación Mundial marles sobre la Asociación Mundial de Medicina Genómica (de Medicina Genómica (World Asso-World Asso-World Asso
ciation of Genomic Medicineciation of Genomic Medicine)(ciation of Genomic Medicine)(ciation of Genomic Medicine WAGEM), creada en el WAGEM), creada en el WAGEM2008 y uno de cuyos compromisos ha sido concluir la World Guide for Drug Use and Pharmacoge-nomics (WG-PGx). Esta guía, que está a punto de nomics (WG-PGx). Esta guía, que está a punto de nomicsver la luz, es el resultado de 5 años de trabajo mul-ver la luz, es el resultado de 5 años de trabajo mul-ver la luz, es el resultado de 5 años de trabajo mul
tidisiciplinar en el que ha participado un nutrido número de personas. La finalidad del proyecto ha sido conseguir un instrumento útil y práctico para el médico prescriptor, para la industria farma-para la industria farma-para la industria farmacéutica, para los farma-céutica, para los farma-céutica, para los farmacéuticos y para las ad-céuticos y para las ad-céuticos y para las administraciones de los estados y las agencias reguladoras.
A continuación, el Dr. Carril, en su A continuación, el Dr. Carril, en su segunda intervención del día, habló segunda intervención del día, habló sobre la “sobre la “Genética en medicina deportiva de alto rendimientode alto rendimiento”. Al igual que para de alto rendimiento”. Al igual que para de alto rendimientoprevenir enfermedades, la genética prevenir enfermedades, la genética
también puede usarse en la búsqueda de los factores también puede usarse en la búsqueda de los factores heredables que afectan a la capacidad física y psicoló-heredables que afectan a la capacidad física y psicoló-heredables que afectan a la capacidad física y psicológica que definen al atleta de élite. Los requisitos para
rendir físicamente al más alto nivel involucran más de 150 genes relacionados con rasgos morfológicos, fi-150 genes relacionados con rasgos morfológicos, fi-150 genes relacionados con rasgos morfológicos, fisiológicos y psicológicos, por lo que la caracterización del fenotipo óptimo del atleta de élite involucrará el estudio de la variabilidad en los genes relacionados con el transporte de oxígeno, el rendimiento mus-con el transporte de oxígeno, el rendimiento mus-con el transporte de oxígeno, el rendimiento muscular y el metabolismo, el control cardiovascular y la función del sistema nervioso central.
A continuación, el Dr. Carril, en su A continuación, el Dr. Carril, en su rendir físicamente al más alto nivel involucran más de
Tras esta ponencia, el Dr. Cacabelos, Tras esta ponencia, el Dr. Cacabelos, tidisiciplinar en el que ha participado un nutrido
gica que definen al atleta de élite. Los requisitos para función del sistema nervioso central.
Tras una breve pausa, tomó la paTras una breve pausa, tomó la pa-Tras una breve pausa, tomó la pa-Tras una breve pausa, tomó la pa áreas técnico-científicas en las que trabaja el Cuerpo
ver la luz, es el resultado de 5 años de trabajo mul reguladoras.
La Conferencia de Clausura la La Conferencia de Clausura la
cía Científica de la Jefatura Superior cía Científica de la Jefatura Superior de Policía de Galicia. En su ponencia, habló de las de Policía de Galicia. En su ponencia, habló de las
de hechos delictivos que de otra forma hubiesen quedado impunes.
ces biotecnológicos y genéticos, europea.ces biotecnológicos y genéticos,

Julio 2011 91
sociedad
Dr. Iván Carrera, como Profesor Dr. Iván Carrera, como Profesor Dr. Iván Carrera, como Profesor Dr. Iván Carrera, como Profesor Adjunto, y el Dr. Pablo Carnota, Adjunto, y el Dr. Pablo Carnota, como Profesor Auxiliar, ambos de como Profesor Auxiliar, ambos de la División de Neurociencias de la la División de Neurociencias de la la División de Neurociencias de la la División de Neurociencias de la Cátedra.
en Neurocienciasen Neurociencias al Profesor al Profesor al Profesor al Profesor Masatoshi Takeda, por más de Masatoshi Takeda, por más de 30 años de investigación en este 30 años de investigación en este campo.
EuroEspes en la expansión de EuroEspes en la expansión de EuroEspes en la expansión de EuroEspes en la expansión de la política internacional del la política internacional del Grupo.
por su eficiencia directiva en la por su eficiencia directiva en la por su eficiencia directiva en la por su eficiencia directiva en la gestión de recursos humanos y gestión de recursos humanos y por su labor al servicio de nues-tra sociedad. tra sociedad.
texto de este discurso en texto de este discurso en texto de este discurso en texto de este discurso en la página web del Grupo: la página web del Grupo: www.euroespes.com/pdf/discurso_www.euroespes.com/pdf/discurso_del_presidente_2010.pdf).
premio fue concedido a Dª Lilia-premio fue concedido a Dª Lilia-premio fue concedido a Dª Lilia-premio fue concedido a Dª Lilia-na Caínzos y a D. Eduardo Casal.na Caínzos y a D. Eduardo Casal.
A continuación, se entregaron las acreditacioA continuación, se entregaron las acreditacio-nes correspondientes a dos nuevos miembros del Staff de la Cátedra EuroEspes de Biotec-nología y Genómicanología y Genómica de la Universidad Camilo de la Universidad Camilo José Cela de Madrid. Dichos miembros son el
Acto seguido, se entregaron premios de la Acto seguido, se entregaron premios de la Fundación EuroEspes a tres destacados per-sonajes presentes en la Cena. En primer lugar, se entregó el Premio a la Educación
En segundo lugar, el Profesor Francesco En segundo lugar, el Profesor Francesco Marotta recibió el Premio a la Colabora-ción Internacional por su cooperación con
El tercer premiado de la noche fue D. Luis El tercer premiado de la noche fue D. Luis García Mañá, Jefe Superior del Cuerpo Na-cional de Policía en Galicia, quien fue agra-ciado con elciado con el Premio a la Excelencia Ejecutiva Premio a la Excelencia Ejecutiva
Por último, y como clausura del acto, el Por último, y como clausura del acto, el Profesor Ramón Cacabelos, Presidente del Grupo EuroEspes, pronunció el mensaje anual de Navidad. Se puede acceder al
Seguidamente, se otorgó credencial a los Me-Seguidamente, se otorgó credencial a los Me-jores Trabajadores del Año. En esta ocasión, el
Tras la entrega de credenciales académicos a los asistentes, se celebró, en el Hotel Hisperia los asistentes, se celebró, en el Hotel Hisperia Finisterre de A Coruña, la tradicional Cena de
Tras la entrega de credenciales académicos a Gala Anual. En el transcurso de la misma, se Gala Anual. En el transcurso de la misma, se entregaron diplomas acreditativos a los ponenentregaron diplomas acreditativos a los ponen-tes de la V Conferencia.

Pharmacogenomics of Metabolic Disease
92
ciencias médicas
Contribución de España al Consorcio Internacional del Genoma del CáncerUn equipo español secuenció el genoma completo de pacientes con leucemia linfática crónica, el más común de los tipos de leucemia en países occidentales, e identificó mutaciones que aportan nuevas claves sobre la enfermedad.
El estudio, publicado en la revista Nature, fue dirigido por los investigadores Elías Campo, del Hospital Clínic y la Universidad de Barcelona, y Carlos López-Otín, de la Universidad de Oviedo. Este trabajo representa la primera contribución de España al Consorcio Internacional del Genoma del Cáncer (ICGC). El ICGC es el mayor proyecto de la historia en la investigación contra el cáncer, y en él participan científicos de 11 países para secuenciar los 50 tipos de cáncer más importantes. En este trabajo utilizaron la más avanzada tecnología para secuenciar los tres mil millones de nucleótidos del genoma completo de las células tumorales de cuatro pacientes, y lo compararon con la secuencia del genoma de las células sanas de los mismos individuos. El investigador López-Otín expuso que “esta aproximación nos ha permitido comprobar que cada tumor ha sufrido unas mil mutaciones en su genoma”. Añadió que “el posterior análisis de los genes mutados en un grupo de más de 300 pacientes permitió identificar cuatro genes cuyas mutaciones provocan el desarrollo de este tipo de leucemia”.
Puente XS, Pinyol M, Quesada V et al. Nature 2011. doi:10.1038/nature10113.
Calbindina, nuevo biomarcador en la enfermedad de AlzheimerEn un trabajo reciente, Rebecca Craig-Schapiro y colaboradores han estudiado diferentes citoquinas, factores de crecimiento y marcadores metabólicos en un elevado número de muestras de líquido cefalorraquídeo pertenecientes a pacientes en estadios iniciales de la enfermedad de Alzheimer. Los resultados analizados mediante una metodología estadística múltiple han evidenciado la presencia de nuevos biomarcadores que pueden mejorar la capacidad de diferenciación de los marcadores convencionales, como la Aβ42 y la proteína tau, para poder determinar el estadio de la enfermedad y predecir el futuro deterioro de los pacientes. Uno de los nuevos marcadores con un potencial pronóstico significativo es la calbindina, una proteína reguladora del metabolismo del calcio intracelular.
Craig-Schapiro R, Kuhn M, Xiong C et al. PLoS One 2011. doi:10.1371/journal.pone.0018850.
Iniciativa europea sobre las demencias
El pasado 19 de enero, el pleno del Parlamento Europeo aprobó, por una amplia mayoría, el texto de la iniciativa europea sobre la enfermedad de Alzheimer y otras demencias.
Esta resolución es un paso clave para avanzar con propuestas concretas de vertebración de las políticas existentes y de las formas de hacer frente a este tipo de enfermedades.
Entre otras cosas, se pide al Consejo que declare la demencia como prioridad de la Unión Europea en materia de salud e insta a los Estados miembros a elaborar planes y estrategias nacionales específicos para abordar las consecuencias sociales y sanitarias de la demencia, y a prestar servicios y apoyo a las personas con demencia y a sus familias.
Destaca el carácter primordial de la prevención y para ello propone a los Estados miembros a que participen en la definición, el desarrollo y la implementación de protocolos para el diagnóstico precoz.
Subraya la importancia de un enfoque multidisciplinario en la coordinación de la investigación sobre la validación de nuevos criterios de diagnóstico, el desarrollo de pruebas de detección precoz y la determinación de los factores de riesgo para el desarrollo de la enfermedad desde la fase de la predemencia hasta fases más avanzadas.
Con esta iniciativa se trata de concienciar a todos los países sobre la importancia de los tratamientos preventivos que ayudan a retrasar la aparición de la demencia.
DOUE 2010/2084(INI).
La materia gris y las personas distraídasUna reciente investigación, encabezada por el neurocientífico Ryota Kanai, del University College de Londres, ha revelado una inesperada University College de Londres, ha revelado una inesperada University Collegerelación entre la tendencia a distraerse con cualquier cosa y el tamaño del cerebro, concretamente con la región del córtex parietal superior izquierdo. Según los resultados de este estudio, las personas que se distraen con más facilidad suelen tener un mayor volumen de materia gris en esta región cerebral. Es decir, tienen más densidad de neuronas en una de las áreas del cerebro que, precisamente, se utiliza en la acción de prestar atención a una tarea. El descubrimiento es contra-intuitivo, como reconocen sus autores, porque tener más células grises debería suponer un mejor resultado a la hora de mantener la concentración. Pero la realidad es justo la contraria.
En cualquier caso, estos resultados obtenidos por Kanai no indican una mayor o menor inteligencia, sino que tan sólo constatan una relación entre la morfología de este área cerebral -lo que no excluye que otras puedan estar también involucradas- y la capacidad de atención.
Kanai R, Dong MY, Bahrami B, Rees G. J Neurosci 2011; 31:6620-6.
Importancia del RNA no codificanteInvestigadores italianos han determinado la función de uno de los fragmentos de RNA, conocido como 38A, que proviene de una parte no codificante del gen que codifica la proteína KCNIP4. KCNIP4 ayuda a asegurar que los impulsos de las neuronas tengan un patrón característico lento y repetitivo. Los investigadores encontraron una variante del RNA 38A, Var IV, que interrumpe esta corriente y podría dar lugar a la neurodegeneración. KCNIP4 normalmente interactúa con la gamma-secretasa, el complejo enzimático que ayuda a generar beta-amiloide, una proteína que se acumula en el cerebro de pacientes con EA. Los investigadores encontraron que los niveles de 38A fueron más de 10 veces mayores en las células cerebrales de pacientes con EA que en los controles.
Massone S, Vassallo I, Castelnuovo M et al. J Cell Biol 2011; 193:851-66.
Los genes de la migrañaLa migraña es un trastorno neurológico común, heterogéneo y hereditario, que afecta a cerca del 20% de la población y es más frecuente entre las mujeres. Su fisiopatología no está del todo definida y las influencias genéticas que la provocan son un misterio para la comunidad científica.
Sin embargo, recientemente se ha publicado un estudio que nos acerca un poco más a la comprensión biológica de esta dolencia en la población general. Las investigaciones, realizadas por científicos del Brigham and Women’s Hospitalde Boston, revelan el descubrimiento de un trío de genes ligados a la migraña. Dos de los genes, PRDM16 y PRDM16 y PRDM16 TRPM8, TRPM8, TRPM8eran específicos para la migraña, y además, el gen TRPM8estaba vinculado sólo a la migraña en las mujeres. Por su parte, el tercer gen implicado, el LRP1, interactúa con algunas de las vías de neurotransmisores, y puede modular la respuesta del nervio que promueve o reprime los ataques de migraña.
Chasman DI, Schürks M, Anttila V et al. Nat Genet 2011. doi:10.1038/ng.856.
Medicina Personalizada en la lucha contra el melanomaLa esperanza de vida de un paciente diagnosticado de melanoma raramente supera el año de vida desde el momento del diagnóstico. La caracterización genética de las mutaciones ocurridas en el gen BRAF en enfermos de melanoma permite BRAF en enfermos de melanoma permite BRAFque un nuevo fármaco (PLX4032), desarrollado por Plexxikon, bloquee el patrón de expresión de BRAF y ralentice la evolución BRAF y ralentice la evolución BRAFde la enfermedad.
Bollag G, Hirth P, Tsai J et al. Nature 2010; 467:596-9.Flaherty KT, Puzanov I, Kim KB et al. N Engl J Med 2010; 363:809-19.Tap WD, Gong KW, Dering J et al. Neoplasia 2010; 12:637-49.
Vacuna comestibleLos principales signos de la enfermedad de Alzheimer (EA) son el deterioro cognitivo y la aparición en determinadas áreas del cerebro de pacientes con EA de placas seniles fundamentalmente compuestas por depósitos de β-amiloide. Por lo tanto, las tendencias terapéuticas actuales se centran principalmente en eliminar estos depósitos. En un estudio reciente realizado en la Universidad de Tokio, el Dr. Nojima y colaboradores han desarrollado una vacuna comestible que utiliza los mecanismos de inmunidad intestinal y no presenta ningún problema de efectos colaterales no deseados. Esta vacuna se ha producido mediante la expresión de GFP-Aβ
42
en arroz. Una vez conseguida la expresión de esta proteína, el arroz transformado fue administrado oralmente a ratones “wild-type” (WT) obteniéndose de esta forma anticuerpos anti- β-amiloide. Los resultados de este estudio demuestran claramente la utilidad de esta metodología, y por otro lado demuestran que el arroz mantenido a temperatura ambiente durante más de un año seguía siendo capaz de inducir una eficaz respuesta inmunogénica. Este estudio representa el primer paso para la obtención de una vacuna frente a la EA; los próximos estudios continuarán con modelos de EA en ratones transgénicos.
Nojima J, Ishii-Katsuno R, Futai E et al. Biosci Biotechnol Biochem 2011; 75:396-400.
Iniciativa europea sobre las demencias

ciencia
Julio 2011 93
ciencias médicas
Caracterización epigenética en diferentes patologíasManel Esteller, director del Programa de Epigenética y Biología del Cáncer del Instituto de Investigación Biomédica de Bellvitge (IDIBELL), profesor de la Universitat de Barcelona e investigador ICREA, ha coordinado un trabajo en el cual se ha caracterizado por primera vez el epigenoma de 1628 personas, gracias al gran avance tecnológico en la genómica. La epigenética estudia los fenómenos que no afectan a la secuencia de ADN de los genes, pero que sí varían su expresión, de tal manera que estas variaciones epigenéticas controlan la actividad de los mismos. Es la herencia de patrones de expresión de genes que no vienen determinados por la secuencia genética (la cadena de pares de bases del ADN de cada individuo). El mecanismo epigenético por excelencia es la metilación del ADN. La epigenética está implicada en numerosas enfermedades, pero es en el cáncer donde se han realizado el mayor número de estudios de investigación. Mediante este estudio, realizado por el grupo de Esteller, se han caracterizado los perfiles de metilación del ADN en tejidos normales procedentes de personas de diferentes edades, y en tejidos asociados a determinadas patologías, fundamentalmente el cáncer. La conclusión principal de este estudio es la existencia de diferentes perfiles de metilación en relación a la mayoría de los casos estudiados, siendo el cáncer la patología donde se han observado cambios epigenéticos más extremos. El estudio demuestra que todos los tumores humanos sufren la inactivación epigenética de genes protectores del cáncer, y además, la célula tumoral pierde su memoria epigenética y no recuerda de qué tejido sano formaba parte.
Fernandez AF, Assenov Y, Martin-Subero J et al. Genome Res 2011. doi:10.1101/gr.119867.110.
Un autobús recorre España para diagnosticar gratis el cáncer de pielLa Fundación de la Academia Española de Dermatología y Venereología (AEDV) ha puesto en marcha un autobús La Fundación de la Academia Española de Dermatología y Venereología (AEDV) ha puesto en marcha un autobús itinerante donde unos especialistas analizarán, de forma gratuita, los lunares de las personas que lo deseen. José Carlos Moreno, itinerante donde unos especialistas analizarán, de forma gratuita, los lunares de las personas que lo deseen. José Carlos Moreno, presidente de la AEDV, explica que para prevenir la aparición del melanoma (la variedad más grave de cáncer de piel), un autobús presidente de la AEDV, explica que para prevenir la aparición del melanoma (la variedad más grave de cáncer de piel), un autobús recorrerá diferentes ciudades españolas con un equipo de dermatólogos que analizará los lunares ‘de riesgo’ de las personas que recorrerá diferentes ciudades españolas con un equipo de dermatólogos que analizará los lunares ‘de riesgo’ de las personas que se acerquen. El melanoma es un tipo de cáncer cutáneo más grave que los cánceres de células basales o escamosas, que son se acerquen. El melanoma es un tipo de cáncer cutáneo más grave que los cánceres de células basales o escamosas, que son más frecuentes. Si bien el melanoma tiene un menor grado de incidencia que otros tipos de cáncer de piel, presenta el mayor índice más frecuentes. Si bien el melanoma tiene un menor grado de incidencia que otros tipos de cáncer de piel, presenta el mayor índice de mortalidad y es responsable del 75% de las muertes por cáncer de piel. Para prevenir el melanoma, es importante examinar la piel de mortalidad y es responsable del 75% de las muertes por cáncer de piel. Para prevenir el melanoma, es importante examinar la piel de forma regular y familiarizarse con los lunares para poder identificar mejor los cambios. Existe un mayor riesgo de que los lunares presentes al nacer y los lunares atípicos se vuelvan malignos. Reconocer los cambios en los lunares siguiendo las normas del ABCD es crucial en la detección del melanoma maligno en sus etapas tempranas. (A: asimetría; B: bordes irregulares; C: color irregular; D: diámetro superior a 6 mm). Los melanomas varían considerablemente de apariencia. Algunos melanomas pueden mostrar todas las características ABCD, mientras que otros únicamente muestran cambios en una o dos características.
http://www.aedv.es/euromelanoma/
Genética, fármacos y cáncerLa caracterización genética de mutaciones en el gen EGFRy en otros que alteran su expresión (FAS, NFKB, PIK3CA) PIK3CA) PIK3CApermiten determinar la eficacia de Gefitinib y Erlotinib en el tratamiento del cáncer de pulmón.
Bivona TG, Hieronymus H, Parker J et al. Nature 2011; 471:523-6.Sequist LV, Waltman BA, Dias-Santagata D et al. Sci Transl Med 2011; 3:75ra26.
47º Congreso Anual de la Sociedad Americana de Oncología Médica Entre los días 3 y 7 de junio de 2011, casi 30.000 oncólogos se han reunido en Chicago con motivo del Congreso Anual de la Sociedad Americana de Oncología Médica (American Society of Clinical Oncology, Congreso Anual de la Sociedad Americana de Oncología Médica (American Society of Clinical Oncology, Congreso Anual de la Sociedad Americana de Oncología Médica (ASCO). Este congreso está considerado como el más importante del mundo en su especialidad.ASCO). Este congreso está considerado como el más importante del mundo en su especialidad.ASCO
A lo largo de estos cinco días, se presentaron los últimos avances en la investigación, la prevención y el tratamiento del cáncer en sus múltiples facetas, destacando un estudio sobre un régimen experimental para los tumores sólidos que ha demostrado tener potencial terapéutico, y los resultados finales de un ensayo de fase I de un régimen prometedor contra el cáncer de páncreas. A destacar también fue el estudio realizado por los doctores Goss, Ingle, Ales-Martínez et al en el que se comprobó que el exemestano puede reducir en un 65% et al en el que se comprobó que el exemestano puede reducir en un 65% et alel riesgo de desarrollar cáncer de mama en las mujeres postmenopáusicas, con unos efectos secundarios mínimos.
http://chicago2011.asco.org/Goss PE, Ingle JN, Ales-Martinez J et al. J Clin Oncol 29: 2011 (suppl; abstr LBA504)
Colitis ulcerosaLa colitis ulcerosa (UC) es una enfermedad crónica inflamatoria del intestino cuya etiología sigue siendo desconocida. Su patogénesis involucra la ruptura de la homeostasis presente en la mucosa intestinal, debida probablemente a factores genéticos que son los causantes de una comunicación errónea entre la flora intestinal y el sistema inmunitario allí presente. Diferentes citoquinas, que representan unos componentes esenciales de la respuesta inmunológica, han sido observadas en las fases inflamatorias activas y crónicas de la UC. Estudios recientes han identificado algunos sistemas de citoquinas que se encuentran muy activados en las mucosas intestinales de los pacientes con UC, y han relacionado su presencia con la patogénesis de esta enfermedad. La presencia de IL-13, TNF e IL-33 y sus receptores en la UC ha sido observada en numerosos pacientes, sugiriendo por un lado la posibilidad de utilizar estos biomarcadores proteómicos para comprobar el estado de la enfermedad, y por otro lado se puede pensar en la posibilidad de desarrollar nuevos fármacos que específicamente podrán neutralizar los mecanismos inflamatorios presentes en la UC.
Bamias G, Kaltsa G, Ladas SD. Discov Med 2011; 11:459-67.
Identificados 6 genes que regulan la progresión del VIHEl equipo de científicos del Instituto de Investigación del SIDA(IrsiCaixa) y del Hospital Universitario de Lausanne (Suiza) ha desarrollado una investigación que analiza el genoma de pacientes infectados por el virus de la inmunodeficiencia humana (VIH). Según este estudio, los genes CASP1, CD38, LAG3, TNFSF13B, SOCS1, y EEF1D están implicados en la EEF1D están implicados en la EEF1Dprogresión rápida de la infección. Además, se compararon estos resultados genéticos con modelos paralelos de infección del virus de inmunodeficiencia de los simios (VIS) en primates no humanos. Este trabajo abre una nueva línea de investigación dentro del proyecto de desarrollo de la vacuna del SIDA HIVACAT. SIDA HIVACAT. SIDA
Rotger M, Dalmau J, Rauch A et al. J Clin Invest 2011; 121:2391-400.
Exploración clínica de las imágenes de depósito de amiloide en la enfermedad de AlzheimerLa hipótesis de que el depósito de amiloide es la principal causa de la enfermedad de Alzheimer (EA) es apoyada por los resultados en modelos animales transgénicos y constituye la base de los ensayos clínicos de agentes anti-amiloide. Según esta teoría, la deposición de amiloide provoca graves daños a las neuronas muchos años antes de la aparición de la demencia a través de una cadena de efectos posteriores. Esta hipótesis, sin embargo, todavía no se ha comprobado directamente en seres humanos debido a la posibilidad muy limitada de diagnóstico de la deposición de amiloide in vivo, lo cual hasta hace poco requería una biopsia cerebral o estudios PET con ciclotrón in situ y de laboratorio de radioquímica. in situ y de laboratorio de radioquímica. in situPor otra parte, un diagnóstico clínico de EA exige que los pacientes ya presenten demencia, y en ese momento cualquier tratamiento eficaz para reducir la deposición de amiloide será probablemente demasiado tarde.
Recientemente se han desarrollado los marcadores radioactivos 18F de depósitos de amiloide flutemetamol, florbetapir y florbetaben para el PET; actualmente están en fase de ensayos clínicos para determinar si pueden ser utilizados para visualizar con precisión imágenes de proteína amiloide fibrilar y para distinguir los pacientes con EA de controles normales y aquellos con otras enfermedades que causan demencia. También podrían utilizarse como biomarcadores para predecir el desarrollo de la EA antes de la aparición de la demencia y para evaluar el efecto de la terapia anti-amiloide. Resultados negativos de amiloide indican ausencia de EA con un alto nivel de precisión, pero en ancianos sanos podría haber resultados positivos de amiloide, por lo que su valor predictivo de forma aislada no está muy claro. En un estudio en fase 3 con florbetapir se ha visto una estrecha relación entre imágenes amiloide in vivo y hallazgos histopatológicos in vivo y hallazgos histopatológicos in vivo post-mortem.
¿Adónde llevan estos resultados preliminares? Los estudios terapéuticos de agentes anti-amiloide que incluyen marcadores de amiloide como marcadores biológicos se espera sean útiles para el desarrollo de fármacos y para aclarar la relación entre la eliminación de amiloide y los efectos clínicos. Una vez que los marcadores 18F estén disponibles para uso clínico, serán necesarios grandes estudios longitudinales para aclarar su poder diagnóstico y pronóstico en relación con la edad, factores de riesgo, y los subtipos de la enfermedad de Alzheimer. En última instancia, estos marcadores se espera que aclaren el papel fisiopatológico de amiloide en la EA y que contribuyan al desarrollo de nuevos tratamientos.
Herholz K, Ebmeier K. Lancet Neurol. 2011 Jul;10(7):667-70.

noticias EuroEspes
El Dr. Cacabelos pronunció la conferencia inaugural del VII Congreso Internacional de Nutrición, Alimentación y DietéticaEl Director General del Centro de Investigación Biomédica EuroEspes participó en la inauguración de la séptima edición del Congreso Internacional de Nutrición, Alimentación y Dietética, con una conferencia titulada “Nutrigenómica y farmacogenómica: de la teoría a la práctica” dentro de las XV Jornadas Nacionales de Nutrición Práctica celebradas durante los días 30 y 31 de marzo y 1 de abril en la Facultad de Medicina de la Universidad Complutense de Madrid.
Programa para la detección precoz de la enfermedad de AlzheimerEl doctor Álvarez, Jefe del Departamento de Farmacología Clínica y Experimental del Centro Médico EuroEspes, explicó durante unas jornadas dedicadas a “La atención al paciente con daño cerebral” celebradas en el Balneario al paciente con daño cerebral” celebradas en el Balneario al paciente con daño cerebralde Guitiriz (Lugo) el nuevo programa puesto en marcha por el CIBE para la detección y el tratamiento precoz de la enfermedad de Alzheimer, dirigido a personas con problemas de memoria: PROMEMORIA.
Ramón Cacabelos destacó la importante presencia de cuatro categorías de genes en los trastornos del Sistema Nervioso CentralEl doctor Ramón Cacabelos, presidente de EuroEspes, participó en el simposio: “Medicina personalizada en trastornos psiquiátricos: Nuevos caminos hacia la farmacogenómica”, en el transcurso del X Congreso Mundial de Psiquiatría Biológica, celebrado en Praga en el mes de junio pasado, con la conferencia “Genómica y farmacogenómica funcional de los trastornos del Sistema Nervioso Central”, donde destacó la importancia de cuatro Nervioso Central”, donde destacó la importancia de cuatro Nervioso Centralcategorías de genes en los trastornos que afectan al Sistema Nervioso Central: los genes pleiotrópicos, genes de trastornos específicos, genes involucrados en el metabolismo de fármacos y los genes asociados con el mecanismo de acción de medicamentos específicos.
EuroEspes Biotecnología participa en el proyecto MAREXLa filial biotecnológica de EuroEspes, Ebiotec, participa en el proyecto comunitario MAREX (Exploración de los recursos marinos en busca de compuestos bioactivos: del descubrimiento a la producción sostenible y las aplicaciones industriales), cuyo objetivo es la búsqueda aplicaciones industriales), cuyo objetivo es la búsqueda aplicaciones industrialesde sustancias biológicamente activas en organismos marinos que se puedan utilizar como base de productos farmacéuticos. Esta iniciativa ha recibido una concesión de 6 millones de euros dentro del contexto “Alimentos, agricultura y pesca, y biotecnología”, perteneciente al VII Programa Marco para recopilar, aislar y clasificar organismos marinos como anémonas, tunicados y micro y macroalgas de los mares Mediterráneo, Báltico y Arábigo y de los océanos Atlántico, Pacífico e Índico. El responsable del proyecto en Ebiotec es el Dr. Valter Lombardi, jefe del Departamento de Biotecnología Aplicada.
EuroEspes firmó un convenio de colaboración con el Círculo de Empresarios de GaliciaA finales de mayo el Centro Médico EuroEspes y el Círculo de Empresarios de Galicia-Club Financiero de Vigo (CFV) firmaron un convenio de colaboración por el que el CIBE prestará sus servicios médicos, sociales y de investigación a los socios del CFV.
Se harán reconocimientos médicos con carácter predictivo y preventivo mediante el desarrollo de los protocolos recogidos en el Programa de Prevención del Riesgo Cerebral y Genético.
EuroEspes participa en el Acto de Clausura del Programa de Formación Continuada para FarmacéuticosEl presidente de EuroEspes, el doctor Ramón Cacabelos, dio la conferencia de clausura del Programa de Formación Continuada para Farmacéuticos para el curso 2010/2011, que se celebró a principios de junio en Barcelona. Dicha conferencia llevaba por título: “Medicina Genómica y Personalización del Tratamiento Farmacológico”.
En esta ponencia, el presidente de EuroEspes relató la experiencia de EuroEspes, centro pionero en farmacogenómica y responsable del lanzamiento de un documento genético único para personalizar el tratamiento farmacológico: la Tarjeta Farmacogenética, que se comercializa en todo el mundo y cuyo objetivo es dar solución a muchos de los problemas causados por una prescripción generalizada con criterios diferenciales adaptados a las características genómicas individuales. La Tarjeta Farmacogenética EuroEspes lleva el perfil genómico de su titular, para adecuar el tipo de medicación y su dosis al perfil del mismo.
EuroEspes cotiza en el Mercado Alternativo Bursátil desde el día 16 de febreroEl MAB ya tiene un nuevo inquilino: EuroEspes. Debutó en el Mercado Alternativo Bursátil el pasado 16 de febrero. El precio de salida de sus títulos se situó en los 2,80 euros, con un free float un poco superior al 2% de su capital. La capitalización float un poco superior al 2% de su capital. La capitalización floatde la compañía a precio de salida superó los 15 millones de euros, lo cual situó el valor de la misma en un rango muy atractivo.
EuroEspes se convirtió así en la decimotercera entidad en cotizar en este mercado diseñado para empresas en expansión. Un debut que coincidió además con el vigésimo aniversario de la fundación de EuroEspes, motivo por el cual se contó en la inauguración con el apoyo del político gallego Manuel Fraga, que inauguró el Centro de Investigación Biomédica EuroEspes el 15 de Diciembre de 1995. Al acto de ingreso en el MAB asistieron el Presidente del MAB, D. Antonio Giralt Serra, el Director General, D. Jesús González Nieto-Márquez, Directivos, Accionistas, Personal de EuroEspes y un selecto colectivo de pacientes y personas vinculadas a la actividad médica y científica del grupo. Tras el tradicional toque de campana, el presidente del Grupo EuroEspes, Ramón Cacabelos, explicó que el objetivo de entrar en el MAB es “participar en un nuevo proyecto” (como es este mercado), y “continuar con el proceso de internacionalización y crecimiento, que lleve a la compañía en los próximos años a situarse en la élite de la biotecnología europea”.
EuroEspes consolida su proceso de reestructuración La Junta General de Accionistas de EuroEspes, celebrada el día 22 de junio en la sede de la compañía en Bergondo (A Coruña), aprobó las cuentas del ejercicio pasado, que reflejan un incremento del beneficio neto del Grupo del 169% en relación al año 2009.
Con un importe neto de la cifra de negocios situado en los 3,33 millones de euros, los resultados suponen un aumento de más del 5% respecto al año anterior.
Estas cifras son la consecuencia de una política de control de gasto riguroso llevada a cabo por EuroEspes, una estrategia que ha permitido absorber el proceso de reestructuración diseñado en el ejercicio 2008 y ha ayudado a generar, si cabe, una mayor fortaleza en el balance.
La empresa pretende seguir reforzando su posición financiera en los próximos ejercicios, a pesar de la situación de crisis que atraviesa la economía mundial.
EuroEspes se convirtió así en la decimotercera entidad


lgo grave está pasando en la so-ciedad, cuando para el común de los mortales sus dirigentes políticos se convierten en un
problema prevalente. La corrupción, el descrédito, la mediocridad, la farsa conti-nua, la escenografía de la confrontación, el maniqueísmo interesado, la judicialización de la vida pública, la falta de compromiso, la indolencia ante las necesidades de la po-blación, la infestación mediática, la incapa-cidad para generar crecimiento y prosperi-dad, la pleitesía al poder oculto del dinero o el esperpento ideológico de los extremos, son elementos suficientes para que una so-ciedad informada interiorice el desencanto y surjan los apáticos convencidos, los anti-sistema, los indignados del movimiento 15-M, los parados por interés (una ofensa a los parados por obligación), los oportunistas, los especuladores, los voyeuristas políticos, los opinadores a sueldo y los redentores de la patria, todos carentes de atractivo para la sociedad civil.
En mis años de carrera en la Facultad de Medicina de la Universidad de Oviedo, con Franco moribundo y la predemocracia en germen, un ilustre amigo mío, Notario de Guipúzcoa, me dijo que “la política es el arte de la mentira”. A aquella generación ansiosa de libertad, sedienta de participación en la vida pública, nos costaba trabajo creer que aquello que perseguíamos, bajo el velo de una democracia incipiente, pudiera ser mentira. Tres décadas después, con bastantes miles de kilómetros a cuestas en diferentes países, y el panorama político del ruedo ibérico actual, me asalta la misma incertidumbre que pu-lula en la mente de muchos ciudadanos. El
No ponga su palabra en boca de quien no sabe hablar ni su pluma en manos de quien no sabe escribir
descontento en política no es nuevo; quizá haya sido un motor evolutivo en los avatares de la historia; un acicate para el cambio so-cial, que se agudiza en momentos de crisis. Pensadores y políticos de oficio nos han de-jado guindas embalsamadas que acreditan la mala reputación de la casta dirigente. Sir Winston Churchill decía que “un buen polí-tico es aquel que, tras haber sido comprado, sigue siendo comprable”. Para Noel Clarasó, “la política es el arte de obtener dinero de los ricos y votos de los pobres, con el fin de proteger a los unos de los otros”. El propio Nikita Kruschev afirmaba que “los políticos son todos iguales; prometen construir puen-tes incluso donde no hay ríos”. En la misma dirección apuntaba Gustave Le Bon: “Uno de los hábitos más peligrosos de los políti-cos mediocres es prometer lo que saben que no pueden cumplir”. El gran Montesquieu tenía claro que “la corrupción raras veces co-mienza por el pueblo”. Napoleón solía decir en sus círculos más próximos que “la política es un lupanar en el que las putas son bas-tante feas”. William M. Ramsay aconsejaba: “vota a quien menos te prometa; será quien menos te decepcione”. El sesudo Bertrand Russell llegó a decir que “los científicos se esfuerzan por hacer posible lo imposible; los políticos, por hacer lo posible imposible”. Al extremista Friedrich Nietzsche no le sonro-jó decir que “la política es un trabajo para cerebros mediocres”. Y el sarcástico Geor-ge Bernard Shaw nos dejó la ironía de que “no es cierto que el poder corrompa, es que hay políticos que corrompen al poder”. Por lo tanto, el hastío y la decepción vienen de lejos. Esta podredumbre es añeja, como la sentencia cáustica de Jean-Lucien Arreat: “Si en la república de las plantas existiera el su-fragio universal, las ortigas desterrarían a las rosas y a los lirios”.
A falta de un nuevo orden social, de un nue-vo modelo de gestión de lo público, y de un sistema económico equilibrado, nos queda el recurso a la reflexión y la apelación a una regeneración moral de los líderes políticos, con la denuncia responsable de quien otor-ga el poder y autoriza al gobierno a través de los comicios electorales. De ahí la perti-nencia de estos “consejos a un presidente”,
96
Pharmacogenomics of Metabolic Disease
Consejos a un Presidente
aún a sabiendas de que quien realmente los necesita los despreciará y hará caso omiso, porque todo tonto bautizado por las urnas pierde repentinamente la capacidad de es-cuchar y ver, le asalta un síndrome de locua-cidad verborreica y una esquizotimia deísta que le lleva a creerse lo que no es y a con-vencerse de que lo que dice tiene sentido, aunque sea una estupidez. Esta enfermedad del homo politicus de Aristóteles represen-ta una grave transgresión del código moral que debe guiar a todo buen político.
Por lo tanto, Presidente (en proyecto o en decadencia, o político santificado por las urnas en el altar del poder), permítame de-dicarle unas líneas con el ánimo de poner en su conocimiento lo que los mortales, sin cetro ni corona, esperamos de nuestros líde-res, de aquellos en cuyas manos depositamos lo que es nuestro.
Lo primero que tiene que tener un político es conciencia de servicio. Está donde está para servir a quien le ha votado y a quien no le ha votado. Ud. es presidente de todos, no sólo de sus acólitos. Ud. nos representa a todos y de Ud. esperamos espíritu de ser-vicio, ejemplaridad, honestidad, austeridad, liderazgo y eficacia. Enrique Tierno Galván dijo que “el triunfo político es la suma del sentido común y la capacidad de liderazgo”. La ejemplaridad emana de un profundo sentido moral. Immanuel Kant decía: “To-dos somos iguales ante el deber moral”. Para Juvenal “el primer castigo del culpable es que no podrá jamás ser absuelto por el tribunal de su conciencia” (si la tiene). Se-gún Blaise Pascal “la conciencia es el me-jor libro de moral que tenemos” (revise su biblioteca, Presidente). Ud. ha elegido un camino que le aboca a luchar por una socie-dad mejor. El objetivo es loable; el destino incierto. Está sometido al capricho de mu-chas vicisitudes, variables incontrolables y factores ajenos a su capacidad de maniobra, aunque el elemento principal es Ud. mismo y el digno ejercicio de su cargo. Asumida la idiosincrasia de la condición humana y la perversión de los maniqueísmos políticos, un cambio social exige una elevación de es-tándares, principalmente en el plano moral,
A
por Ramón [email protected]
Res Sacra Consilium (el buen consejo no tiene precio)

Hoy el mundo no es de izquierdas ni de
derechas, sino de los que tienen de más y de
los que tienen menos; un desequilibrio estable
que pocos quieren cambiar
Julio 2011 97
Consejos a un Presidenteen la educación, en el equilibrio de las rela-en la educación, en el equilibrio de las rela-en la educación, en el equilibrio de las relaciones humanas y en el saneamiento de las estructuras de poder para que tenga cabida un nuevo orden programático. “Todo está perdido cuando los malos sirven de ejemplo y los buenos de mofa”, decía Demócrates. Tenga cuidado con quien se alía, a quien vende su alma, con quien fabula, en quien deposita su confianza, con quien duerme, a quien designa para puestos clave, a quien contrata para asesorarle. Los gobernantes más ilustres han tenido círculos pequeños y horizontes externos amplios. Cúidese de los que dicen “amén” a todo y se pasan la vida alimentando su egolatría, para comprar sus favores; serán los primeros en apuñalarle por la espalda; serán los primeros en aban-donarle cuando no puedan disfrutar de sus victorias. Nunca les crea del todo. A ellos no les interesa que Ud. encuentre nuevas com-pañías, escuche nuevas voces, le nutran de nuevas ideas o le enciendan luces de futuro. Muchas cosas y personas que a Ud. podrían beneficiarle serían un atentado contra los que han montado la tienda a la sombra de su figura. Agudice el sentido ante los ecos le-janos. Escuche a los distantes. La equidistan-cia política e ideológica es como un telesco-pio con gran angular que permite ver lo que no quieren mirar sus termitas. Vea sin mirar y escuche sin oír. Los equidistantes debieran ser como un fulcro en su balanza, como un referente de equidad. No desprecie a los neutros, a los que no piden nada, a los que no rinden pleitesía, a los que respetan el silencio, a los que tienen sentido de estado creando futuro con su trabajo, a los que no se esconden en las ideologías, a los que cul-tivan la simiente de la sabiduría (aunque no piensen como Ud., aunque le aconsejen que los ignore). Le serán más útiles que los dog-los ignore). Le serán más útiles que los dog-los ignore). Le serán más útiles que los dogmáticos babosos que viven del favor ajeno y de la concupiscencia. Rodéese de los mede la concupiscencia. Rodéese de los me-jores. Es imposible que una sola persona jores. Es imposible que una sola persona sea capaz de abarcar sea capaz de abarcar todos los frentes de todos los frentes de la vida pública o enla vida pública o en-tender con suficiencia tender con suficiencia los diversos problemas los diversos problemas que afectan a la socieque afectan a la socie-dad. Necesita tener a su dad. Necesita tener a su lado el complemento lado el complemento perfecto, el símbolo de perfecto, el símbolo de la fidelidad, las mejores la fidelidad, las mejores cabezas, los más adustos, cabezas, los más adustos, los más innovadores, los los más innovadores, los menos influenciables menos influenciables ante los cantos de sirena ante los cantos de sirena de la tentación, los más vade la tentación, los más va-de la tentación, los más va-de la tentación, los más valientes, los que se visten por la lientes, los que se visten por la mañana con el traje del deber y mañana con el traje del deber y se ponen por la noche el pijama se ponen por la noche el pijama
de la responsabilidad, los que dan testimo-nio con su vida y no con su lengua. En su corte (o cortijo) se infiltrarán oportunistas y aprovechados, supervivientes camaleónicos que han flirteado con sus predecesores en beneficio propio. Esté prevenido frente a los que sirven (o han servido) a varios señores. En las alcantarillas de palacio se esconden harpías que sólo aspiran a verle pasar, a que llegue el siguiente, instalados en el inmovi-lismo de lo vitalicio. Burócratas y tecnócratas son máquinas expendedoras de servicios en el aparato de la administración para quie-nes Ud. es sólo un inquilino temporal. De-cía Manuel Fraga Iribarne que “en política todas las victorias son efímeras, y todas las derrotas son provisionales”; pero el subsuelo del poder está sembrado de minas colocadas por los permanentes, por los que tiran la pie-dra y esconden la mano, por los que ocultan su rostro tras las cortinas del cargo, sin expo-nerse, por los que compran voluntades, por los que engrasan la máquina de la locomo-tora tributaria, por los que están acostum-brados a mandar a la infantería ladera abajo mientras ellos se escudan en guarniciones de la colina; todos ellos buscarán en Ud. un caudillo utilizable o un caudillo mutilado. La frescura está en el exterior, Presidente; no en las estancias lúgubres de la corte, donde conspiran aquellos a los que nunca debiera dar crédito para no caer como una mosca en su telaraña. Mire hacia afuera. El éxito está en el horizonte de las ideas. “No pensar más que en sí mismo y en el presente es un error en política”, decía Jean de La Bruyère. Un gran hombre de estado, como Otto von Bis-marck, fue quien delineó la frontera entre el político y el estadista: “El político piensa en la próxima elección; el estadista, en la próxima generación”. Los inútiles a los que alimenta con su victoria electoral no pueden
ayudarle a pensar como un hombre de ayudarle a pensar como un hombre de estado porque ellos viven la preestado porque ellos viven la pre-
ocupación del momento, ocupación del momento, centrados en su centrados en su
propia superpropia super-propia super-propia supervivencia, con vivencia, con la mirada fija la mirada fija en la próxima en la próxima confrontación confrontación electoral. Para electoral. Para ellos el futuro ellos el futuro es sólo presenes sólo presen-te. Cuando Ud. te. Cuando Ud. se vaya, ellos dese vaya, ellos de-jarán de existir y jarán de existir y nadie les echará nadie les echará de menos; pero de menos; pero a Ud. le juzgará a Ud. le juzgará
la historia, para bien o la historia, para bien o para mal. Abra la ventana y respara mal. Abra la ventana y res-
pire aire fresco no infectado por la ideología o el vicio de la conveniencia. Si desea que la historia le respete, salga de la ciénaga en la que se instala el poder. El líder está conde-nado a la soledad si quiere conservar su pu-reza; tiene que saber nadar en mar abierto, cabalgar por el campo verde ajeno a la con-taminación orgánica, beber del manantial de la sabiduría, y descansar sobre el pecho de las ninfas impolutas.
Un hombre de honor es aquel al que reco-nocen por el cumplimiento de su palabra. Decía Aldous Huxley que “cuanto más si-niestros son los designios de un político, más estentórea se hace la nobleza de su len-guaje”. Tome ejemplo de Abraham Lincoln, entre cuyos pensamientos había uno que rezaba: “Hay momentos en la vida de todo político en que lo mejor que puede hacerse es no despegar los labios”. La gente llana, la gente de buen corazón, detesta oírle hablar mal de sus adversarios, hacer promesas in-cumplibles, profetizar éxitos de calamidad, abusar de la desgracia para obtener venta-abusar de la desgracia para obtener venta-abusar de la desgracia para obtener ventajas, primar el descompromiso, justificar la inconsecuencia, subirse al potro del verbo para esconder su debilidad; lo que la gente quiere es que hable el líder, el ser humano
comprometido, el capaz de penetrar en el corazón de sus paisanos para explicarles sus ideas, su forma de hacer frente a la realidad económica, social, laboral, educativa, sanita-económica, social, laboral, educativa, sanita-económica, social, laboral, educativa, sanitaria, industrial, judicial e internacional de la gran aldea global en la que vivimos. Decía Dwight W. Morrow que “si un partido políti-co se atribuye el mérito de la lluvia, no debe extrañarse de que sus adversarios lo hagan culpable de la sequía”. Alexander Pope afir-culpable de la sequía”. Alexander Pope afir-culpable de la sequía”. Alexander Pope afirmaba que “un partido es la locura de mu-chos en beneficio de unos pocos”. Pero Ud. ya es Presidente, mucho más que un líder sectario; ya no toca hostigar, remover el lodo de la charca preelectoral, enardecer

Seis honrados servidores me enseñaron cuanto sé; sus nombres son cómo, cuándo, dónde, qué, quién y por qué
Rudyard Kipling
98
Consejos a un Presidente
a sus huestes. A Ud. le corresponde ahora apacentar a un rebaño que desea confiar en su pastor. Tiene que saber ahuyentar a los lobos y a las alimañas. Tiene que saber conducir, proteger, administrar, instruir, y ejemplarizar. Todavía quedan ingenuos que, en el silencio de la noche, en una conversa-en el silencio de la noche, en una conversa-en el silencio de la noche, en una conversación fraternal, en el secreto de una oración, o en el mensaje a un hijo, piensan que de-trás de un político es posible la existencia de un hombre honrado, aunque al segundo convengan con Max Weber que “quien hace política pacta con los poderes diabólicos que acechan a todo poder”.
Al estilo de Rudyard Kipling, trate al éxito y al fracaso de igual forma, y recuerde aquello de: “Seis honrados servidores me enseñaron cuanto sé; sus nombres son cómo, cuándo, dónde, qué, quién y por qué”. Valore a las personas por lo que son más que por lo que tienen o por lo que le han dado. Un ciuda-tienen o por lo que le han dado. Un ciuda-tienen o por lo que le han dado. Un ciudadano no es un voto; es un ser humano que deposita su confianza en Ud., que paga su sueldo y el de sus colaboradores con sus im-puestos, que a cambio le pide responsabili-dad y eficacia. Un país no es un cardumen de peces asustados a los que se les bombar-de peces asustados a los que se les bombar-de peces asustados a los que se les bombardea con consignas para que caigan en la red de sus promesas. Un país es el mar; y Ud., como mucho, es un simple pescador que vive de lo que el mar quiera darle.
Adórnese de virtud para cosechar bondad, comprensión y respaldo a sus decisiones. Mantenga la confianza siempre un peldaño por encima de la suspicacia. No ponga su pa-por encima de la suspicacia. No ponga su pa-por encima de la suspicacia. No ponga su palabra en boca de quien no sabe hablar ni su pluma en manos de quien no sabe escribir. Predique con el ejemplo y cobre con la mis-ma moneda con la que paga. No use su po-sición de privilegio para ofender, desterrar, arruinar, destruir o exterminar. No prohíba, Presidente; eduque, conciencie, sensibilice; pero no prohíba. Las políticas punitivas son propias de los que para gobernar necesitan del castigo y la fuerza con el fin de imponer sus ideas. Controle a sus perros para que
no muerdan la mano de quien les da de co-mer. No judicialice la política ni politice la justicia; ambos vicios generan descrédito y conducen al ostracismo (ganado a pulso por jueces y políticos). No caiga en la tentación de llevar a los tribunales a sus enemigos o a aquellos que no comulgan con sus precep-tos, para desacreditarlos y destruirlos; tarde o temprano le pasarán por el filo de la mis-ma espada.
Un Presidente tiene que ser valiente, firme y convincente en sus decisiones, sin renun-ciar nunca a la flexibilidad de la inteligencia. Necesitamos dar contenido y credibilidad a esta democracia pusilánime y sindicada, en la que se educa sin autoridad, en la que se convive y cohabita con el terrorismo, en la que no se vota a las personas sino a la mas-carada de los partidos, en la que las mino-rías condicionan decisiones de estado con fórmulas de chantaje, en la que la justicia practica una independencia controlada (y cómplice), en la que todos los ciudadanos no son iguales ni ante la ley ni ante las ins-tituciones, en la que se busca el trabajo fácil y descomprometido, en la que se prefiere la beneficencia al premio por el esfuerzo, en la que cualquiera puede acusar sin pruebas, en la que se practican juicios mediáticos pa-en la que se practican juicios mediáticos pa-en la que se practican juicios mediáticos paralelos a diario para dañar la reputación del adversario, en la que se vive en bandos, en la que se desentierra a los muertos en función del color de su boina, en la que la confrontación ha suplantado a la cooperación, en la que 17 reinos de Taifas han despedazado la unidad nacional.
Regenerar la vida política equivale a pretender modificar la condición humana. Nos enfrentamos a una utopía. Hoy el mundo no es de izquierdas ni de derechas, sino de los que tienen de más y de los que tienen menos; un desequilibrio estable que pocos quieren cambiar. Sea utópico hasta donde la razón y la equidad se lo permitan. Muévase al filo de lo imposible para conseguir lo posible. Alíese con el conocimiento cuando tenga que diseñar la estrategia. Súbase al tren del progreso, en el que viajan los que creen que una sociedad avanza cuando el bienestar compromete a la mayoría. Sacúdase los complejos para cambiar la historia, si desea que algún día la historia le respete y reconozca.
El poder en exceso intoxica, Presidente. Lo importante de su historia personal no es como empieza sino como acaba. Lo normal es que emerja con laureles y acabe con cenizas, como ha ocurrido con todos los presidentes de la Hispania democrática, in
misericorde con todos sus líderes. No olvide que los mortales sufren amnesia para pagar el éxito ajeno y tienen una memoria prodi-giosa para cobrar la venganza. El pobre está curtido en la paciencia, en el sufrimiento, en la espera. La dureza de la vida nos ha pulido las aristas y nos ha hecho tan simples que creemos que a todo cerdo le llega su San Martín. Cuide su siembra para no cosechar desprecio y olvido cuando empiece la caída. Y en ese momento, sepa retirarse a tiempo, con honor, con dignidad, con la elegancia de los espíritus grandes. No se aferre al más-til de un barco que se hunde. No se arrastre entre los escombros de un edificio derruido. Que no se le vea buscando residuos en los contenedores de basura.
Es obvio que la Sofocracia de Platón (el go-bierno de los sabios) no está de moda; en realidad, nunca lo estuvo, salvo en tiempos de Pericles, el primer ciudadano de Atenas, como lo definía Tucídides; y aún el propio Pericles, quizá el más honorable gobernan-te de la meritoria democracia griega, tuvo que sufrir la ingratitud de su tiempo. El gobernante y el genio viven siempre bajo la espada de Damocles; no se puede satisfacer a todos; seguidores y detractores son bandos separados por una línea visceral; la gloria es efímera, como la flor de sakura en la filosoralelos a diario para dañar la reputación del
adversario, en la que se vive en bandos, en la que se desentierra a los muertos en función del color de su boina, en la que la confron-tación ha suplantado a la cooperación, en la que 17 reinos de Taifas han despedazado la
Regenerar la vida política equivale a preten-der modificar la condición humana. Nos enfrentamos a una utopía. Hoy el mundo no es de izquierdas ni de derechas, sino de los que tienen de más y de los que tienen menos; un desequilibrio estable que pocos quieren cambiar. Sea utópico hasta donde la razón y la equidad se lo permitan. Mué-vase al filo de lo imposible para conseguir lo posible. Alíese con el conocimiento cuan-do tenga que diseñar la estrategia. Súbase al tren del progreso, en el que viajan los que creen que una sociedad avanza cuando el bienestar compromete a la mayoría. Sacúda-bienestar compromete a la mayoría. Sacúda-bienestar compromete a la mayoría. Sacúdase los complejos para cambiar la historia, si desea que algún día la historia le respete y
El poder en exceso intoxica, Presidente. Lo importante de su historia personal no es como empieza sino como acaba. Lo nor-es como empieza sino como acaba. Lo nor-es como empieza sino como acaba. Lo normal es que emerja con laureles y acabe con cenizas, como ha ocurrido con todos los presidentes de la Hispania democrática, in-
efímera, como la flor de sakura en la filoso-fía samurái. La única recompensa del líder puro es la tranquilidad de su conciencia; el reconocimiento lo dará el tiempo, cuando el coro de los eunucos entonen odas a su fé-retro o cuando algún anónimo agradecido ponga flores sobre la lápida de su tumba.
Antes de que todo esto ocurra, llegará el otoño de la vida. Espero que con la caída de la hoja, libre de complejos megalómanos, en un arrebato de humildad, despojado de todo hábito regio, tenga ocasión de valorar el pensamiento noble que vuela en esta brisa distante. Pero ya será tarde…

LIDERANDO A TRAVÉS DEL CONOCIMIENTOEDUCACIÓN - CIENCIA - PROGRESO - FUTURO
FORMACIÓN CONTINUADAFUNDACIÓN EUROESPES
Fundación EuroEspes - EuroEspes Foundation - Santa Marta de Babío s/n, 15165 Bergondo, Coruña, Spain[T] +34 981 780 505 [F] +34 981 780 511 www.fundacioneuroespes.org - [email protected]
EUROESPES
FUNDACIÓN

THE ESSENTIAL GUIDE TO PHARMACOGENOMICS FOR MEDICAL PRACTICE
Coming in 2011 !!!More Information: www.wgpgx.com
THE WORLD GUIDEFOR DRUG USE AND PHARMACOGENOMICS


















